
Whole Exome Sequencing in Multi-Incident Families Identifies Novel Candidate Genes for Multiple Sclerosis

 , , ,
, , ,
Abstract
1. Introduction
2. Results
2.1. Participants
2.2. Whole Exome Sequencing and Analysis
2.3. Selection of Genetic Variants
2.4. Validation of Genetic Variants and Co-Segregation Analyses
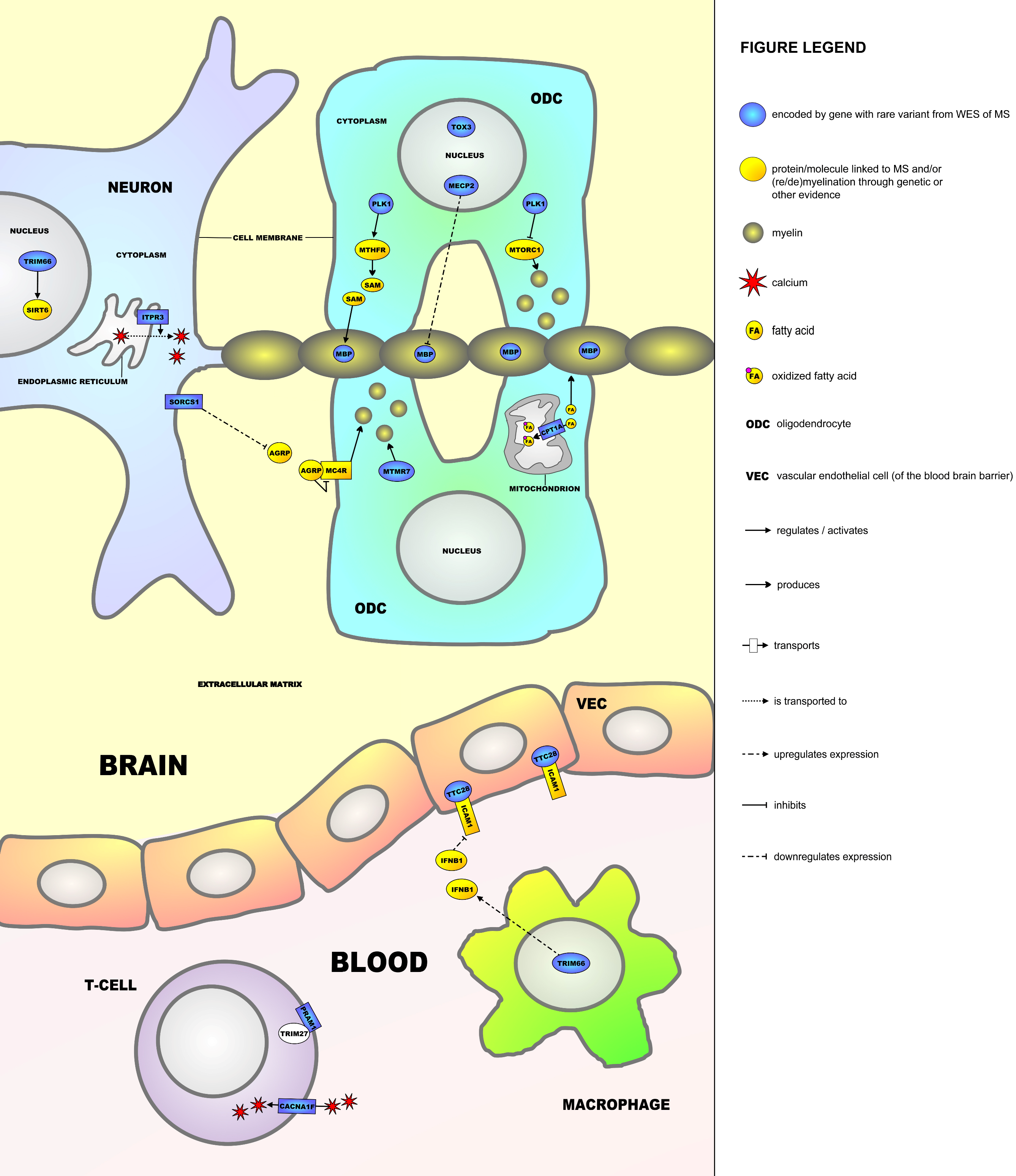
2.5. Molecular Framework
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. Whole Exome Sequencing and Analysis
4.3. Selection of Candidate Single Nucleotide Variants
4.4. Validation of SNVs
4.5. Construction of Molecular Framework
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of multiple sclerosis 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 269–285. [Google Scholar] [CrossRef]
- McFarland, H.F.; Martin, R. Multiple sclerosis: A complicated picture of autoimmunity. Nat. Immunol. 2007, 8, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, B.; Archelos, J.J.; Hartung, H.P. New concepts in the immunopathogenesis of multiple sclerosis. Nat. Rev. Neurosci. 2002, 3, 291–301. [Google Scholar] [CrossRef]
- Ciccarelli, O.; Barkhof, F.; Bodini, B.; De Stefano, N.; Golay, X.; Nicolay, K.; Pelletier, D.; Pouwels, P.J.; Smith, S.A.; Wheeler-Kingshott, C.A.; et al. Pathogenesis of multiple sclerosis: Insights from molecular and metabolic imaging. Lancet Neurol. 2014, 13, 807–822. [Google Scholar] [CrossRef]
- Bogie, J.F.; Stinissen, P.; Hendriks, J.J. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol. 2014, 128, 191–213. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.S.; Lees, J.G.; Moalem-Taylor, G. The contribution of immune and glial cell types in experimental autoimmune encephalomyelitis and multiple sclerosis. Mult. Scler. Int. 2014, 2014, 285245. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Ysrraelit, M.C. Multiple Sclerosis and Aging: The Dynamics of Demyelination and Remyelination. ASN Neuro 2022, 14, 17590914221118502. [Google Scholar] [CrossRef]
- Chu, F.; Shi, M.; Zheng, C.; Shen, D.; Zhu, J.; Zheng, X.; Cui, L. The roles of macrophages and microglia in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2018, 318, 1–7. [Google Scholar] [CrossRef]
- Miron, V.E.; Boyd, A.; Zhao, J.-W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef]
- Zettl, U.K.; Stüve, O.; Patejdl, R. Immune-mediated CNS diseases: A review on nosological classification and clinical features. Autoimmun. Rev. 2012, 11, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Karussis, D. The diagnosis of multiple sclerosis and the various related demyelinating syndromes: A critical review. J. Autoimmun. 2014, 48–49, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Goodin, D.S.; Khankhanian, P.; Gourraud, P.A.; Vince, N. The nature of genetic and environmental susceptibility to multiple sclerosis. PLoS ONE 2021, 16, e0246157. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Amato, M.P.; Derfuss, T.; Hemmer, B.; Liblau, R.; Montalban, X.; Soelberg Sørensen, P.; Miller, D.H. Environmental modifiable risk factors for multiple sclerosis: Report from the 2016 ECTRIMS focused workshop. Mult. Scler. J. 2018, 24, 590–603. [Google Scholar] [CrossRef]
- Orton, S.M.; Herrera, B.M.; Yee, I.M.; Valdar, W.; Ramagopalan, S.V.; Sadovnick, A.D.; Ebers, G.C. Sex ratio of multiple sclerosis in Canada: A longitudinal study. Lancet Neurol. 2006, 5, 932–936. [Google Scholar] [CrossRef]
- Sadovnick, A.D.; Ebers, G.C.; Dyment, D.A.; Risch, N.J. Evidence for genetic basis of multiple sclerosis. The Canadian Collaborative Study Group. Lancet 1996, 347, 1728–1730. [Google Scholar] [CrossRef]
- Canto, E.; Oksenberg, J.R. Multiple sclerosis genetics. Mult. Scler. J. 2018, 24, 75–79. [Google Scholar] [CrossRef]
- Hollenbach, J.A.; Oksenberg, J.R. The immunogenetics of multiple sclerosis: A comprehensive review. J. Autoimmun. 2015, 64, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Mescheriakova, J.Y.; Verkerk, A.J.; Amin, N.; Uitterlinden, A.G.; van Duijn, C.M.; Hintzen, R.Q. Linkage analysis and whole exome sequencing identify a novel candidate gene in a Dutch multiple sclerosis family. Mult. Scler. J. 2019, 25, 909–917. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium. Low-Frequency and Rare-Coding Variation Contributes to Multiple Sclerosis Risk. Cell 2018, 175, 1679–1687.e7. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Gourraud, P.A.; McElroy, J.P.; Caillier, S.J.; Johnson, B.A.; Santaniello, A.; Hauser, S.L.; Oksenberg, J.R. Aggregation of multiple sclerosis genetic risk variants in multiple and single case families. Ann. Neurol. 2011, 69, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Ramagopalan, S.V.; Dyment, D.A.; Cader, M.Z.; Morrison, K.M.; Disanto, G.; Morahan, J.M.; Berlanga-Taylor, A.J.; Handel, A.; De Luca, G.C.; Sadovnick, A.D.; et al. Rare variants in the CYP27B1 gene are associated with multiple sclerosis. Ann. Neurol. 2011, 70, 881–886. [Google Scholar] [CrossRef]
- Dyment, D.A.; Cader, M.Z.; Chao, M.J.; Lincoln, M.R.; Morrison, K.M.; Disanto, G.; Morahan, J.M.; De Luca, G.C.; Sadovnick, A.D.; Lepage, P.; et al. Exome sequencing identifies a novel multiple sclerosis susceptibility variant in the TYK2 gene. Neurology 2012, 79, 406–411. [Google Scholar] [CrossRef]
- Wang, Z.; Sadovnick, A.D.; Traboulsee, A.L.; Ross, J.P.; Bernales, C.Q.; Encarnacion, M.; Yee, I.M.; de Lemos, M.; Greenwood, T.; Lee, J.D.; et al. Nuclear Receptor NR1H3 in Familial Multiple Sclerosis. Neuron 2016, 90, 948–954. [Google Scholar] [CrossRef]
- Maver, A.; Lavtar, P.; Ristić, S.; Stopinšek, S.; Simčič, S.; Hočevar, K.; Sepčić, J.; Drulović, J.; Pekmezović, T.; Novaković, I.; et al. Identification of rare genetic variation of NLRP1 gene in familial multiple sclerosis. Sci. Rep. 2017, 7, 3715. [Google Scholar] [CrossRef]
- Sadovnick, A.D.; Gu, B.J.; Traboulsee, A.L.; Bernales, C.Q.; Encarnacion, M.; Yee, I.M.; Criscuoli, M.G.; Huang, X.; Ou, A.; Milligan, C.J.; et al. Purinergic receptors P2RX4 and P2RX7 in familial multiple sclerosis. Hum. Mutat. 2017, 38, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Zrzavy, T.; Leutmezer, F.; Kristoferitsch, W.; Kornek, B.; Schneider, C.; Rommer, P.; Berger, T.; Zimprich, A. Exome-Sequence Analyses of Four Multi-Incident Multiple Sclerosis Families. Genes 2020, 11, 988. [Google Scholar] [CrossRef] [PubMed]
- Ban, M.; Caillier, S.; Mero, I.L.; Myhr, K.M.; Celius, E.G.; Aarseth, J.; Torkildsen, Ø.; Harbo, H.F.; Oksenberg, J.; Hauser, S.L.; et al. No evidence of association between mutant alleles of the CYP27B1 gene and multiple sclerosis. Ann. Neurol. 2013, 73, 430–432. [Google Scholar] [CrossRef]
- Barizzone, N.; Pauwels, I.; Luciano, B.; Franckaert, D.; Guerini, F.R.; Cosemans, L.; Hilven, K.; Salviati, A.; Dooley, J.; Danso-Abeam, D.; et al. No evidence for a role of rare CYP27B1 functional variations in multiple sclerosis. Ann. Neurol. 2013, 73, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Antel, J.; Ban, M.; Baranzini, S.; Barcellos, L.; Barizzone, N.; Beecham, A.; Berge, T.; Bernardinelli, L.; Booth, D.; Bos, S.; et al. NR1H3 p.Arg415Gln Is Not Associated to Multiple Sclerosis Risk. Neuron 2016, 92, 333–335. [Google Scholar] [CrossRef][Green Version]
- Minikel, E.V.; MacArthur, D.G. Publicly Available Data Provide Evidence against NR1H3 R415Q Causing Multiple Sclerosis. Neuron 2016, 92, 336–338. [Google Scholar] [CrossRef]
- Berger, T.; Rubner, P.; Schautzer, F.; Egg, R.; Ulmer, H.; Mayringer, I.; Dilitz, E.; Deisenhammer, F.; Reindl, M. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N. Engl. J. Med. 2003, 349, 139–145. [Google Scholar] [CrossRef]
- Zhou, Y.; Simpson, S., Jr.; Charlesworth, J.C.; van der Mei, I.; Lucas, R.M.; Ponsonby, A.L.; Taylor, B.V. Variation within MBP gene predicts disease course in multiple sclerosis. Brain Behav. 2017, 7, e00670. [Google Scholar] [CrossRef]
- Khorshid Ahmad, T.; Zhou, T.; AlTaweel, K.; Cortes, C.; Lillico, R.; Lakowski, T.M.; Gozda, K.; Namaka, M.P. Experimental Autoimmune Encephalomyelitis (EAE)-Induced Elevated Expression of the E1 Isoform of Methyl CpG Binding Protein 2 (MeCP2E1): Implications in Multiple Sclerosis (MS)-Induced Neurological Disability and Associated Myelin Damage. Int. J. Mol. Sci. 2017, 18, 1254. [Google Scholar] [CrossRef]
- Sharma, K.; Singh, J.; Pillai, P.P. MeCP2 Differentially Regulate the Myelin MBP and PLP Protein Expression in Oligodendrocytes and C6 Glioma. J. Mol. Neurosci. 2018, 65, 343–350. [Google Scholar] [CrossRef]
- Mørkholt, A.S.; Trabjerg, M.S.; Oklinski, M.K.E.; Bolther, L.; Kroese, L.J.; Pritchard, C.E.J.; Huijbers, I.J.; Nieland, J.D.V. CPT1A plays a key role in the development and treatment of multiple sclerosis and experimental autoimmune encephalomyelitis. Sci. Rep. 2019, 9, 13299. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.L.; Ifergan, I.; Miller, S.D. Experimental Autoimmune Encephalomyelitis in Mice. Methods Mol. Biol. 2016, 1304, 145–160. [Google Scholar] [PubMed]
- Ponomarenko, N.A.; Durova, O.M.; Vorobiev, I.I.; Belogurov, A.A., Jr.; Kurkova, I.N.; Petrenko, A.G.; Telegin, G.B.; Suchkov, S.V.; Kiselev, S.L.; Lagarkova, M.A.; et al. Autoantibodies to myelin basic protein catalyze site-specific degradation of their antigen. Proc. Natl. Acad. Sci. USA 2006, 103, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nai, S.; Ding, Y.; Geng, Q.; Zhu, B.; Yu, K.; Zhu, W.-G.; Dong, M.-Q.; Su, X.-D.; Xu, X.; et al. Polo-like kinase 1 (PLK1)-dependent phosphorylation of methylenetetrahydrofolate reductase (MTHFR) regulates replication via histone methylation. Cell Cycle 2017, 16, 1933–1942. [Google Scholar] [CrossRef]
- Froese, D.S.; Kopec, J.; Rembeza, E.; Bezerra, G.A.; Oberholzer, A.E.; Suormala, T.; Lutz, S.; Chalk, R.; Borkowska, O.; Baumgartner, M.R.; et al. Structural basis for the regulation of human 5,10-methylenetetrahydrofolate reductase by phosphorylation and S-adenosylmethionine inhibition. Nat. Commun. 2018, 9, 2261. [Google Scholar] [CrossRef]
- Kim, S.; Lim, I.K.; Park, G.-H.; Paik, W.K. Biological methylation of myelin basic protein: Enzymology and biological significance. Int. J. Biochem. Cell Biol. 1997, 29, 743–751. [Google Scholar] [CrossRef]
- Klotz, L.; Farkas, M.; Bain, N.; Keskitalo, S.; Semmler, A.; Ineichen, B.; Jelcic, J.; Klockgether, T.; Kölsch, H.; Weller, M.; et al. The variant methylenetetrahydrofolate reductase c.1298A>C (p.E429A) is associated with multiple sclerosis in a German case-control study. Neurosci. Lett. 2010, 468, 183–185. [Google Scholar] [CrossRef]
- Ineichen, B.V.; Keskitalo, S.; Farkas, M.; Bain, N.; Kallweit, U.; Weller, M.; Klotz, L.; Linnebank, M. Genetic variants of homocysteine metabolism and multiple sclerosis: A case-control study. Neurosci. Lett. 2014, 562, 75–78. [Google Scholar] [CrossRef]
- Cevik, B.; Yigit, S.; Karakus, N.; Aksoy, D.; Kurt, S.; Ates, O. Association of methylenetetrahydrofolate reductase gene C677T polymorphism with multiple sclerosis in Turkish patients. J. Investig. Med. 2014, 62, 980–984. [Google Scholar] [CrossRef]
- Naghibalhossaini, F.; Ehyakonandeh, H.; Nikseresht, A.; Kamali, E. Association between MTHFR Genetic Variants and Multiple Sclerosis in a Southern Iranian Population. Int. J. Mol. Cell. Med. 2015, 4, 87–93. [Google Scholar]
- Lebrun-Julien, F.; Bachmann, L.; Norrmén, C.; Trötzmüller, M.; Köfeler, H.; Rüegg, M.A.; Hall, M.N.; Suter, U. Balanced mTORC1 activity in oligodendrocytes is required for accurate CNS myelination. J. Neurosci. 2014, 34, 8432–8448. [Google Scholar] [CrossRef] [PubMed]
- Ruf, S.; Heberle, A.M.; Langelaar-Makkinje, M.; Gelino, S.; Wilkinson, D.; Gerbeth, C.; Schwarz, J.J.; Holzwarth, B.; Warscheid, B.; Meisinger, C.; et al. PLK1 (polo like kinase 1) inhibits MTOR complex 1 and promotes autophagy. Autophagy 2017, 13, 486–505. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Singh, J.; Pillai, P.P.; Frost, E.E. Involvement of MeCP2 in Regulation of Myelin-Related Gene Expression in Cultured Rat Oligodendrocytes. J. Mol. Neurosci. 2015, 57, 176–184. [Google Scholar] [CrossRef] [PubMed]
- KhorshidAhmad, T.; Acosta, C.; Cortes, C.; Lakowski, T.M.; Gangadaran, S.; Namaka, M. Transcriptional Regulation of Brain-Derived Neurotrophic Factor (BDNF) by Methyl CpG Binding Protein 2 (MeCP2): A Novel Mechanism for Re-Myelination and/or Myelin Repair Involved in the Treatment of Multiple Sclerosis (MS). Mol. Neurobiol. 2016, 53, 1092–1107. [Google Scholar] [CrossRef]
- Kippert, A.; Trajkovic, K.; Fitzner, D.; Opitz, L.; Simons, M. Identification of Tmem10/Opalin as a novel marker for oligodendrocytes using gene expression profiling. BMC Neurosci. 2008, 9, 40. [Google Scholar] [CrossRef]
- Bastien, D.; Landete, V.B.; Lessard, M.; Vallières, N.; Champagne, M.; Takashima, A.; Tremblay, M.; Doyon, Y.; Lacroix, S. IL-1α Gene Deletion Protects Oligodendrocytes after Spinal Cord Injury through Upregulation of the Survival Factor Tox3. J. Neurosci. 2015, 35, 10715–10730. [Google Scholar] [CrossRef]
- Sedel, F.; Bernard, D.; Mock, D.M.; Tourbah, A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology 2016, 110, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Hermey, G.; Riedel, I.B.; Rezgaoui, M.; Westergaard, U.B.; Schaller, C.; Hermans-Borgmeyer, I. SorCS1, a member of the novel sorting receptor family, is localized in somata and dendrites of neurons throughout the murine brain. Neurosci. Lett. 2001, 313, 83–87. [Google Scholar] [CrossRef]
- Subkhangulova, A.; Malik, A.R.; Hermey, G.; Popp, O.; Dittmar, G.; Rathjen, T.; Poy, M.N.; Stumpf, A.; Beed, P.S.; Schmitz, D.; et al. SORCS1 and SORCS3 control energy balance and orexigenic peptide production. EMBO Rep. 2018, 19, e44810. [Google Scholar] [CrossRef]
- Steinman, L.; Zamvil, S. Transcriptional analysis of targets in multiple sclerosis. Nat. Rev. Immunol. 2003, 3, 483–492. [Google Scholar] [CrossRef]
- Matarese, G.; Carrieri, P.B.; Montella, S.; De Rosa, V.; La Cava, A. Leptin as a metabolic link to multiple sclerosis. Nat. Rev. Neurol. 2010, 6, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Zachmann, J.; Kritsi, E.; Tapeinou, A.; Zoumpoulakis, P.; Tselios, T.; Matsoukas, M.T. Combined Computational and Structural Approach into Understanding the Role of Peptide Binding and Activation of Melanocortin Receptor 4. J. Chem. Inf. Model. 2020, 60, 1461–1468. [Google Scholar] [CrossRef]
- Benjamins, J.A.; Nedelkoska, L.; Lisak, R.P. Melanocortin receptor subtypes are expressed on cells in the oligodendroglial lineage and signal ACTH protection. J. Neurosci. Res. 2018, 96, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Sadovnick, A.D.; Traboulsee, A.L.; Zhao, Y.; Bernales, C.Q.; Encarnacion, M.; Ross, J.P.; Yee, I.M.; Criscuoli, M.G.; Vilariño-Güell, C. Genetic modifiers of multiple sclerosis progression, severity and onset. Clin. Immunol. 2017, 180, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Z.; Guo, X.; Li, F.; Wei, Q.; Chen, X.; Gong, D.; Xu, Y.; Chen, W.; Liu, Y.; et al. TRIM66 reads unmodified H3R2K4 and H3K56ac to respond to DNA damage in embryonic stem cells. Nat. Commun. 2019, 10, 4273. [Google Scholar] [CrossRef] [PubMed]
- Foolad, F.; Khodagholi, F.; Javan, M. Sirtuins in Multiple Sclerosis: The crossroad of neurodegeneration, autoimmunity and metabolism. Mult. Scler. Relat. Disord. 2019, 34, 47–58. [Google Scholar] [CrossRef]
- Versteeg, G.A.; Rajsbaum, R.; Sánchez-Aparicio, M.T.; Maestre, A.M.; Valdiviezo, J.; Shi, M.; Inn, K.S.; Fernandez-Sesma, A.; Jung, J.; García-Sastre, A. The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 2013, 38, 384–398. [Google Scholar] [CrossRef]
- Cohan, S.L.; Hendin, B.A.; Reder, A.T.; Smoot, K.; Avila, R.; Mendoza, J.P.; Weinstock-Guttman, B. Interferons and Multiple Sclerosis: Lessons from 25 Years of Clinical and Real-World Experience with Intramuscular Interferon Beta-1a (Avonex). CNS Drugs 2021, 35, 743–767. [Google Scholar] [CrossRef]
- Mo, M.; Hoang, H.T.; Schmidt, S.; Clark, R.B.; Ehrlich, B.E. The role of chromogranin B in an animal model of multiple sclerosis. Mol. Cell. Neurosci. 2013, 56, 102–114. [Google Scholar] [CrossRef]
- Woo, M.S.; Ufer, F.; Rothammer, N.; Di Liberto, G.; Binkle, L.; Haferkamp, U.; Sonner, J.K.; Engler, J.B.; Hornig, S.; Bauer, S.; et al. Neuronal metabotropic glutamate receptor 8 protects against neurodegeneration in CNS inflammation. J. Exp. Med. 2021, 218, e20201290. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Abadier, M.; Haghayegh Jahromi, N.; Cardoso Alves, L.; Boscacci, R.; Vestweber, D.; Barnum, S.; Deutsch, U.; Engelhardt, B.; Lyck, R. Cell surface levels of endothelial ICAM-1 influence the transcellular or paracellular T-cell diapedesis across the blood-brain barrier. Eur. J. Immunol. 2015, 45, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Defazio, G.; Livrea, P.; Giorelli, M.; Martino, D.; Roselli, F.; Ricchiuti, F.; Trojano, M. Interferon β-1a downregulates TNFα-induced intercellular adhesion molecule 1 expression on brain microvascular endothelial cells through a tyrosine kinase-dependent pathway. Brain Res. 2000, 881, 227–230. [Google Scholar] [CrossRef]
- Omilusik, K.; Priatel, J.J.; Chen, X.; Wang, Y.T.; Xu, H.; Choi, K.B.; Gopaul, R.; McIntyre-Smith, A.; Teh, H.-S.; Tan, R.; et al. The CaV1.4 calcium channel is a critical regulator of T cell receptor signaling and naive T cell homeostasis. Immunity 2011, 35, 349–360. [Google Scholar] [CrossRef]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef]
- Fenninger, F.; Han, J.; Stanwood, S.R.; Nohara, L.L.; Arora, H.; Choi, K.B.; Munro, L.; Pfeifer, C.G.; Shanina, I.; Horwitz, M.S.; et al. Mutation of an L-Type Calcium Channel Gene Leads to T Lymphocyte Dysfunction. Front. Immunol. 2019, 10, 2473. [Google Scholar] [CrossRef]
- Cao, T.; Duprez, E.; Borden, K.L.; Freemont, P.S.; Etkin, L.D. Ret finger protein is a normal component of PML nuclear bodies and interacts directly with PML. J. Cell Sci. 1998, 111 Pt 10, 1319–1329. [Google Scholar] [CrossRef]
- Cai, X.; Srivastava, S.; Sun, Y.; Li, Z.; Wu, H.; Zuvela-Jelaska, L.; Li, J.; Salamon, R.S.; Backer, J.M.; Skolnik, E.Y. Tripartite motif containing protein 27 negatively regulates CD4 T cells by ubiquitinating and inhibiting the class II PI3K-C2β. Proc. Natl. Acad. Sci. USA 2011, 108, 20072–20077. [Google Scholar] [CrossRef]
- de Seze, J.; Bigaut, K. Multiple sclerosis diagnostic criteria: From poser to the 2017 revised McDonald criteria. Presse Med. 2021, 50, 104089. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef] [PubMed]
- Klemann, C.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated molecular landscape of Parkinson’s disease. NPJ Park. Dis. 2017, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Klemann, C.; Visser, J.E.; Van Den Bosch, L.; Martens, G.J.M.; Poelmans, G. Integrated molecular landscape of amyotrophic lateral sclerosis provides insights into disease etiology. Brain Pathol. 2018, 28, 203–211. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Family | Participant | Gender | MS Diagnosis | MS Subtype | Age at Diagnosis (Years) | Age at DNA Collection (Years) | WES Performed |
|---|---|---|---|---|---|---|---|
| 1 | I.2 | F | No | - | - | 62 | No |
| 1 | II.4 | F | No | - | - | 40 | Yes |
| 1 | II.5 | M | Yes | RRMS | 29 | 38 | Yes |
| 1 | II.11 | F | Yes | RRMS | 27 | 34 | No |
| 1 | II.13 | F | Yes | RRMS | 26 | 29 | Yes |
| 2 | II.1 | M | No | - | - | 81 | Yes |
| 2 | III.1 | M | No | - | - | 60 | No |
| 2 | III.2 | F | Yes | SPMS | 46 | 57 | Yes |
| 2 | III.10 | F | No | - | - | 51 | No |
| 2 | III.12 | F | Yes | SPMS | 16 | 44 | Yes |
| 2 | IV.1 | F | No | - | - | 27 | No |
| 2 | IV.2 | M | Yes | RRMS | 24 | 25 | No |
| 3 | II.1 | M | No | - | - | 60 | Yes |
| 3 | II.2 | F | Yes | RRMS | 32 | 57 | Yes |
| 3 | II.3 | M | No | - | - | 51 | No |
| 3 | III.1 | M | Yes | RRMS | 24 | 34 | Yes |
| 3 | III.3 | F | No | - | - | 31 | No |
| 4 | III.2 | M | Yes | SPMS | 51 | 71 | Yes |
| 4 | III.3 | F | No | - | - | 68 | Yes |
| 4 | III.6 | F | No | - | - | 61 | No |
| 4 | III.8 | F | Yes | NA | 22 | 56 | No |
| 4 | IV.1 | F | No | - | - | 35 | No |
| 4 | IV.2 | F | Yes | RRMS | 29 | 34 | Yes |
| 4 | IV.4 | F | Yes | RRMS | 31 | 32 | Yes |
| 4 | IV.5 | M | No | - | - | 48 | No |
| 4 | IV.6 | M | No | - | - | 27 | No |
| 4 | IV.8 | F | No | - | - | 22 | No |
| 5 | I.1 | M | Yes | SPMS | 29 | 68 | Yes |
| 5 | I.2 | F | No | - | - | 64 | No |
| 5 | II.1 | F | No | - | - | 37 | No |
| 5 | II.2 | M | Yes | RRMS | 36 | 40 | Yes |
| 5 | II.3 | F | Yes | RRMS | 26 | 38 | Yes |
| 5 | II.4 | M | No | - | - | 39 | Yes |
| 6 | I.2 | F | No | - | - | 65 | Yes |
| 6 | II.1 | F | Yes | RRMS | 27 | 31 | Yes |
| 6 | II.2 | F | No | - | - | 28 | No |
| 6 | II.3 | F | Yes | RRMS | 23 | 26 | Yes |
| 7 | II.2 | F | Yes | SPMS | 39 | 66 | Yes |
| 7 | II.5 | M | No | - | - | 63 | No |
| 7 | II.7 | F | Yes | SPMS | 35 | 62 | No |
| 7 | II.9 | F | No | - | - | 61 | No |
| 7 | II.10 | M | No | - | - | 56 | No |
| 7 | III.1 | F | Yes | PPMS | 38 | 40 | Yes |
| 7 | III.2 | F | Yes | NA | 24 | 38 | Yes |
| 7 | III.3 | F | No | - | - | 36 | Yes |
| 8 | I.1 | M | No | - | 70 | No | |
| 8 | I.2 | F | No | - | 68 | No | |
| 8 | II.1 | M | Yes | NA | 31 | 42 | Yes |
| 8 | II.2 | F | Yes | NA | 25 | 40 | Yes |
| 8 | II.3 | M | No | - | 38 | Yes | |
| 8 | II.4 | F | Yes | RRMS | 34 | 36 | Yes |
| 9 | II.7 | F | Yes | NA | 42 | 76 | Yes |
| 9 | II.8 | F | Yes | PPMS | 52 | 71 | Yes |
| 9 | III.1 | M | No | - | - | 56 | No |
| 9 | III.2 | Male | No | - | - | 54 | Yes |
| Family | Chromosome | Gene | Variant ID | MAF (%) | Nucleotide Change | Amino Acid Change | CADD |
|---|---|---|---|---|---|---|---|
| 1 | chr6 | EXOC2 | rs760365995 | ≤0.05 | G>A | p.Pro134Leu | 25.0 |
| 1 | chr16 | PLK1 | rs35056440 | ≤1 | C>T | p.Leu261Phe | 22.9 |
| 1 | chr18 | MBP | 18:74696828 | NA | C>A | p.Gly151Val | 25.4 |
| 2 | chr12 | ZNF641 | rs200502528 | ≤0.05 | G>A | p.His342Tyr | 27.7 |
| 3 | chr4 | CCNI | rs139547927 | ≤0.05 | T>C | p.Tyr334Cys | 29.1 |
| 3 | chr6 | ITPR3 | 6:33648383–33648394 | NA | DEL (a) | - | 26.6 |
| 3 | chr10 | SORCS1 | 10:108439392 | NA | G>A | p.Ser554Leu | 34.0 |
| 3 | chr16 | PLK1 | rs35056440 | ≤1 | C>T | p.Leu261Phe | 22.9 |
| 3 | chr19 | PRAM1 | rs138042924 | ≤1 | A>G | p.Ser387Pro | 22.0 |
| 3 | chrX | CACNA1F | X:49067910 | NA | T>A | p.Met1324Leu | 23.4 |
| 3 | chrX | MECP2 | rs61751445 | ≤0.05 | G>A | p.Thr311Met | 26.1 |
| 4 | chr1 | USH2A | rs200802261 | ≤0.05 | G>A | p.Pro2870Leu | 31.0 |
| 5 | chr11 | CPT1A | rs140958507 | ≤1 | C>T | p.Arg288Gln | 23.7 |
| 6 | chr8 | MTMR7 | rs760803217 | ≤0.05 | G>A | p.Thr6Met | 22.5 |
| 6 | chr11 | TRIM66 | rs138444298 | ≤1 | G>A | p.Arg1037Trp | 21.5 |
| 6 | chr16 | CNGB1 | rs776392588 | ≤0.05 | DEL (b) | - | 51.0 |
| 7 | chrX | CACNA1F | X:49067910 | NA | T>A | p.Met1324Leu | 23.4 |
| 8 | chr22 | TTC28 | 22:28501278 | NA | T>C | p.Tyr1099Cys | 32.0 |
| 9 | chr4 | MANBA | rs1283991082 | ≤0.05 | DEL (c) | - | 30.0 |
| 9 | chr11 | TRIM66 | rs138444298 | ≤1 | G>A | p.Arg1037Trp | 21.5 |
| 9 | chr11 | CPT1A | rs140958507 | ≤1 | C>T | p.Arg288Gln | 23.7 |
| 9 | chr16 | TOX3 | 16:52580565 | NA | T>C | p.Tyr24Cys | 23.8 |
| 9 | chr19 | PRAM1 | rs138042924 | ≤1 | A>G | p.Ser387Pro | 22.0 |
| Gene | Variant ID | Amino Acid Change | Co-Segregation MS Cases/Controls (%) |
|---|---|---|---|
| EXOC2 | rs760365995 | p.Pro134Leu | 100/50 |
| PLK1 | rs35056440 | p.Leu261Phe | 100/0 |
| MBP | 18:74696828 | p.Gly151Val | 100/0 |
| ZNF641 | rs200502528 | p.His342Tyr | 66/25 |
| CCNI | rs139547927 | p.Tyr334Cys | 100/66 |
| ITPR3 | 6:33648383–33648394 | -(DEL (a)) | 100/33 |
| SORCS1 | 10:108439392 | p.Ser554Leu | 100/0 |
| PRAM1 | rs138042924 | p.Ser387Pro | 100/20 |
| CACNA1F | X:49067910 | p.Met1324Leu | 100/29 |
| MECP2 | rs61751445 | p.Thr311Met | 100/33 |
| USH2A | rs200802261 | p.Pro2870Leu | 75/17 |
| CPT1A | rs140958507 | p.Arg288Gln | 100/20 |
| MTMR7 | rs760803217 | p.Thr6Met | 100/0 |
| TRIM66 | rs138444298 | p.Arg1037Trp | 100/25 |
| CNGB1 | rs776392588 | -(DEL (b)) | 100/50 |
| TTC28 | 22:28501278 | p.Tyr1099Cys | 100/33 |
| MANBA | rs1283991082 | -(DEL (c)) | 100/50 |
| TOX3 | 16:52580565 | p.Tyr24Cys | 100/0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horjus, J.; van Mourik-Banda, T.; Heerings, M.A.P.; Hakobjan, M.; De Witte, W.; Heersema, D.J.; Jansen, A.J.; Strijbis, E.M.M.; de Jong, B.A.; Slettenaar, A.E.J.; et al. Whole Exome Sequencing in Multi-Incident Families Identifies Novel Candidate Genes for Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 11461. https://doi.org/10.3390/ijms231911461
Horjus J, van Mourik-Banda T, Heerings MAP, Hakobjan M, De Witte W, Heersema DJ, Jansen AJ, Strijbis EMM, de Jong BA, Slettenaar AEJ, et al. Whole Exome Sequencing in Multi-Incident Families Identifies Novel Candidate Genes for Multiple Sclerosis. International Journal of Molecular Sciences. 2022; 23(19):11461. https://doi.org/10.3390/ijms231911461
Chicago/Turabian StyleHorjus, Julia, Tineke van Mourik-Banda, Marco A. P. Heerings, Marina Hakobjan, Ward De Witte, Dorothea J. Heersema, Anne J. Jansen, Eva M. M. Strijbis, Brigit A. de Jong, Astrid E. J. Slettenaar, and et al. 2022. "Whole Exome Sequencing in Multi-Incident Families Identifies Novel Candidate Genes for Multiple Sclerosis" International Journal of Molecular Sciences 23, no. 19: 11461. https://doi.org/10.3390/ijms231911461
APA StyleHorjus, J., van Mourik-Banda, T., Heerings, M. A. P., Hakobjan, M., De Witte, W., Heersema, D. J., Jansen, A. J., Strijbis, E. M. M., de Jong, B. A., Slettenaar, A. E. J., Zeinstra, E. M. P. E., Hoogervorst, E. L. J., Franke, B., Kruijer, W., Jongen, P. J., Visser, L. J., & Poelmans, G. (2022). Whole Exome Sequencing in Multi-Incident Families Identifies Novel Candidate Genes for Multiple Sclerosis. International Journal of Molecular Sciences, 23(19), 11461. https://doi.org/10.3390/ijms231911461