

Safeguarding DNA Replication: A Golden Touch of MiDAS and Other Mechanisms
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. DNA Replication and Replication Stress
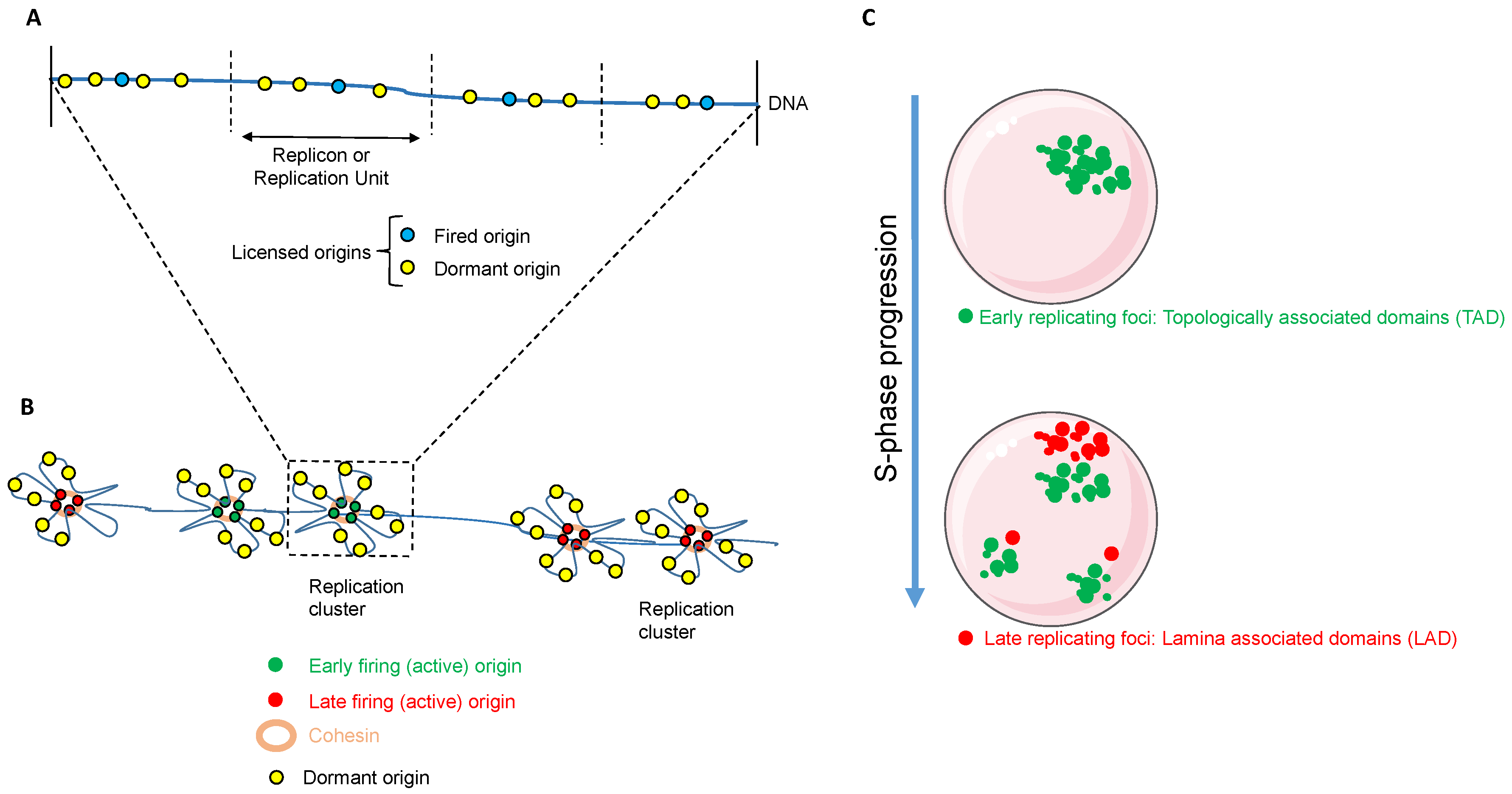
2.1. Origin Licensing and Firing
2.2. Regulation of DNA Replication in Space and Time
3. Deregulation in Origin Licensing
3.1. Origin Under-Licensing
3.2. Unscheduled (Anticipated) Origin Licensing
3.3. Origin Re-Licensing
4. Replication Fork Progression
4.1. Alteration in the Nucleotides Pool
4.2. Barriers to Fork Progression
4.2.1. G-Quadruplexes
4.2.2. Replication–Transcription Conflicts
4.2.3. DNA Damage
5. Mechanisms for Managing Replication Stress
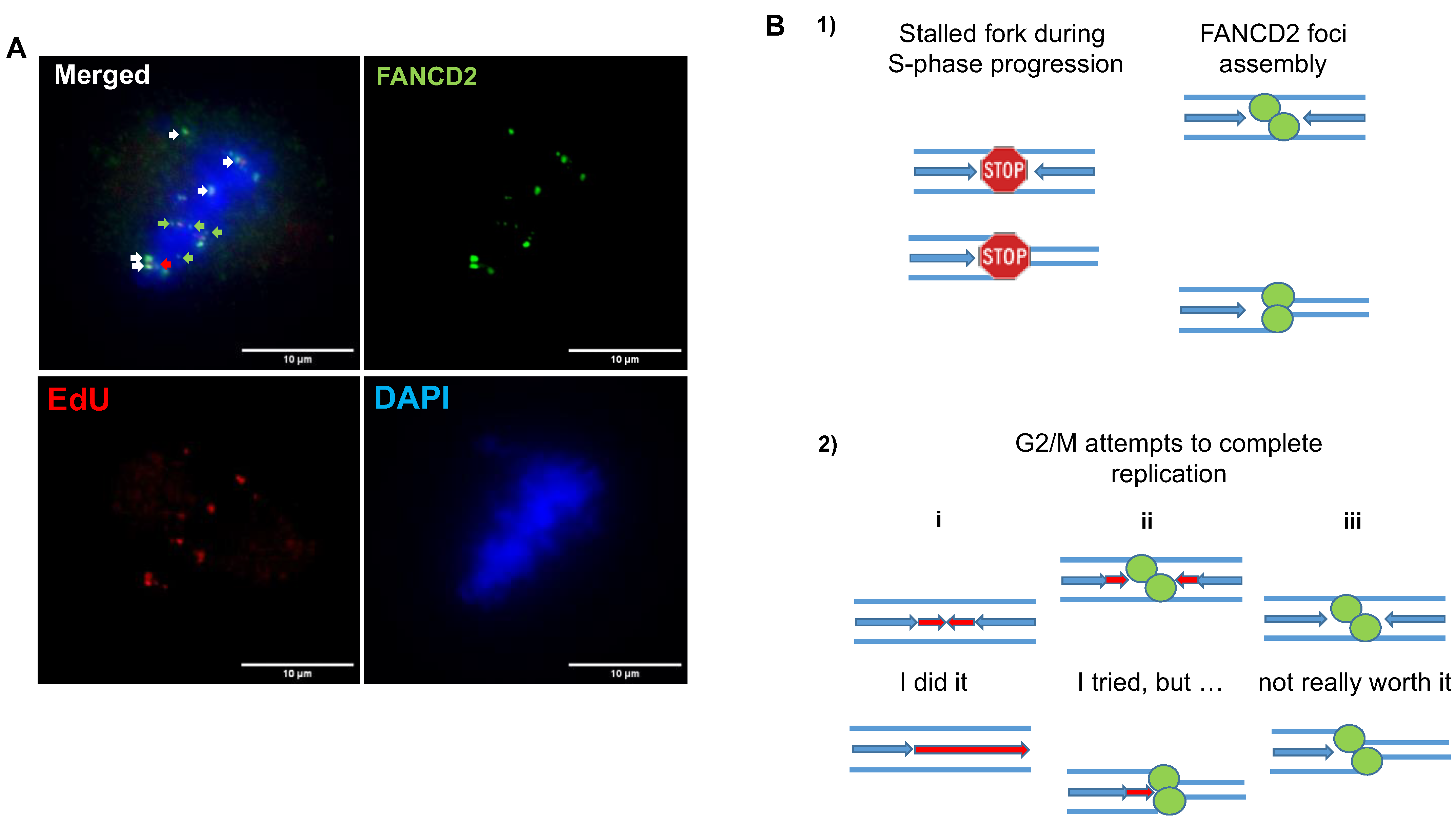
5.1. FANC/BRCA Pathway-Mediated Replication Rescue during S Phase
5.2. Replication Rescue beyond S Phase
5.2.1. During G2/Mitosis
5.2.2. G1 of the Next Cell Cycle
5.2.3. Mitotic DNA Synthesis: A Fact or an Artifact
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bacolla, A.; Tainer, J.A.; Vasquez, K.M.; Cooper, D.N. Translocation and Deletion Breakpoints in Cancer Genomes Are Associated with Potential Non-B DNA-Forming Sequences. Nucleic Acids Res. 2016, 44, 5673–5688. [Google Scholar] [CrossRef]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between Replication and Transcription Complexes Cause Common Fragile Site Instability at the Longest Human Genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef]
- Alexander, J.L.; Orr-Weaver, T.L. Replication Fork Instability and the Consequences of Fork Collisions from Rereplication. Genes Dev. 2016, 30, 2241–2252. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, S.; Méndez, J. DNA Replication Stress: From Molecular Mechanisms to Human Disease. Chromosoma 2017, 126, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. Intragenic Origins Due to Short G1 Phases Underlie Oncogene-Induced DNA Replication Stress. Nature 2018, 555, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.K.; Wang, X.; Li, Y.; Ekholm-Reed, S.; Wu, X.; Wang, P.; Reed, S.I. Cyclin E Deregulation Promotes Loss of Specific Genomic Regions. Curr. Biol. 2015, 25, 1327–1333. [Google Scholar] [CrossRef]
- Lengronne, A.; Schwob, E. The Yeast CDK Inhibitor Sic1 Prevents Genomic Instability by Promoting Replication Origin Licensing in Late G(1). Mol. Cell 2002, 9, 1067–1078. [Google Scholar] [CrossRef]
- Tanaka, S.; Diffley, J.F.X. Deregulated G1-Cyclin Expression Induces Genomic Instability by Preventing Efficient Pre-RC Formation. Genes Dev. 2002, 16, 2639–2649. [Google Scholar] [CrossRef]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide Deficiency Promotes Genomic Instability in Early Stages of Cancer Development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef]
- Beck, H.; Nähse-Kumpf, V.; Larsen, M.S.Y.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuåsen, R.G.; et al. Cyclin-Dependent Kinase Suppression by WEE1 Kinase Protects the Genome through Control of Replication Initiation and Nucleotide Consumption. Mol. Cell. Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef]
- Tsegay, P.S.; Lai, Y.; Liu, Y. Replication Stress and Consequential Instability of the Genome and Epigenome. Mol. Basel Switz. 2019, 24, E3870. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81-EME1 Promote Sister Chromatid Separation by Processing Late Replication Intermediates at Common Fragile Sites during Mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication Stress Activates DNA Repair Synthesis in Mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef]
- Bialic, M.; Al Ahmad Nachar, B.; Koźlak, M.; Coulon, V.; Schwob, E. Measuring S-Phase Duration from Asynchronous Cells Using Dual EdU-BrdU Pulse-Chase Labeling Flow Cytometry. Genes 2022, 13, 408. [Google Scholar] [CrossRef]
- Sawyer, S.L.; Tian, L.; Kähkönen, M.; Schwartzentruber, J.; University of Washington Centre for Mendelian Genomics; FORGE Canada Consortium; Kircher, M.; Majewski, J.; Dyment, D.A.; Innes, A.M.; et al. Biallelic Mutations in BRCA1 Cause a New Fanconi Anemia Subtype. Cancer Discov. 2015, 5, 135–142. [Google Scholar] [CrossRef]
- Alter, B.P.; Rosenberg, P.S.; Brody, L.C. Clinical and Molecular Features Associated with Biallelic Mutations in FANCD1/BRCA2. J. Med. Genet. 2007, 44, 1–9. [Google Scholar] [CrossRef]
- Renaudin, X.; Rosselli, F. The FANC/BRCA Pathway Releases Replication Blockades by Eliminating DNA Interstrand Cross-Links. Genes 2020, 11, E585. [Google Scholar] [CrossRef]
- Debatisse, M.; Rosselli, F. A Journey with Common Fragile Sites: From S Phase to Telophase. Genes. Chromosomes Cancer 2019, 58, 305–316. [Google Scholar] [CrossRef]
- Gueiderikh, A.; Rosselli, F.; Neto, J.B.C. A Never-Ending Story: The Steadily Growing Family of the FA and FA-like Genes. Genet. Mol. Biol. 2017, 40, 398–407. [Google Scholar] [CrossRef]
- Gueiderikh, A.; Maczkowiak-Chartois, F.; Rosselli, F. A New Frontier in Fanconi Anemia: From DNA Repair to Ribosome Biogenesis. Blood Rev. 2022, 52, 100904. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, G.; Clapp, D.W. Fanconi Anaemia and Cancer: An Intricate Relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca, R.; Andreassen, P.R.; Margossian, S.P.; Gregory, R.C.; Taniguchi, T.; Wang, X.; Houghtaling, S.; Grompe, M.; D’Andrea, A.D. Regulated Interaction of the Fanconi Anemia Protein, FANCD2, with Chromatin. Blood 2005, 105, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Knipscheer, P.; Räschle, M.; Smogorzewska, A.; Enoiu, M.; Ho, T.V.; Schärer, O.D.; Elledge, S.J.; Walter, J.C. The Fanconi Anemia Pathway Promotes Replication-Dependent DNA Interstrand Cross-Link Repair. Science 2009, 326, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.-C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Alver, R.C.; Chadha, G.S.; Blow, J.J. The Contribution of Dormant Origins to Genome Stability: From Cell Biology to Human Genetics. DNA Repair 2014, 19, 182–189. [Google Scholar] [CrossRef]
- Blow, J.J.; Ge, X.Q.; Jackson, D.A. How Dormant Origins Promote Complete Genome Replication. Trends Biochem. Sci. 2011, 36, 405–414. [Google Scholar] [CrossRef]
- Strehl, S.; LaSalle, J.M.; Lalande, M. High-Resolution Analysis of DNA Replication Domain Organization across an R/G-Band Boundary. Mol. Cell. Biol. 1997, 17, 6157–6166. [Google Scholar] [CrossRef]
- Ferreira, J.; Paolella, G.; Ramos, C.; Lamond, A.I. Spatial Organization of Large-Scale Chromatin Domains in the Nucleus: A Magnified View of Single Chromosome Territories. J. Cell Biol. 1997, 139, 1597–1610. [Google Scholar] [CrossRef]
- Bell, S.P.; Stillman, B. ATP-Dependent Recognition of Eukaryotic Origins of DNA Replication by a Multiprotein Complex. Nature 1992, 357, 128–134. [Google Scholar] [CrossRef]
- Cocker, J.H.; Piatti, S.; Santocanale, C.; Nasmyth, K.; Diffley, J.F. An Essential Role for the Cdc6 Protein in Forming the Pre-Replicative Complexes of Budding Yeast. Nature 1996, 379, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.F.; Beach, D. Cdt1 Is an Essential Target of the Cdc10/Sct1 Transcription Factor: Requirement for DNA Replication and Inhibition of Mitosis. EMBO J. 1994, 13, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.P.J.; Mahbubani, H.M.; Khoo, C.-Y.; Blow, J.J. Purification of an MCM-Containing Complex as a Component of the DNA Replication Licensing System. Nature 1995, 375, 418–421. [Google Scholar] [CrossRef]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant Origins Licensed by Excess Mcm2–7 Are Required for Human Cells to Survive Replicative Stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef]
- Ibarra, A.; Schwob, E.; Méndez, J. Excess MCM Proteins Protect Human Cells from Replicative Stress by Licensing Backup Origins of Replication. Proc. Natl. Acad. Sci. USA 2008, 105, 8956–8961. [Google Scholar] [CrossRef]
- Xu, X.; Rochette, P.J.; Feyissa, E.A.; Su, T.V.; Liu, Y. MCM10 Mediates RECQ4 Association with MCM2-7 Helicase Complex during DNA Replication. EMBO J. 2009, 28, 3005–3014. [Google Scholar] [CrossRef] [PubMed]
- Ilves, I.; Petojevic, T.; Pesavento, J.J.; Botchan, M.R. Activation of the MCM2-7 Helicase by Association with Cdc45 and GINS Proteins. Mol. Cell 2010, 37, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Ottiger, H.P.; Hübscher, U. Mammalian DNA Polymerase Alpha Holoenzymes with Possible Functions at the Leading and Lagging Strand of the Replication Fork. Proc. Natl. Acad. Sci. USA 1984, 81, 3993–3997. [Google Scholar] [CrossRef] [PubMed]
- Podust, L.M.; Podust, V.N.; Sogo, J.M.; Hübscher, U. Mammalian DNA Polymerase Auxiliary Proteins: Analysis of Replication Factor C-Catalyzed Proliferating Cell Nuclear Antigen Loading onto Circular Double-Stranded DNA. Mol. Cell Biol. 1995, 15, 3072–3081. [Google Scholar] [CrossRef]
- Fukui, T.; Yamauchi, K.; Muroya, T.; Akiyama, M.; Maki, H.; Sugino, A.; Waga, S. Distinct Roles of DNA Polymerases Delta and Epsilon at the Replication Fork in Xenopus Egg Extracts. Genes Cells Dev. Mol. Cell Mech. 2004, 9, 179–191. [Google Scholar] [CrossRef]
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA Replication Origin Activation in Space and Time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Gineau, L.; Cognet, C.; Kara, N.; Lach, F.P.; Dunne, J.; Veturi, U.; Picard, C.; Trouillet, C.; Eidenschenk, C.; Aoufouchi, S.; et al. Partial MCM4 Deficiency in Patients with Growth Retardation, Adrenal Insufficiency, and Natural Killer Cell Deficiency. J. Clin. Investig. 2012, 122, 821–832. [Google Scholar] [CrossRef]
- Ekholm-Reed, S.; Méndez, J.; Tedesco, D.; Zetterberg, A.; Stillman, B.; Reed, S.I. Deregulation of Cyclin E in Human Cells Interferes with Prereplication Complex Assembly. J. Cell Biol. 2004, 165, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Huberman, J.A.; Riggs, A.D. On the Mechanism of DNA Replication in Mammalian Chromosomes. J. Mol. Biol. 1968, 32, 327–341. [Google Scholar] [CrossRef]
- Lebofsky, R.; Heilig, R.; Sonnleitner, M.; Weissenbach, J.; Bensimon, A. DNA Replication Origin Interference Increases the Spacing between Initiation Events in Human Cells. Mol. Biol. Cell 2006, 17, 5337–5345. [Google Scholar] [CrossRef]
- Marheineke, K.; Hyrien, O. Control of Replication Origin Density and Firing Time in Xenopus Egg Extracts: Role of a Caffeine-Sensitive, ATR-Dependent Checkpoint. J. Biol. Chem. 2004, 279, 28071–28081. [Google Scholar] [CrossRef]
- Guillou, E.; Ibarra, A.; Coulon, V.; Casado-Vela, J.; Rico, D.; Casal, I.; Schwob, E.; Losada, A.; Méndez, J. Cohesin Organizes Chromatin Loops at DNA Replication Factories. Genes Dev. 2010, 24, 2812–2822. [Google Scholar] [CrossRef]
- Kumagai, A.; Dunphy, W.G. Binding of the Treslin-MTBP Complex to Specific Regions of the Human Genome Promotes the Initiation of DNA Replication. Cell Rep. 2020, 32, 108178. [Google Scholar] [CrossRef]
- Dellino, G.I.; Cittaro, D.; Piccioni, R.; Luzi, L.; Banfi, S.; Segalla, S.; Cesaroni, M.; Mendoza-Maldonado, R.; Giacca, M.; Pelicci, P.G. Genome-Wide Mapping of Human DNA-Replication Origins: Levels of Transcription at ORC1 Sites Regulate Origin Selection and Replication Timing. Genome Res. 2013, 23, 1–11. [Google Scholar] [CrossRef]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically Associating Domains Are Stable Units of Replication-Timing Regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef]
- Xiang, W.; Roberti, M.J.; Hériché, J.-K.; Huet, S.; Alexander, S.; Ellenberg, J. Correlative Live and Super-Resolution Imaging Reveals the Dynamic Structure of Replication Domains. J. Cell Biol. 2018, 217, 1973–1984. [Google Scholar] [CrossRef] [PubMed]
- Alver, R.C.; Chadha, G.S.; Gillespie, P.J.; Blow, J.J. Reversal of DDK-Mediated MCM Phosphorylation by Rif1-PP1 Regulates Replication Initiation and Replisome Stability Independently of ATR/Chk1. Cell Rep. 2017, 18, 2508–2520. [Google Scholar] [CrossRef] [PubMed]
- Debatisse, M.; Le Tallec, B.; Letessier, A.; Dutrillaux, B.; Brison, O. Common Fragile Sites: Mechanisms of Instability Revisited. Trends Genet. TIG 2012, 28, 22–32. [Google Scholar] [CrossRef]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.-M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-Type-Specific Replication Initiation Programs Set Fragility of the FRA3B Fragile Site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef]
- Symeonidou, I.-E.; Kotsantis, P.; Roukos, V.; Rapsomaniki, M.-A.; Grecco, H.E.; Bastiaens, P.; Taraviras, S.; Lygerou, Z. Multi-Step Loading of Human Minichromosome Maintenance Proteins in Live Human Cells. J. Biol. Chem. 2013, 288, 35852–35867. [Google Scholar] [CrossRef] [PubMed]
- Keyomarsi, K.; Tucker, S.L.; Buchholz, T.A.; Callister, M.; Ding, Y.; Hortobagyi, G.N.; Bedrosian, I.; Knickerbocker, C.; Toyofuku, W.; Lowe, M.; et al. Cyclin E and Survival in Patients with Breast Cancer. N. Engl. J. Med. 2002, 347, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Porter, P.L.; Malone, K.E.; Heagerty, P.J.; Alexander, G.M.; Gatti, L.A.; Firpo, E.J.; Daling, J.R.; Roberts, J.M. Expression of Cell-Cycle Regulators P27Kip1 and Cyclin E, Alone and in Combination, Correlate with Survival in Young Breast Cancer Patients. Nat. Med. 1997, 3, 222–225. [Google Scholar] [CrossRef]
- Dong, Y.; Sui, L.; Tai, Y.; Sugimoto, K.; Hirao, T.; Tokuda, M. Prognostic Significance of Cyclin E Overexpression in Laryngeal Squamous Cell Carcinomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 4253–4258. [Google Scholar]
- Fukuse, T.; Hirata, T.; Naiki, H.; Hitomi, S.; Wada, H. Prognostic Significance of Cyclin E Overexpression in Resected Non-Small Cell Lung Cancer. Cancer Res. 2000, 60, 242–244. [Google Scholar]
- Hwang, H.C.; Clurman, B.E. Cyclin E in Normal and Neoplastic Cell Cycles. Oncogene 2005, 24, 2776–2786. [Google Scholar] [CrossRef]
- Ahn, M.J.; Kim, B.H.; Jang, S.J.; Hong, E.K.; Lee, W.M.; Baik, H.K.; Park, H.K.; Lee, C.B.; Ki, M. Expression of Cyclin D1 and Cyclin E in Human Gastric Carcinoma and Its Clinicopathologic Significance. J. Korean Med. Sci. 1998, 13, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Bani-Hani, K.E.; Almasri, N.M.; Khader, Y.S.; Sheyab, F.M.; Karam, H.N. Combined Evaluation of Expressions of Cyclin E and P53 Proteins as Prognostic Factors for Patients with Gastric Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 1447–1453. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keum, J.S.; Kong, G.; Yang, S.C.; Shin, D.H.; Park, S.S.; Lee, J.H.; Lee, J.D. Cyclin D1 Overexpression Is an Indicator of Poor Prognosis in Resectable Non-Small Cell Lung Cancer. Br. J. Cancer 1999, 81, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Terunuma, A.; Putluri, N.; Mishra, P.; Mathé, E.A.; Dorsey, T.H.; Yi, M.; Wallace, T.A.; Issaq, H.J.; Zhou, M.; Killian, J.K.; et al. MYC-Driven Accumulation of 2-Hydroxyglutarate Is Associated with Breast Cancer Prognosis. J. Clin. Investig. 2014, 124, 398–412. [Google Scholar] [CrossRef]
- Zhou, J.; Hui, X.; Mao, Y.; Fan, L. Identification of Novel Genes Associated with a Poor Prognosis in Pancreatic Ductal Adenocarcinoma via a Bioinformatics Analysis. Biosci. Rep. 2019, 39, BSR20190625. [Google Scholar] [CrossRef]
- Muñoz, S.; Búa, S.; Rodríguez-Acebes, S.; Megías, D.; Ortega, S.; de Martino, A.; Méndez, J. In Vivo DNA Re-Replication Elicits Lethal Tissue Dysplasias. Cell Rep. 2017, 19, 928–938. [Google Scholar] [CrossRef]
- Mukherjee, S.; Conrad, S.E. C-Myc Suppresses P21WAF1/CIP1 Expression during Estrogen Signaling and Antiestrogen Resistance in Human Breast Cancer Cells. J. Biol. Chem. 2005, 280, 17617–17625. [Google Scholar] [CrossRef]
- Bukholm, I.K.; Nesland, J.M. Protein Expression of P53, P21 (WAF1/CIP1), Bcl-2, Bax, Cyclin D1 and PRb in Human Colon Carcinomas. Virchows Arch. Int. J. Pathol. 2000, 436, 224–228. [Google Scholar] [CrossRef]
- Edmonston, T.B.; Cuesta, K.H.; Burkholder, S.; Barusevicius, A.; Rose, D.; Kovatich, A.J.; Boman, B.; Fry, R.; Fishel, R.; Palazzo, J.P. Colorectal Carcinomas with High Microsatellite Instability: Defining a Distinct Immunologic and Molecular Entity with Respect to Prognostic Markers. Hum. Pathol. 2000, 31, 1506–1514. [Google Scholar] [CrossRef]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Ogawa, A.; Dorfman, I.; Loda, M.; Fuchs, C.S. Down-Regulation of P21 (CDKN1A/CIP1) Is Inversely Associated with Microsatellite Instability and CpG Island Methylator Phenotype (CIMP) in Colorectal Cancer. J. Pathol. 2006, 210, 147–154. [Google Scholar] [CrossRef]
- Singh, A.; Xu, Y.-J. The Cell Killing Mechanisms of Hydroxyurea. Genes 2016, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Magdalou, I.; Barascu, A.; Técher, H.; Debatisse, M.; Lopez, B.S. Spontaneous Slow Replication Fork Progression Elicits Mitosis Alterations in Homologous Recombination-Deficient Mammalian Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, J.M.; Kehrli, K.; Mao, F.; Monnat, R. Distinct Functions of Human RECQ Helicases WRN and BLM in Replication Fork Recovery and Progression after Hydroxyurea-Induced Stalling. DNA Repair 2013, 12, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Ercilla, A.; Feu, S.; Aranda, S.; Llopis, A.; Brynjólfsdóttir, S.H.; Sørensen, C.S.; Toledo, L.I.; Agell, N. Acute Hydroxyurea-Induced Replication Blockade Results in Replisome Components Disengagement from Nascent DNA without Causing Fork Collapse. Cell. Mol. Life Sci. 2020, 77, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Gellert, M.; Lipsett, M.N.; Davies, D.R. HELIX FORMATION BY GUANYLIC ACID. Proc. Natl. Acad. Sci. USA 1962, 48, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.M.; Ryan, M.; Kim, R.; Zakas, A.L.; Fu, H.; Lin, C.M.; Reinhold, W.C.; Davis, S.R.; Bilke, S.; Liu, H.; et al. Genome-Wide Depletion of Replication Initiation Events in Highly Transcribed Regions. Genome Res. 2011, 21, 1822–1832. [Google Scholar] [CrossRef]
- Valton, A.-L.; Hassan-Zadeh, V.; Lema, I.; Boggetto, N.; Alberti, P.; Saintomé, C.; Riou, J.-F.; Prioleau, M.-N. G4 Motifs Affect Origin Positioning and Efficiency in Two Vertebrate Replicators. EMBO J. 2014, 33, 732–746. [Google Scholar] [CrossRef]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative Visualization of DNA G-Quadruplex Structures in Human Cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef]
- Papadopoulou, C.; Guilbaud, G.; Schiavone, D.; Sale, J.E. Nucleotide Pool Depletion Induces G-Quadruplex-Dependent Perturbation of Gene Expression. Cell Rep. 2015, 13, 2491–2503. [Google Scholar] [CrossRef]
- King, J.J.; Irving, K.L.; Evans, C.W.; Chikhale, R.V.; Becker, R.; Morris, C.J.; Peña Martinez, C.D.; Schofield, P.; Christ, D.; Hurley, L.H.; et al. DNA G-Quadruplex and i-Motif Structure Formation Is Interdependent in Human Cells. J. Am. Chem. Soc. 2020, 142, 20600–20604. [Google Scholar] [CrossRef]
- Cluett, T.J.; Akman, G.; Reyes, A.; Kazak, L.; Mitchell, A.; Wood, S.R.; Spinazzola, A.; Spelbrink, J.N.; Holt, I.J. Transcript Availability Dictates the Balance between Strand-Asynchronous and Strand-Coupled Mitochondrial DNA Replication. Nucleic Acids Res. 2018, 46, 10771–10781. [Google Scholar] [CrossRef] [PubMed]
- Boque-Sastre, R.; Soler, M.; Oliveira-Mateos, C.; Portela, A.; Moutinho, C.; Sayols, S.; Villanueva, A.; Esteller, M.; Guil, S. Head-to-Head Antisense Transcription and R-Loop Formation Promotes Transcriptional Activation. Proc. Natl. Acad. Sci. USA 2015, 112, 5785–5790. [Google Scholar] [CrossRef] [PubMed]
- Kireeva, M.L.; Komissarova, N.; Kashlev, M. Overextended RNA:DNA Hybrid as a Negative Regulator of RNA Polymerase II Processivity. J. Mol. Biol. 2000, 299, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskii, B.P.; Liu, R.; Tornaletti, S.; Krasilnikova, M.M.; Mirkin, S.M.; Hanawalt, P.C. Mechanisms and Implications of Transcription Blockage by Guanine-Rich DNA Sequences. Proc. Natl. Acad. Sci. USA 2010, 107, 12816–12821. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human Senataxin Resolves RNA/DNA Hybrids Formed at Transcriptional Pause Sites to Promote Xrn2-Dependent Termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J. A Double-Edged Sword: R Loops as Threats to Genome Integrity and Powerful Regulators of Gene Expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef]
- Zhao, D.Y.; Gish, G.; Braunschweig, U.; Li, Y.; Ni, Z.; Schmitges, F.W.; Zhong, G.; Liu, K.; Li, W.; Moffat, J.; et al. SMN and Symmetric Arginine Dimethylation of RNA Polymerase II C-Terminal Domain Control Termination. Nature 2016, 529, 48–53. [Google Scholar] [CrossRef]
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.; Chédin, F. Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol. Cell 2016, 63, 167–178. [Google Scholar] [CrossRef]
- Sollier, J.; Cimprich, K.A. Breaking Bad: R-Loops and Genome Integrity. Trends Cell Biol. 2015, 25, 514–522. [Google Scholar] [CrossRef]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017, 170, 774–786.e19. [Google Scholar] [CrossRef]
- Tresini, M.; Warmerdam, D.O.; Kolovos, P.; Snijder, L.; Vrouwe, M.G.; Demmers, J.A.A.; van IJcken, W.F.J.; Grosveld, F.G.; Medema, R.H.; Hoeijmakers, J.H.J.; et al. The Core Spliceosome as Target and Effector of Non-Canonical ATM Signalling. Nature 2015, 523, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Santos-Pereira, J.M.; Aguilera, A. R Loops: New Modulators of Genome Dynamics and Function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Barroso, S.; Herrera-Moyano, E.; Muñoz, S.; García-Rubio, M.; Gómez-González, B.; Aguilera, A. The DNA Damage Response Acts as a Safeguard against Harmful DNA-RNA Hybrids of Different Origins. EMBO Rep. 2019, 20, e47250. [Google Scholar] [CrossRef]
- Chappidi, N.; Nascakova, Z.; Boleslavska, B.; Zellweger, R.; Isik, E.; Andrs, M.; Menon, S.; Dobrovolna, J.; Balbo Pogliano, C.; Matos, J.; et al. Fork Cleavage-Religation Cycle and Active Transcription Mediate Replication Restart after Fork Stalling at Co-Transcriptional R-Loops. Mol. Cell 2020, 77, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Prado, F. Homologous Recombination: To Fork and Beyond. Genes 2018, 9, E603. [Google Scholar] [CrossRef]
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for Stalled Replication Fork Stabilization: New Targets for Synthetic Lethality Strategies in Cancer Treatments. EMBO Rep. 2018, 19, e46263. [Google Scholar] [CrossRef]
- García-Gómez, S.; Reyes, A.; Martínez-Jiménez, M.I.; Chocrón, E.S.; Mourón, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J.; et al. PrimPol, an Archaic Primase/Polymerase Operating in Human Cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef]
- Bianchi, J.; Rudd, S.G.; Jozwiakowski, S.K.; Bailey, L.J.; Soura, V.; Taylor, E.; Stevanovic, I.; Green, A.J.; Stracker, T.H.; Lindsay, H.D.; et al. PrimPol Bypasses UV Photoproducts during Eukaryotic Chromosomal DNA Replication. Mol. Cell 2013, 52, 566–573. [Google Scholar] [CrossRef]
- Mourón, S.; Rodriguez-Acebes, S.; Martínez-Jiménez, M.I.; García-Gómez, S.; Chocrón, S.; Blanco, L.; Méndez, J. Repriming of DNA Synthesis at Stalled Replication Forks by Human PrimPol. Nat. Struct. Mol. Biol. 2013, 20, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Lou, J.; Xia, Y.; Su, B.; Liu, T.; Cui, J.; Sun, Y.; Lou, H.; Huang, J. HPrimpol1/CCDC111 Is a Human DNA Primase-Polymerase Required for the Maintenance of Genome Integrity. EMBO Rep. 2013, 14, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Guilliam, T.A.; Jozwiakowski, S.K.; Ehlinger, A.; Barnes, R.P.; Rudd, S.G.; Bailey, L.J.; Skehel, J.M.; Eckert, K.A.; Chazin, W.J.; Doherty, A.J. Human PrimPol Is a Highly Error-Prone Polymerase Regulated by Single-Stranded DNA Binding Proteins. Nucleic Acids Res. 2015, 43, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- González-Acosta, D.; Blanco-Romero, E.; Ubieto-Capella, P.; Mutreja, K.; Míguez, S.; Llanos, S.; García, F.; Muñoz, J.; Blanco, L.; Lopes, M.; et al. PrimPol-Mediated Repriming Facilitates Replication Traverse of DNA Interstrand Crosslinks. EMBO J. 2021, 40, e106355. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.S.; Tonomura, A. A High Susceptibility of Fanconi’s Anemia to Chromosome Breakage by DNA Cross-Linking Agents. Cancer Res. 1973, 33, 1829–1836. [Google Scholar] [PubMed]
- Porfirio, B.; Dallapiccola, B.; Gandini, E. The Effect of Aphidicolin on Fanconi’s Anemia Lymphocyte Chromosomes. Mutat. Res. 1985, 144, 257–263. [Google Scholar] [CrossRef]
- Renaud, E.; Rosselli, F. FANC Pathway Promotes UV-Induced Stalled Replication Forks Recovery by Acting Both Upstream and Downstream Polη and Rev1. PLoS ONE 2013, 8, e53693. [Google Scholar] [CrossRef]
- Andreassen, P.R.; D’Andrea, A.D.; Taniguchi, T. ATR Couples FANCD2 Monoubiquitination to the DNA-Damage Response. Genes Dev. 2004, 18, 1958–1963. [Google Scholar] [CrossRef]
- O’Brien, P.J.; Siraki, A.G.; Shangari, N. Aldehyde Sources, Metabolism, Molecular Toxicity Mechanisms, and Possible Effects on Human Health. Crit. Rev. Toxicol. 2005, 35, 609–662. [Google Scholar] [CrossRef]
- Seitz, H.K.; Stickel, F. Acetaldehyde as an Underestimated Risk Factor for Cancer Development: Role of Genetics in Ethanol Metabolism. Genes Nutr. 2010, 5, 121–128. [Google Scholar] [CrossRef]
- Tagaino, R.; Washio, J.; Abiko, Y.; Tanda, N.; Sasaki, K.; Takahashi, N. Metabolic Property of Acetaldehyde Production from Ethanol and Glucose by Oral Streptococcus and Neisseria. Sci. Rep. 2019, 9, 10446. [Google Scholar] [CrossRef] [PubMed]
- Vaca, C.E.; Fang, J.L.; Schweda, E.K. Studies of the Reaction of Acetaldehyde with Deoxynucleosides. Chem. Biol. Interact. 1995, 98, 51–67. [Google Scholar] [CrossRef]
- Baldacci, G.; Hoffmann, J.-S.; Cadoret, J.-C. Impact of the DNA Polymerase Theta on the DNA Replication Program. Genom. Data 2014, 3, 90–93. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, M.; McIntee, E.J.; Cheng, G.; Shi, Y.; Villalta, P.W.; Hecht, S.S. Identification of DNA Adducts of Acetaldehyde. Chem. Res. Toxicol. 2000, 13, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Witt, E.; Huber, P.A.J.; Medhurst, A.L.; Ashworth, A.; Mathew, C.G. Direct Interaction of the Fanconi Anaemia Protein FANCG with BRCA2/FANCD1. Hum. Mol. Genet. 2003, 12, 2503–2510. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ishiai, M.; Matsushita, N.; Arakawa, H.; Lamerdin, J.E.; Buerstedde, J.-M.; Tanimoto, M.; Harada, M.; Thompson, L.H.; Takata, M. Fanconi Anemia FANCG Protein in Mitigating Radiation- and Enzyme-Induced DNA Double-Strand Breaks by Homologous Recombination in Vertebrate Cells. Mol. Cell Biol. 2003, 23, 5421–5430. [Google Scholar] [CrossRef]
- Kim, H.; Yang, K.; Dejsuphong, D.; D’Andrea, A.D. Regulation of Rev1 by the Fanconi Anemia Core Complex. Nat. Struct. Mol. Biol. 2012, 19, 164–170. [Google Scholar] [CrossRef]
- Ge, X.Q.; Blow, J.J. Chk1 Inhibits Replication Factory Activation but Allows Dormant Origin Firing in Existing Factories. J. Cell Biol. 2010, 191, 1285–1297. [Google Scholar] [CrossRef]
- Schwab, R.A.; Blackford, A.N.; Niedzwiedz, W. ATR Activation and Replication Fork Restart Are Defective in FANCM-Deficient Cells. EMBO J. 2010, 29, 806–818. [Google Scholar] [CrossRef]
- Sobeck, A.; Stone, S.; Landais, I.; de Graaf, B.; Hoatlin, M.E. The Fanconi Anemia Protein FANCM Is Controlled by FANCD2 and the ATR/ATM Pathways. J. Biol. Chem. 2009, 284, 25560–25568. [Google Scholar] [CrossRef]
- Singh, T.R.; Ali, A.M.; Paramasivam, M.; Pradhan, A.; Wahengbam, K.; Seidman, M.M.; Meetei, A.R. ATR-Dependent Phosphorylation of FANCM at Serine 1045 Is Essential for FANCM Functions. Cancer Res. 2013, 73, 4300–4310. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Lee, W.T.C.; Gupta, D.; Xue, H.; Tonzi, P.; Borowiec, J.A.; Huang, T.T.; Modesti, M.; Rothenberg, E. A Basal-Level Activity of ATR Links Replication Fork Surveillance and Stress Response. Mol. Cell 2021, 81, 4243–4257.e6. [Google Scholar] [CrossRef] [PubMed]
- Lüscher-Firzlaff, J.M.; Lilischkis, R.; Lüscher, B. Regulation of the Transcription Factor FOXM1c by Cyclin E/CDK2. FEBS Lett. 2006, 580, 1716–1722. [Google Scholar] [CrossRef]
- Lee, J.; Kumagai, A.; Dunphy, W.G. Positive Regulation of Wee1 by Chk1 and 14-3-3 Proteins. Mol. Biol. Cell 2001, 12, 551–563. [Google Scholar] [CrossRef]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 Checkpoint Pathway in Mammals: Linkage of DNA Damage to Cdk Regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef]
- Ju, J.-Q.; Li, X.-H.; Pan, M.-H.; Xu, Y.; Sun, M.-H.; Xu, Y.; Sun, S.-C. CHK1 Monitors Spindle Assembly Checkpoint and DNA Damage Repair during the First Cleavage of Mouse Early Embryos. Cell Prolif. 2020, 53, e12895. [Google Scholar] [CrossRef]
- Tang, J.; Erikson, R.L.; Liu, X. Checkpoint Kinase 1 (Chk1) Is Required for Mitotic Progression through Negative Regulation of Polo-like Kinase 1 (Plk1). Proc. Natl. Acad. Sci. USA 2006, 103, 11964–11969. [Google Scholar] [CrossRef]
- Patil, M.; Pabla, N.; Dong, Z. Checkpoint Kinase 1 in DNA Damage Response and Cell Cycle Regulation. Cell. Mol. Life Sci. CMLS 2013, 70, 4009–4021. [Google Scholar] [CrossRef]
- Taniguchi, T.; Garcia-Higuera, I.; Xu, B.; Andreassen, P.R.; Gregory, R.C.; Kim, S.-T.; Lane, W.S.; Kastan, M.B.; D’Andrea, A.D. Convergence of the Fanconi Anemia and Ataxia Telangiectasia Signaling Pathways. Cell 2002, 109, 459–472. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, W.; Sy, S.M.H.; Huen, M.S.Y. ATM-Dependent Phosphorylation of the Fanconi Anemia Protein PALB2 Promotes the DNA Damage Response. J. Biol. Chem. 2015, 290, 27545–27556. [Google Scholar] [CrossRef] [PubMed]
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 Kinase in the DNA Damage Response and Beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Renaud, E.; Barascu, A.; Rosselli, F. Impaired TIP60-Mediated H4K16 Acetylation Accounts for the Aberrant Chromatin Accumulation of 53BP1 and RAP80 in Fanconi Anemia Pathway-Deficient Cells. Nucleic Acids Res. 2016, 44, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Collis, S.J.; Adelman, C.A.; Silva, N.; Horejsi, Z.; Ward, J.D.; Martinez-Perez, E.; Boulton, S.J.; La Volpe, A. Preventing Nonhomologous End Joining Suppresses DNA Repair Defects of Fanconi Anemia. Mol. Cell 2010, 39, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Pace, P.; Mosedale, G.; Hodskinson, M.R.; Rosado, I.V.; Sivasubramaniam, M.; Patel, K.J. Ku70 Corrupts DNA Repair in the Absence of the Fanconi Anemia Pathway. Science 2010, 329, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Pichierri, P.; Averbeck, D.; Rosselli, F. DNA Cross-Link-Dependent RAD50/MRE11/NBS1 Subnuclear Assembly Requires the Fanconi Anemia C Protein. Hum. Mol. Genet. 2002, 11, 2531–2546. [Google Scholar] [CrossRef]
- Ruff, P.; Donnianni, R.A.; Glancy, E.; Oh, J.; Symington, L.S. RPA Stabilization of Single-Stranded DNA Is Critical for Break-Induced Replication. Cell Rep. 2016, 17, 3359–3368. [Google Scholar] [CrossRef]
- Giaccherini, C.; Gaillard, P. Control of Structure-Specific Endonucleases during Homologous Recombination in Eukaryotes. Curr. Opin. Genet. Dev. 2021, 71, 195–205. [Google Scholar] [CrossRef]
- Mirchandani, K.D.; McCaffrey, R.M.; D’Andrea, A.D. The Fanconi Anemia Core Complex Is Required for Efficient Point Mutagenesis and Rev1 Foci Assembly. DNA Repair 2008, 7, 902–911. [Google Scholar] [CrossRef]
- Sun, H.; Karow, J.K.; Hickson, I.D.; Maizels, N. The Bloom’s Syndrome Helicase Unwinds G4 DNA. J. Biol. Chem. 1998, 273, 27587–27592. [Google Scholar] [CrossRef]
- Kamath-Loeb, A.S.; Loeb, L.A.; Johansson, E.; Burgers, P.M.J.; Fry, M. Interactions between the Werner Syndrome Helicase and DNA Polymerase δ Specifically Facilitate Copying of Tetraplex and Hairpin Structures of the d(CGG) n Trinucleotide Repeat Sequence. J. Biol. Chem. 2001, 276, 16439–16446. [Google Scholar] [CrossRef] [PubMed]
- Sarkies, P.; Murat, P.; Phillips, L.G.; Patel, K.J.; Balasubramanian, S.; Sale, J.E. FANCJ Coordinates Two Pathways That Maintain Epigenetic Stability at G-Quadruplex DNA. Nucleic Acids Res. 2012, 40, 1485–1498. [Google Scholar] [CrossRef] [PubMed]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The Regulation and Functions of DNA and RNA G-Quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef]
- Piatti, S.; Lengauer, C.; Nasmyth, K. Cdc6 Is an Unstable Protein Whose de Novo Synthesis in G1 Is Important for the Onset of S Phase and for Preventing a “reductional” Anaphase in the Budding Yeast Saccharomyces Cerevisiae. EMBO J. 1995, 14, 3788–3799. [Google Scholar] [CrossRef]
- Ruiz-Herrera, A.; Castresana, J.; Robinson, T.J. Is Mammalian Chromosomal Evolution Driven by Regions of Genome Fragility? Genome Biol. 2006, 7, R115. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Zou, L. Hallmarks of DNA Replication Stress. Mol. Cell 2022, 82, 2298–2314. [Google Scholar] [CrossRef]
- Howlett, N.G.; Taniguchi, T.; Durkin, S.G.; D’Andrea, A.D.; Glover, T.W. The Fanconi Anemia Pathway Is Required for the DNA Replication Stress Response and for the Regulation of Common Fragile Site Stability. Hum. Mol. Genet. 2005, 14, 693–701. [Google Scholar] [CrossRef]
- Madireddy, A.; Kosiyatrakul, S.T.; Gerhardt, J.; Boisvert, R.A.; Vuono, E.A.; Moyano, E.H.; Garcia Rubio, M.L.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef]
- Naim, V.; Rosselli, F. The FANC Pathway and BLM Collaborate during Mitosis to Prevent Micro-Nucleation and Chromosome Abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef]
- Naim, V.; Rosselli, F. The FANC Pathway and Mitosis: A Replication Legacy. Cell Cycle Georget. Tex 2009, 8, 2907–2911. [Google Scholar] [CrossRef]
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication Stress Induces Sister-Chromatid Bridging at Fragile Site Loci in Mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Sølvhøj Pedersen, R.; Grøfte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 Nuclear Bodies Form around DNA Lesions Generated by Mitotic Transmission of Chromosomes under Replication Stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Spies, J.; Lukas, C.; Somyajit, K.; Rask, M.-B.; Lukas, J.; Neelsen, K.J. 53BP1 Nuclear Bodies Enforce Replication Timing at Under-Replicated DNA to Limit Heritable DNA Damage. Nat. Cell Biol. 2019, 21, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Lezaja, A.; Panagopoulos, A.; Wen, Y.; Carvalho, E.; Imhof, R.; Altmeyer, M. RPA Shields Inherited DNA Lesions for Post-Mitotic DNA Synthesis. Nat. Commun. 2021, 12, 3827. [Google Scholar] [CrossRef]
- Macheret, M.; Bhowmick, R.; Sobkowiak, K.; Padayachy, L.; Mailler, J.; Hickson, I.D.; Halazonetis, T.D. High-Resolution Mapping of Mitotic DNA Synthesis Regions and Common Fragile Sites in the Human Genome through Direct Sequencing. Cell Res. 2020, 30, 997–1008. [Google Scholar] [CrossRef]
- Wassing, I.E.; Graham, E.; Saayman, X.; Rampazzo, L.; Ralf, C.; Bassett, A.; Esashi, F. The RAD51 Recombinase Protects Mitotic Chromatin in Human Cells. Nat. Commun. 2021, 12, 5380. [Google Scholar] [CrossRef]
- Groelly, F.J.; Dagg, R.A.; Petropoulos, M.; Rossetti, G.G.; Prasad, B.; Panagopoulos, A.; Paulsen, T.; Karamichali, A.; Jones, S.E.; Ochs, F.; et al. Mitotic DNA Synthesis Is Caused by Transcription-Replication Conflicts in BRCA2-Deficient Cells. Mol. Cell 2022, 82, 3382–3397.e7. [Google Scholar] [CrossRef]
- Mocanu, C.; Karanika, E.; Fernández-Casañas, M.; Herbert, A.; Olukoga, T.; Özgürses, M.E.; Chan, K.-L. DNA Replication Is Highly Resilient and Persistent under the Challenge of Mild Replication Stress. Cell Rep. 2022, 39, 110701. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Ahmad Nachar, B.; Rosselli, F. Safeguarding DNA Replication: A Golden Touch of MiDAS and Other Mechanisms. Int. J. Mol. Sci. 2022, 23, 11331. https://doi.org/10.3390/ijms231911331
Al Ahmad Nachar B, Rosselli F. Safeguarding DNA Replication: A Golden Touch of MiDAS and Other Mechanisms. International Journal of Molecular Sciences. 2022; 23(19):11331. https://doi.org/10.3390/ijms231911331
Chicago/Turabian StyleAl Ahmad Nachar, Baraah, and Filippo Rosselli. 2022. "Safeguarding DNA Replication: A Golden Touch of MiDAS and Other Mechanisms" International Journal of Molecular Sciences 23, no. 19: 11331. https://doi.org/10.3390/ijms231911331
APA StyleAl Ahmad Nachar, B., & Rosselli, F. (2022). Safeguarding DNA Replication: A Golden Touch of MiDAS and Other Mechanisms. International Journal of Molecular Sciences, 23(19), 11331. https://doi.org/10.3390/ijms231911331