
Cytokine Imbalance as a Biomarker of Treatment-Resistant Schizophrenia

 ,
,  ,
,  , ,
, ,
Abstract
1. Introduction
2. Pathogenetic Aspect of Inflammation in Treatment Resistance Schizophrenia
2.1. Changes in the Functional Activity of Microglia in Treatment Resistance Schizophrenia
2.2. Sensitization or Kindling in Treatment Resistance Schizophrenia
2.3. Vulnerability-Stress-Inflammation in Treatment Resistance Schizophrenia
2.4. Prenatal, Perinatal and Postnatal Infection in Treatment Resistance Schizophrenia
2.5. Cytokine Imbalance in Treatment Resistance Schizophrenia
3. Cytokines Alteration in Treatment-Resistant Schizophrenia
3.1. Pro-Inflammatory Cytokines
3.1.1. Interleukin 1 β
3.1.2. Tumor Necrosis Factor Alpha
3.1.3. Interferon Gamma
3.1.4. Interleukin 12
3.1.5. Interleukin 18
3.1.6. Interleukin 8
3.1.7. Interleukin 17
3.2. Anti-Inflammatory Cytokines
3.2.1. Interleukin 4
3.2.2. Interleukin 6
3.2.3. Interleukin 10
4. Correction of Cytokine Status Imbalance as a Promising Therapeutic Strategy for Treatment-Resistant Schizophrenia
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saha, S.; Chant, D.; McGrath, J. A systematic review of mortality in schizophrenia: Is the differential mortality gap worsening over time? Arch. Gen. Psychiatry 2007, 64, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Laursen, T.M. Life expectancy among persons with schizophrenia or bipolar affective disorder. Schizophr. Res. 2011, 131, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Chesney, E.; Goodwin, G.M.; Fazel, S. Risks of all-cause and suicide mortality in mental disorders: A meta-review. World Psychiatry 2014, 13, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Khasanova, A.K.; Dobrodeeva, V.S.; Shnayder, N.A.; Petrova, M.M.; Pronina, E.A.; Bochanova, E.N.; Lareva, N.V.; Garganeeva, N.P.; Smirnova, D.A.; Nasyrova, R.F. Blood and urinary biomarkers of antipsychotic-induced metabolic syndrome. Metabolites 2022, 12, 726. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Thase, M.E.; Pillinger, T. Treatment resistance in psychiatry: State of the art and new directions. Mol. Psychiatry 2022, 27, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Vaiman, E.E.; Shnayder, N.A.; Khasanova, A.K.; Strelnik, A.I.; Gayduk, A.J.; Al-Zamil, M.; Sapronova, M.R.; Zhukova, N.G.; Smirnova, D.A.; Nasyrova, R.F. Pathophysiological mechanisms of antipsychotic-induced parkinsonism. Biomedicines 2022, 10, 2010. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, S.; Dencker, S.J.; Malm, U.; Dahl, M.-L.; Svetnson, J.-O.; Halldin, C.; Naskashima, Y.; Farde, L. D(2)- and 5-Ht(2) receptor occupancy in high-dose neuroleptictreated patients. Int. J. Neuropsychopharmacol. 1998, 1, 95–101. [Google Scholar] [CrossRef]
- Grunder, G.; Carlsson, A.; Wong, D.F. Mechanism of new antipsychotic medications: Occupancy is not just antagonism. Arch. Gen. Psychiatry 2003, 60, 974–977. [Google Scholar] [CrossRef]
- Kane, J.M.; Agid, O.; Baldwin, M.L.; Howes, O.; Lindenmayer, J.P.; Marder, S.; Olfson, M.; Potkin, S.G.; Correll, C.U. Clinical Guidance on the Identification and Management of Treatment-Resistant Schizophrenia. J. Clin. Psychiatry 2019, 80, 2783. [Google Scholar] [CrossRef]
- Suzuki, T.; Remington, G.; Mulsant, B.H.; Rajji, T.K.; Uchida, H.; Graff-Guerrero, A.; Mamo, D.C. Treatment resistant schizophrenia and response to antipsychotics: A review. Schizophr. Res. 2011, 133, 54–62. [Google Scholar] [CrossRef]
- Work Group on Schizophrenia. Practice Guideline for the Treatment of Patients with Schizophrenia, 2nd ed.; American Psychiatric Press: Arlington, VA, USA, 2004. [Google Scholar]
- Hasan, A.; Falkai, P.; Wobrock, T.; Lieberman, J.; Glenthoj, B.; Gattaz, W.F.; Thibaut, F.; Möller, H.J.; World Federation of Societies of Biological Psychiatry (WFSBP) Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) Guidelines for Biological Treatment of Schizophrenia, part 1: Update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J. Biol. Psychiatry Off. J. World Fed. Soc. Biol. Psychiatry 2012, 13, 318–378. [Google Scholar] [CrossRef] [PubMed]
- National Collaborating Centre for Mental Health (UK). Psychosis and Schizophrenia in Adults: Treatment and Management; National Institute for Health and Care Excellence (UK): London, UK, 2014. [Google Scholar]
- Howes, O.D.; McCutcheon, R.; Agid, O.; de Bartolomeis, A.; van Beveren, N.J.; Birnbaum, M.L.; Bloomfield, M.A.; Bressan, R.A.; Buchanan, R.W.; Carpenter, W.T.; et al. Treatment-Resistant Schizophrenia: Treatment Response and Resistance in Psychosis (TRRIP) Working Group Consensus Guidelines on Diagnosis and Terminology. Am. J. Psychiatry 2017, 174, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.A.; Buchanan, R.W.; Buckley, P.F.; Chiles, J.A.; Conley, R.R.; Crismon, M.L.; Essock, S.M.; Finnerty, M.; Marder, S.R.; Miller, D.D.; et al. The Texas Medication Algorithm Project antipsychotic algorithm for schizophrenia: 2006 update. J. Clin. Psychiatry 2007, 68, 1751–1762. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, R.W.; Kreyenbuhl, J.; Kelly, D.L.; Noel, J.M.; Boggs, D.L.; Fischer, B.A.; Himelhoch, S.; Fang, B.; Peterson, E.; Aquino, P.R.; et al. The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr. Bull. 2010, 36, 71–93. [Google Scholar] [CrossRef]
- The International Psychopharmacology Algorithm Project. Available online: http://www.ipap.org (accessed on 15 August 2022).
- Mørup, M.F.; Kymes, S.M.; Oudin Åström, D. A modelling approach to estimate the prevalence of treatment-resistant schizophrenia in the United States. PLoS ONE 2020, 15, e0234121. [Google Scholar] [CrossRef]
- Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm (accessed on 15 August 2022).
- Leung, C.C.; Gadelrab, R.; Ntephe, C.U.; McGuire, P.K.; Demjaha, A. Clinical Course, Neurobiology and Therapeutic Approaches to Treatment Resistant Schizophrenia. Toward an Integrated View. Front. Psychiatry 2019, 10, 601. [Google Scholar] [CrossRef]
- Kinon, B.J. The Group of Treatment Resistant Schizophrenias. Heterogeneity in Treatment Resistant Schizophrenia (TRS). Front. Psychiatry 2019, 9, 757. [Google Scholar] [CrossRef]
- Pisanu, C.; Squassina, A. Treatment-Resistant Schizophrenia: Insights From Genetic Studies and Machine Learning Approaches. Front. Pharmacol. 2019, 10, 617. [Google Scholar] [CrossRef]
- Vita, A.; Minelli, A.; Barlati, S.; Deste, G.; Giacopuzzi, E.; Valsecchi, P.; Turrina, C.; Gennarelli, M. Treatment-Resistant Schizophrenia: Genetic and Neuroimaging Correlates. Front. Pharmacol. 2019, 10, 402. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Folsom, T.D. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr. Bull. 2009, 35, 528–548. [Google Scholar] [CrossRef]
- Potkin, S.G.; Kane, J.M.; Correll, C.U.; Lindenmayer, J.P.; Agid, O.; Marder, S.R.; Olfson, M.; Howes, O.D. The neurobiology of treatment-resistant schizophrenia: Paths to antipsychotic resistance and a roadmap for future research. NPJ Schizophr. 2020, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Takao, N.; Murai, T.; Fujiwara, H. Treatment-resistant schizophrenia characterised by dopamine supersensitivity psychosis and efficacy of asenapine. BMJ Case Rep. 2021, 14, e242495. [Google Scholar] [CrossRef] [PubMed]
- Lowe, P.; Krivoy, A.; Porffy, L.; Henriksdottir, E.; Eromona, W.; Shergill, S.S. When the drugs don’t work: Treatment-resistant schizophrenia, serotonin and serendipity. Ther. Adv. Psychopharmacol. 2018, 8, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.C.; Lin, S.H.; Tseng, H.H.; Chen, K.C.; Yang, Y.K. The integrated model of glutamate and dopamine hypothesis for schizophrenia: Prediction and personalized medicine for prevent potential treatment-resistant patients. Med. Hypotheses 2020, 143, 110159. [Google Scholar] [CrossRef] [PubMed]
- Shnayder, N.A.; Abdyrakhmanova, A.K.; Nasyrova, R.F. Oxidation of antipsychotics. Encyclopedia 2022, 2, 974–989. [Google Scholar] [CrossRef]
- Abdyrakhmanova, A.K.; Nasyrova, R.F. Pharmacogenetic testing of cytochrome P450 metabolizing enzymes in 28-year-old man with treatment-resistant schizophrenia. Pers. Psychiatry Neurol. 2022, 2, 81–88. [Google Scholar] [CrossRef]
- Linova, L.P.; Torgovtsev, A.A.; Limankin, O.V.; Nasyrova, R.F. Clinical case of a 36-year-old man with treatment-resistant paranoid shizophrenia: Personalized therapy selection. Pers. Psychiatry Neurol. 2022, 2, 89–97. [Google Scholar]
- Moons, T.; de Roo, M.; Claes, S.; Dom, G. Relationship between P-glycoprotein and second-generation antipsychotics. Pharmacogenomics 2011, 12, 1193–1211. [Google Scholar] [CrossRef]
- Osipova, S.M.; Shnayder, N.A. Pharmacogenetic testing of antipsychotic transporter proteins: A case report in a 32-year-old woman with treatment-resistant schizophrenia. Pers. Psychiatry Neurol. 2022, 2, 98–106. [Google Scholar] [CrossRef]
- Hoosain, F.G.; Choonara, Y.E.; Tomar, L.K.; Kumar, P.; Tyagi, C.; du Toit, L.C.; Pillay, V. Bypassing P-Glycoprotein drug efflux mechanisms: Possible applications in pharmacoresistant schizophrenia therapy. BioMed Res. Int. 2015, 2015, 84963. [Google Scholar] [CrossRef]
- Bošković, M.; Vovk, T.; Kores Plesničar, B.; Grabnar, I. Oxidative stress in schizophrenia. Curr. Neuropharmacol. 2011, 9, 301–312. [Google Scholar] [PubMed]
- Buosi, P.; Borghi, F.A.; Lopes, A.M.; Facincani, I.; Fernandes-Ferreira, R.; Oliveira-Brancati, C.; do Carmo, T.S.; Souza, D.; da Silva, D.; de Almeida, E.A.; et al. Oxidative stress biomarkers in treatment-responsive and treatment-resistant schizophrenia patients. Trends Psychiatry Psychother. 2021, 43, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Ermakov, E.A.; Dmitrieva, E.M.; Parshukova, D.A.; Kazantseva, D.V.; Vasilieva, A.R.; Smirnova, L.P. Oxidative stress-related mechanisms in schizophrenia pathogenesis and new treatment perspectives. Oxid. Med. Cell. Longev. 2021, 2021, 8881770. [Google Scholar] [CrossRef] [PubMed]
- Leboyer, M.; Godin, O.; Terro, E.; Boukouaci, W.; Lu, C.L.; Andre, M.; Aouizerate, B.; Berna, F.; Barau, C.; Capdevielle, D.; et al. Immune signatures of treatment-resistant schizophrenia: A FondaMental Academic Centers of Expertise for Schizophrenia (FACE-SZ) Study. Schizophr. Bull. Open 2021, 2, sgab012. [Google Scholar] [CrossRef]
- Miller, B.J.; Goldsmith, D.R. Evaluating the hypothesis that schizophrenia is an inflammatory disorder. Focus (Am. Psychiatr. Publ.) 2020, 18, 391–401. [Google Scholar] [CrossRef]
- Labonté, C.; Zhand, N.; Park, A.; Harvey, P.D. Complete blood count inflammatory markers in treatment-resistant schizophrenia: Evidence of association between treatment responsiveness and levels of inflammation. Psychiatry Res. 2022, 308, 114382. [Google Scholar] [CrossRef]
- Manchia, M.; Fontana, A.; Panebianco, C.; Paribello, P.; Arzedi, C.; Cossu, E.; Garzilli, M.; Montis, M.A.; Mura, A.; Pisanu, C.; et al. Involvement of Gut Microbiota in Schizophrenia and Treatment Resistance to Antipsychotics. Biomedicines 2021, 9, 875. [Google Scholar] [CrossRef]
- Seeman, M.V. The Gut Microbiome and Treatment-Resistance in Schizophrenia. Psychiatr. Q. 2020, 91, 127–136. [Google Scholar] [CrossRef]
- Manchia, M.; Squassina, A.; Tozzi, F.; Antoniades, A.; Carpiniello, B. Gut microbiota and treatment-resistant schizophrenia: Many questions, fewer answers. Pharmacogenomics 2022, 23, 277–280. [Google Scholar] [CrossRef]
- Teasdale, S.; Mörkl, S.; Müller-Stierlin, A.S. Nutritional Psychiatry in the treatment of psychotic disorders: Current hypotheses and research challenges. Brain Behav. Immun. Health 2020, 5, 100070. [Google Scholar] [CrossRef]
- Marx, W.; Moseley, G.; Berk, M.; Jacka, F. Nutritional psychiatry: The present state of the evidence. Proc. Nutr. Soc. 2017, 76, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Onaolapo, O.J.; Onaolapo, A.Y. Nutrition, nutritional deficiencies, and schizophrenia: An association worthy of constant reassessment. World J. Clin. Cases 2021, 9, 8295–8311. [Google Scholar] [CrossRef] [PubMed]
- Borst, K.; Dumas, A.A.; Prinz, M. Microglia: Immune and non-immune functions. Immunity 2021, 54, 2194–2208. [Google Scholar] [CrossRef] [PubMed]
- Lasselin, J. Back to the future of psychoneuroimmunology: Studying inflammation-induced sickness behavior. Brain Behav. Immun. Health 2021, 18, 100379. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cué, C.; Rueda, N. Cellular Senescence in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 16. [Google Scholar] [CrossRef] [PubMed]
- Lecours, C.; Bordeleau, M.; Cantin, L.; Parent, M.; Paolo, T.D.; Tremblay, M.È. Microglial implication in Parkinson’s disease: Loss of beneficial physiological roles or gain of inflammatory functions? Front. Cell. Neurosci. 2018, 12, 282. [Google Scholar] [CrossRef]
- Fonken, L.K.; Frank, M.G.; Gaudet, A.D.; Maier, S.F. Stress and aging act through common mechanisms to elicit neuroinflammatory priming. Brain Behav. Immun. 2018, 73, 133–148. [Google Scholar] [CrossRef]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflamm. 2021, 18, 258. [Google Scholar] [CrossRef]
- Dantzer, R. Neuroimmune interactions: From the brain to the immune system and vice versa. Physiol. Rev. 2018, 98, 477–504. [Google Scholar] [CrossRef]
- Mondelli, V.; Vernon, A.C.; Turkheimer, F.; Dazzan, P.; Pariante, C.M. Brain microglia in psychiatric disorders. Lancet Psychiatry 2017, 4, 563–572. [Google Scholar] [CrossRef]
- Amato, D.; Beasley, C.L.; Hahn, M.K.; Vernon, A.C. Neuroadaptations to antipsychotic drugs: Insights from pre-clinical and human post-mortem studies. Neurosci. Biobehav. Rev. 2017, 76, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Stapel, B.; Sieve, I.; Falk, C.S.; Bleich, S.; Hilfiker-Kleiner, D.; Kahl, K.G. Second generation atypical antipsychotics olanzapine and aripiprazole reduce expression and secretion of inflammatory cytokines in human immune cells. J. Psychiatr. Res. 2018, 105, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Fonken, L.K.; Annis, J.L.; Watkins, L.R.; Maier, S.F. Stress disinhibits microglia via down-regulation of CD200R: A mechanism of neuroinflammatory priming. Brain Behav. Immun. 2018, 69, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.W.; Kim, Y.K. The role of neuroinflammation and neurovascular dysfunction in major depressive disorder. J. Inflamm. Res. 2018, 11, 179. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.J. Cytokines, neurophysiology, neuropsychology, and psychiatric symptoms. Dialogues Clin. Neurosci. 2022, 5, 139–153. [Google Scholar] [CrossRef]
- Réus, G.Z.; Abelaira, H.M.; Coutellier, L.D.; Manosso, L.M.; Pavlovic, Z.M. Role of Glutamatergic Neurotransmission in the Pathophysiology of Stress-Related Disorders and Chronic Stress Response. In Glutamate and Neuropsychiatric Disorders; Springer International Publishing: Berlin/Heidelberg, Germany, 2022; pp. 65–112. [Google Scholar]
- Kumar, V.; Manchegowda, S.; Jacob, A.; Rao, N.P. Glutamate metabolites in treatment resistant schizophrenia: A meta-analysis and systematic review of 1H-MRS studies. Psychiatry Res. Neuroimaging 2020, 300, 111080. [Google Scholar] [CrossRef]
- Zubin, J.; Spring, B. Vulnerability-a new view of schizophrenia. J. Abnorm. Psychol. 1977, 86, 103–126. [Google Scholar] [CrossRef]
- Müller, N. Inflammation in Schizophrenia: Pathogenetic Aspects and Therapeutic Considerations. Schizophr. Bull. 2018, 44, 973–982. [Google Scholar] [CrossRef]
- Lumertz, F.S.; Kestering-Ferreira, E.; Orso, R.; Creutzberg, K.C.; Tractenberg, S.G.; Stocchero, B.A.; Viola, T.W.; Grassi-Oliveira, R. Effects of early life stress on brain cytokines: A systematic review and meta-analysis of rodent studies. Neurosci. Biobehav. Rev. 2022, 139, 104746. [Google Scholar] [CrossRef]
- Cheslack-Postava, K.; Brown, A.S. Prenatal infection and schizophrenia: A decade of further progress. Schizophr. Res. 2022, 247, 7–15. [Google Scholar] [CrossRef]
- Rovira, P.; Gutiérrez, B.; Sorlózano-Puerto, A.; Gutiérrez-Fernández, J.; Molina, E.; Rivera, M.; Martínez-Leal, R.; Ibanez-Casas, I.; Martín-Laguna, M.V.; Rosa, A.; et al. Toxoplasma gondii Seropositivity Interacts with Catechol-O-methyltransferase Val105/158Met Variation Increasing the Risk of Schizophrenia. Genes 2022, 13, 1088. [Google Scholar] [CrossRef] [PubMed]
- Stilo, S.A.; Murray, R.M. Non-genetic factors in schizophrenia. Curr. Psychiatry Rep. 2019, 21, 100. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, M.B.; Cavalcante, C.T.D.M.B.; Sarno, M.; Barini, R.; Kwak-Kim, J. Maternal immune responses and obstetrical outcomes of pregnant women with COVID-19 and possible health risks of offspring. J. Reprod. Immunol. 2021, 143, 103250. [Google Scholar] [CrossRef] [PubMed]
- Minakova, E.; Warner, B.B. Maternal immune activation, central nervous system development and behavioral phenotypes. Birth Defects Res. 2018, 110, 1539–1550. [Google Scholar] [CrossRef]
- Benros, M.E.; Mortensen, P.B. Role of Infection, Autoimmunity, Atopic Disorders, and the Immune System in Schizophrenia: Evidence from Epidemiological and Genetic Studies. Curr. Top. Behav. Neurosci. 2020, 44, 141–159. [Google Scholar]
- Lee, Y.H.; Cherkerzian, S.; Seidman, L.J.; Papandonatos, G.D.; Savitz, D.A.; Tsuang, M.T.; Goldstein, J.M.; Buka, S.L. Maternal Bacterial Infection During Pregnancy and Offspring Risk of Psychotic Disorders: Variation by Severity of Infection and Offspring Sex. Am. J. Psychiatry 2020, 177, 66–75. [Google Scholar] [CrossRef]
- Vasistha, N.A.; Pardo-Navarro, M.; Gasthaus, J.; Weijers, D.; Müller, M.K.; García-González, D.; Malwade, S.; Korshunova, I.; Pfisterer, U.; von Engelhardt, J.; et al. Maternal inflammation has a profound effect on cortical interneuron development in a stage and subtype-specific manner. Mol. Psychiatry 2020, 25, 2313–2329. [Google Scholar] [CrossRef]
- Lydholm, C.N.; Köhler-Forsberg, O.; Nordentoft, M.; Yolken, R.H.; Mortensen, P.B.; Petersen, L.; Benros, M.E. Parental Infections Before, During, and After Pregnancy as Risk Factors for Mental Disorders in Childhood and Adolescence: A Nationwide Danish Study. Biol. Psychiatry 2019, 85, 317–325. [Google Scholar] [CrossRef]
- Chaves Filho, A.; Mottin, M.; Lós, D.B.; Andrade, C.H.; Macedo, D.S. The tetrapartite synapse in neuropsychiatric disorders: Matrix metalloproteinases (MMPs) as promising targets for treatment and rational drug design. Biochimie 2022, 201, 79–99. [Google Scholar] [CrossRef]
- Meehan, C.; Harms, L.; Frost, J.D.; Barreto, R.; Todd, J.; Schall, U.; Shannon Weickert, C.; Zavitsanou, K.; Michie, P.T.; Hodgson, D.M. Effects of immune activation during early or late gestation on schizophrenia-related behaviour in adult rat offspring. Brain Behav. Immun. 2017, 63, 8–20. [Google Scholar] [CrossRef]
- Müller, N. Inflammation and Immunity in Schizophrenia. Immuno-Psychiatry 2021, 2, 227–240. [Google Scholar]
- Metcalf, S.A.; Jones, P.B.; Nordstrom, T.; Timonen, M.; Mäki, P.; Miettunen, J.; Jääskeläinen, E.; Järvelin, M.R.; Stochl, J.; Murray, G.K.; et al. Serum C-reactive protein in adolescence and risk of schizophrenia in adulthood: A prospective birth cohort study. Brain Behav. Immun. 2017, 59, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Orlovska-Waast, S.; Benros, M.E. Autoimmune Diseases and Infections as Risk Factors for Mental Disorders. In Immuno-Psychiatry; Springer: Berlin/Heidelberg, Germany, 2021; pp. 3–16. [Google Scholar]
- Momtazmanesh, S.; Zare-Shahabadi, A.; Rezaei, N. Cytokine alterations in schizophrenia: An updated review. Front. Psychiatry 2019, 10, 892. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.R.; Rapaport, M.H.; Miller, B.J. A meta-analysis of blood cytokine network alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.J.; Buckley, P.; Seabolt, W.; Mellor, A.; Kirkpatrick, B. Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry 2011, 70, 663–671. [Google Scholar] [CrossRef]
- Orlovska-Waast, S.; Köhler-Forsberg, O.; Brix, S.W.; Nordentoft, M.; Kondziella, D.; Krogh, J.; Benros, M.E. Cerebrospinal fluid markers of inflammation and infections in schizophrenia and affective disorders: A systematic review and meta-analysis. Mol. Psychiatry 2019, 24, 869–887. [Google Scholar] [CrossRef]
- Wang, A.K.; Miller, B.J. Meta-analysis of Cerebrospinal Fluid Cytokine and Tryptophan Catabolite Alterations in Psychiatric Patients: Comparisons Between Schizophrenia, Bipolar Disorder, and Depression. Schizophr. Bull. 2018, 44, 75–83. [Google Scholar] [CrossRef]
- Brandon, A.; Cui, X.; Luan, W.; Ali, A.A.; Pertile, R.A.N.; Alexander, S.A.; Eyles, D.W. Prenatal hypoxia alters the early ontogeny of dopamine neurons. Transl. Psychiatry 2022, 12, 238. [Google Scholar] [CrossRef]
- Wang, M.; Ling, K.H.; Tan, J.J.; Lu, C.B. Development and differentiation of midbrain dopaminergic neuron: From bench to bedside. Cells 2020, 9, 1489. [Google Scholar] [CrossRef]
- Jarskog, L.F.; Xiao, H.; Wilkie, M.B.; Lauder, J.M.; Gilmore, J.H. Cytokine regulation of embryonic rat dopamine and serotonin neuronal survival in vitro. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 1997, 15, 711–716. [Google Scholar] [CrossRef]
- Romeo, B.; Brunet-Lecomte, M.; Martelli, C.; Benyamina, A. Kinetics of cytokine levels during antipsychotic treatment in schizophrenia: A meta-analysis. Int. J. Neuropsychopharmacol. 2018, 21, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Scheiber, C.; Schulz, T.; Schneider, J.M.; Bechter, K.; Schneider, E.M. Old and New Biomarkers for Infection, Inflammation, and Autoimmunity in Treatment-Resistant Affective and Schizophrenic Spectrum Disorders. Pharmaceuticals 2022, 15, 299. [Google Scholar] [CrossRef] [PubMed]
- Dziurkowska, E.; Wesolowski, M. Cortisol as a Biomarker of Mental Disorder Severity. J. Clin. Med. 2021, 10, 5204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Zhou, D.F.; Cao, L.Y.; Wu, G.Y.; Shen, Y.C. Cortisol and cytokines in chronic and treatment-resistant patients with schizophrenia: Association with psychopathology and response to antipsychotics. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2005, 30, 1532–1538. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef]
- Dawidowski, B.; Górniak, A.; Podwalski, P.; Lebiecka, Z.; Misiak, B.; Samochowiec, J. The Role of Cytokines in the Pathogenesis of Schizophrenia. J. Clin. Med. 2021, 10, 3849. [Google Scholar] [CrossRef]
- Kravtsov, V.V.; Shnayder, N.A.; Neznanov, N.G.; Krivopalov, A.A.; Yanov, Y.K.; Nasyrova, R.F.; Shamkina, P.A.; Gavrilyuk, O.A. Genetic predictors of cytokine response in ENT-associated encephalitis. Pers. Psychiatry Neurol. 2021, 1, 18–36. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L.; Verdugo-Rodriguez, A.; Rodriguez, L.L.; Borca, M.V. The role of interleukin 6 during viral infections. Front. Microbiol. 2019, 10, 1057. [Google Scholar] [CrossRef]
- Chauhan, P.; Nair, A.; Patidar, A.; Dandapat, J.; Sarkar, A.; Saha, B. A primer on cytokines. Cytokine 2021, 145, 155458. [Google Scholar] [CrossRef]
- Zádor, F.; Joca, S.; Nagy-Grócz, G.; Dvorácskó, S.; Szűcs, E.; Tömböly, C.; Benyhe, S.; Vécsei, L. Pro-Inflammatory Cytokines: Potential Links between the Endocannabinoid System and the Kynurenine Pathway in Depression. Int. J. Mol. Sci. 2021, 22, 5903. [Google Scholar] [CrossRef]
- Vallée, A. Neuroinflammation in Schizophrenia: The Key Role of the WNT/β-Catenin Pathway. Int. J. Mol. Sci. 2022, 23, 2810. [Google Scholar] [CrossRef] [PubMed]
- Boiko, A.S.; Mednova, I.A.; Kornetova, E.G.; Gerasimova, V.I.; Kornetov, A.N.; Loonen, A.J.M.; Bokhan, N.A.; Ivanova, S.A. Cytokine level changes in schizophrenia patients with and without metabolic syndrome treated with atypical antipsychotics. Pharmaceuticals 2021, 14, 446. [Google Scholar] [CrossRef] [PubMed]
- Mendiola, A.S.; Cardona, A.E. The IL-1β phenomena in neuroinflammatory diseases. J. Neural Transm. 2018, 125, 781–795. [Google Scholar] [CrossRef]
- Wooff, Y.; Man, S.M.; Aggio-Bruce, R.; Natoli, R.; Fernando, N. IL-1 family members mediate cell death, inflammation and angiogenesis in retinal degenerative diseases. Front. Immunol. 2019, 10, 1618. [Google Scholar] [CrossRef] [PubMed]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Enache, D.; Nikkheslat, N.; Fathalla, D.; Morgan, B.P.; Lewis, S.; Drake, R.; Deakin, B.; Walters, J.; Lawrie, S.M.; Egerton, A.; et al. Peripheral immune markers and antipsychotic non-response in psychosis. Schizophr. Res. 2021, 230, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, S.R.; Al-Rawi, K.F.; Stoyanov, D.; Al-Dujaili, A.H.; Supasitthumrong, T.; Al-Hakeim, H.K.; Maes, M. The endogenous opioid system in schizophrenia and treatment resistant schizophrenia: Increased plasma endomorphin 2, and κ and μ opioid receptors are associated with interleukin-6. Diagnostics 2020, 10, 633. [Google Scholar] [CrossRef]
- Subedi, L.; Lee, S.E.; Madiha, S.; Gaire, B.P.; Jin, M.; Yumnam, S.; Kim, S.Y. Phytochemicals against TNFα-Mediated Neuroinflammatory Diseases. Int. J. Mol. Sci. 2020, 21, 764. [Google Scholar] [CrossRef]
- Desu, H.L.; Illiano, P.; Choi, J.S.; Ascona, M.C.; Gao, H.; Lee, J.K.; Brambilla, R. TNFR2 signaling regulates the immunomodulatory function of oligodendrocyte precursor cells. Cells 2021, 10, 1785. [Google Scholar] [CrossRef]
- Inoubli, O.; Jemli, A.; Ben Fredj, S.; Mechri, A.; Gaha, L.; Bel Hadj Jrad, B. Haplotypes of TNFα/β Genes Associated with Sex-Specific Paranoid Schizophrenic Risk in Tunisian Population. Dis. Markers 2018, 2018, 3502564. [Google Scholar] [CrossRef]
- Aytac, H.M.; Ozdilli, K.; Tuncel, F.C.; Pehlivan, M.; Pehlivan, S. Tumor Necrosis Factor-alpha (TNF-α) -238 G/A Polymorphism Is Associated with the Treatment Resistance and Attempted Suicide in Schizophrenia. Immunol. Investig. 2022, 51, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Noto, C.; Maes, M.; Ota, V.K.; Teixeira, A.L.; Bressan, R.A.; Gadelha, A.; Brietzke, E. High predictive value of immune-inflammatory biomarkers for schizophrenia diagnosis and association with treatment resistance. World J. Biol. Psychiatry Off. J. World Fed. Soc. Biol. Psychiatry 2015, 16, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Ta, T.T.; Dikmen, H.O.; Schilling, S.; Chausse, B.; Lewen, A.; Hollnagel, J.O.; Kann, O. Priming of microglia with IFN-γ slows neuronal gamma oscillations in situ. Proc. Natl. Acad. Sci. USA 2019, 116, 4637–4642. [Google Scholar] [CrossRef] [PubMed]
- Upthegrove, R.; Khandaker, G.M. Cytokines, Oxidative Stress and Cellular Markers of Inflammation in Schizophrenia. Curr. Top. Behav. Neurosci. 2020, 44, 49–66. [Google Scholar] [PubMed]
- Kaplanski, G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol. Rev. 2018, 281, 138–153. [Google Scholar] [CrossRef]
- Syed, A.A.S.; He, L.; Shi, Y.; Mahmood, S. Elevated levels of IL-18 associated with schizophrenia and first episode psychosis: A systematic review and meta-analysis. Early Interv. Psychiatry 2021, 15, 896–905. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef]
- Dunleavy, C.; Elsworthy, R.J.; Upthegrove, R.; Wood, S.J.; Aldred, S. Inflammation in first-episode psychosis: The contribution of inflammatory biomarkers to the emergence of negative symptoms, a systematic review and meta-analysis. Acta Psychiatr. Scand. 2022, 146, 6–20. [Google Scholar] [CrossRef]
- Fang, X.; Zhang, Y.; Fan, W.; Tang, W.; Zhang, C. Interleukin-17 Alteration in First-Episode Psychosis: A Meta-Analysis. Mol. Neuropsychiatry 2017, 3, 135–140. [Google Scholar] [CrossRef]
- Marcinowicz, P.; Więdłocha, M.; Zborowska, N.; Dębowska, W.; Podwalski, P.; Misiak, B.; Tyburski, E.; Szulc, A. A Meta-Analysis of the Influence of Antipsychotics on Cytokines Levels in First Episode Psychosis. J. Clin. Med. 2021, 10, 2488. [Google Scholar] [CrossRef]
- Pandolfo, G.; Genovese, G.; Casciaro, M.; Muscatello, M.; Bruno, A.; Pioggia, G.; Gangemi, S. IL-33 in Mental Disorders. Medicina 2021, 57, 315. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Chen, H.Y.; Lin, J.J.; Lu, M.K.; Tan, H.P.; Jang, F.L.; Lin, S.H. Alterations of plasma cytokine biomarkers for identifying age at onset of schizophrenia with neurological soft signs. Int. J. Med. Sci. 2020, 17, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Kenis, G.; Bignotti, S.; Tura, G.J.; De Jong, R.; Bosmans, E.; Pioli, R.; Altamura, C.; Scharpé, S.; Maes, M. The inflammatory response system in treatment-resistant schizophrenia: Increased serum interleukin-6. Schizophr. Res. 1998, 32, 9–15. [Google Scholar] [CrossRef]
- Kartalcı, Ş.; Gönenir Erbay, L.; Porgalı Zayman, E.; Otlu, Ö.; Bay Karabulut, A.; Kartalcı, G. IL-4, TGF-β, NF-κB and MPO levels in Patients with Treatment Resistant Schizophrenia. Tedaviye Dirençli Şizofreni Hastalarında IL-4, TGF-β, NF-κB ve MPO Düzeyleri. Turk psikiyatri dergisi = Turk. J. Psychiatry 2016, 27, 170–175. [Google Scholar]
- Luo, Y.; He, H.; Zhang, J.; Ou, Y.; Fan, N. Changes in serum TNF-α, IL-18, and IL-6 concentrations in patients with chronic schizophrenia at admission and at discharge. Compr. Psychiatry 2019, 90, 82–87. [Google Scholar] [CrossRef]
- Mongan, D.; Ramesar, M.; Föcking, M.; Cannon, M.; Cotter, D. Role of inflammation in the pathogenesis of schizophrenia: A review of the evidence, proposed mechanisms and implications for treatment. Early Interv. Psychiatry 2020, 14, 385–397. [Google Scholar] [CrossRef]
- Schwieler, L.; Larsson, M.K.; Skogh, E.; Kegel, M.E.; Orhan, F.; Abdelmoaty, S.; Finn, A.; Bhat, M.; Samuelsson, M.; Lundberg, K.; et al. Increased levels of IL-6 in the cerebrospinal fluid of patients with chronic schizophrenia--significance for activation of the kynurenine pathway. J. Psychiatry Neurosci. JPN 2015, 40, 126–133. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, Y.; Li, C.; Zhou, C.; Tang, X.; Zhu, Q. Analysis of interleukin levels in patients with treatment-resistant schizophrenia. J. Clin. Med. Pract. 2020, 24, 96–98. [Google Scholar]
- Reale, M.; Costantini, E.; Greig, N.H. Cytokine Imbalance in Schizophrenia. From Research to Clinic: Potential Implications for Treatment. Front. Psychiatry 2021, 12, 536257. [Google Scholar] [CrossRef]
- Wilbers, R.; van Raaij, D.R.; Westerhof, L.B.; Bakker, J.; Smant, G.; Schots, A. Re-evaluation of IL-10 signaling reveals novel insights on the contribution of the intracellular domain of the IL-10R2 chain. PLoS ONE 2017, 12, e0186317. [Google Scholar] [CrossRef]
- Saxton, R.A.; Tsutsumi, N.; Su, L.L.; Abhiraman, G.C.; Mohan, K.; Henneberg, L.T.; Aduri, N.G.; Gati, C.; Garcia, K.C. Structure-based decoupling of the pro- and anti-inflammatory functions of interleukin-10. Science 2021, 371, eabc8433. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.R.; Cherian, J.; Gohil, K.; Atkinson, D. Schizophrenia: Overview and treatment options. P T: A Peer-Rev. J. Formul. Manag. 2014, 39, 638–645. [Google Scholar]
- Zubov, D.S.; Dorofeikova, M.V.; Petrova, N.N.; Dorofeykov, V.V.; Ivanov, M.V. Changes in levels of neuromarkers and cognitive functioning of patients with treatment-resistant schizophrenia. V.M. Bekhterev Rev. Psychiatry Med. Psychol. 2016, 3, 51–56. [Google Scholar]
- Melbourne, J.K.; Feiner, B.; Rosen, C.; Sharma, R.P. Targeting the Immune System with Pharmacotherapy in Schizophrenia. Curr. Treat. Options Psychiatry 2017, 4, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Kroken, R.A.; Sommer, I.E.; Steen, V.M.; Dieset, I.; Johnsen, E. Constructing the Immune Signature of Schizophrenia for Clinical Use and Research; An Integrative Review Translating Descriptives Into Diagnostics. Front. Psychiatry 2019, 9, 753. [Google Scholar] [CrossRef]
- Sommer, I.E.; van Westrhenen, R.; Begemann, M.J.; de Witte, L.D.; Leucht, S.; Kahn, R.S. Efficacy of anti-inflammatory agents to improve symptoms in patients with schizophrenia: An update. Schizophr. Bull. 2014, 40, 181–191. [Google Scholar] [CrossRef]
- Akhondzadeh, S.; Tabatabaee, M.; Amini, H.; Ahmadi Abhari, S.A.; Abbasi, S.H.; Behnam, B. Celecoxib as adjunctive therapy in schizophrenia: A double-blind, randomized and placebo-controlled trial. Schizophr. Res. 2007, 90, 179–185. [Google Scholar] [CrossRef]
- Müller, N.; Riedel, M.; Scheppach, C.; Brandstätter, B.; Sokullu, S.; Krampe, K.; Ulmschneider, M.; Engel, R.R.; Möller, H.J.; Schwarz, M.J. Beneficial antipsychotic effects of celecoxib add-on therapy compared to risperidone alone in schizophrenia. Am. J. Psychiatry 2002, 159, 1029–1034. [Google Scholar] [CrossRef]
- Attari, A.; Mojdeh, A.; Soltani, F.A.S.K.; Najarzadegan, M.R. Aspirin inclusion in antipsychotic treatment on severity of symptoms in schizophrenia: A randimized clinical trial. Iran. J. Psychiatry Behav. Sci. 2017, 11, e5848. [Google Scholar]
- Laan, W.; Grobbee, D.E.; Selten, J.P.; Heijnen, C.J.; Kahn, R.S.; Burger, H. Adjuvant aspirin therapy reduces symptoms of schizophrenia spectrum disorders: Results from a randomized, double-blind, placebo-controlled trial. J. Clin. Psychiatry 2010, 71, 520–527. [Google Scholar] [CrossRef]
- Shen, H.; Li, R.; Yan, R.; Zhou, X.; Feng, X.; Zhao, M.; Xiao, H. Adjunctive therapy with statins in schizophrenia patients: A meta-analysis and implications. Psychiatry Res. 2018, 262, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, B.; Stock, S.; Borba, C.P.; Cleary, S.M.; Oppenheim, C.E.; Petruzzi, L.J.; Fan, X.; Copeland, P.M.; Freudenreich, O.; Cather, C.; et al. A randomized placebo-controlled pilot study of pravastatin as an adjunctive therapy in schizophrenia patients: Effect on inflammation, psychopathology, cognition and lipid metabolism. Schizophr. Res. 2014, 159, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Rees, L.; King, G.M. Intensive Cortisone Therapy in Schizophrenia. J. Ment. Sci. 1956, 102, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Nasib, L.G.; Sommer, I.E.; Winter-van Rossum, I.; de Vries, J.; Gangadin, S.S.; Oomen, P.P.; Judge, G.; Blom, R.E.; Luykx, J.J.; van Beveren, N.; et al. Prednisolone versus placebo addition in the treatment of patients with recent-onset psychotic disorder: A trial design. Trials 2020, 21, 492. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.J.; Dias, J.K.; Lemos, H.P.; Buckley, P.F. An open-label, pilot trial of adjunctive tocilizumab in schizophrenia. J. Clin. Psychiatry 2016, 77, 275–276. [Google Scholar] [CrossRef]
- Girgis, R.R.; Ciarleglio, A.; Choo, T.; Haynes, G.; Bathon, J.M.; Cremers, S.; Kantrowitz, J.T.; Lieberman, J.A.; Brown, A.S. A Randomized, Double-Blind, Placebo-Controlled Clinical Trial of Tocilizumab, An Interleukin-6 Receptor Antibody, For Residual Symptoms in Schizophrenia. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2018, 43, 1317–1323. [Google Scholar] [CrossRef]
- Grüber, L.; Bunse, T.; Weidinger, E.; Reichard, H.; Müller, N. Adjunctive recombinant human interferon gamma-1b for treatment-resistant schizophrenia in 2 patients. J. Clin. Psychiatry 2014, 75, 1266–1267. [Google Scholar] [CrossRef]
- Lennox, B.R.; Tomei, G.; Vincent, S.A.; Yeeles, K.; Pollard, R.; Palmer-Cooper, E.; Jones, P.; Zandi, M.S.; Coles, A. Study of immunotherapy in antibody positive psychosis: Feasibility and acceptability (SINAPPS1). J. Neurol. Neurosurg. Psychiatry 2019, 90, 365–367. [Google Scholar] [CrossRef]
- Zandi, M.S.; Deakin, J.B.; Morris, K.; Buckley, C.; Jacobson, L.; Scoriels, L.; Cox, A.L.; Coles, A.J.; Jones, P.B.; Vincent, A.; et al. Immunotherapy for patients with acute psychosis and serum N-Methyl D-Aspartate receptor (NMDAR) antibodies: A description of a treated case series. Schizophr. Res. 2014, 160, 193–195. [Google Scholar] [CrossRef]
- Berk, M.; Copolov, D.; Dean, O.; Lu, K.; Jeavons, S.; Schapkaitz, I.; Anderson-Hunt, M.; Judd, F.; Katz, F.; Katz, P.; et al. N-acetyl cysteine as a glutathione precursor for schizophrenia--a double-blind, randomized, placebo-controlled trial. Biol. Psychiatry 2008, 64, 361–368. [Google Scholar] [CrossRef]
- Farokhnia, M.; Azarkolah, A.; Adinehfar, F.; Khodaie-Ardakani, M.R.; Hosseini, S.M.; Yekehtaz, H.; Tabrizi, M.; Rezaei, F.; Salehi, B.; Sadeghi, S.M.; et al. N-acetylcysteine as an adjunct to risperidone for treatment of negative symptoms in patients with chronic schizophrenia: A randomized, double-blind, placebo-controlled study. Clin. Neuropharmacol. 2013, 36, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.Q.; Zheng, W.; Wang, S.B.; Yang, X.H.; Cai, D.B.; Ng, C.H.; Ungvari, G.S.; Kelly, D.L.; Xu, W.Y.; Xiang, Y.T. Adjunctive minocycline for schizophrenia: A meta-analysis of randomized controlled trials. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2017, 27, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.; Lee, T.Y.; Kwak, Y.B.; Yoon, Y.B.; Kim, M.; Kwon, J.S. Adjunctive use of anti-inflammatory drugs for schizophrenia: A meta-analytic investigation of randomized controlled trials. Aust. N. Z. J. Psychiatry 2019, 53, 742–759. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Bang, M. Anti-inflammatory Strategies for Schizophrenia: A Review of Evidence for Therapeutic Applications and Drug Repurposing. Clin. Psychopharmacol. Neurosci. Off. Sci. J. Korean Coll. Neuropsychopharmacol. 2020, 18, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Saredy, J.; Zhang, R.; Shao, Y.; Sun, Y.; Yang, W.Y.; Wang, J.; Liu, L.; Drummer, C., 4th; Johnson, C.; et al. Approaching Inflammation Paradoxes-Proinflammatory Cytokine Blockages Induce Inflammatory Regulators. Front. Immunol. 2020, 11, 554301. [Google Scholar] [CrossRef]
- de Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.G.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef]
- Fuster, J.J.; Walsh, K. Somatic Mutations and Clonal Hematopoiesis: Unexpected Potential New Drivers of Age-Related Cardiovascular Disease. Circ. Res. 2018, 122, 523–532. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Bennett, J.M.; Reeves, G.; Billman, G.E.; Sturmberg, J.P. Inflammation-Nature’s Way to Efficiently Respond to All Types of Challenges: Implications for Understanding and Managing “the Epidemic” of Chronic Diseases. Front. Med. 2018, 5, 316. [Google Scholar] [CrossRef]
- Johnson, C.; Drummer, C., 4th; Virtue, A.; Gao, T.; Wu, S.; Hernandez, M.; Singh, L.; Wang, H.; Yang, X.F. Increased Expression of Resistin in MicroRNA-155-Deficient White Adipose Tissues May Be a Possible Driver of Metabolically Healthy Obesity Transition to Classical Obesity. Front. Physiol. 2018, 9, 1297. [Google Scholar] [CrossRef]





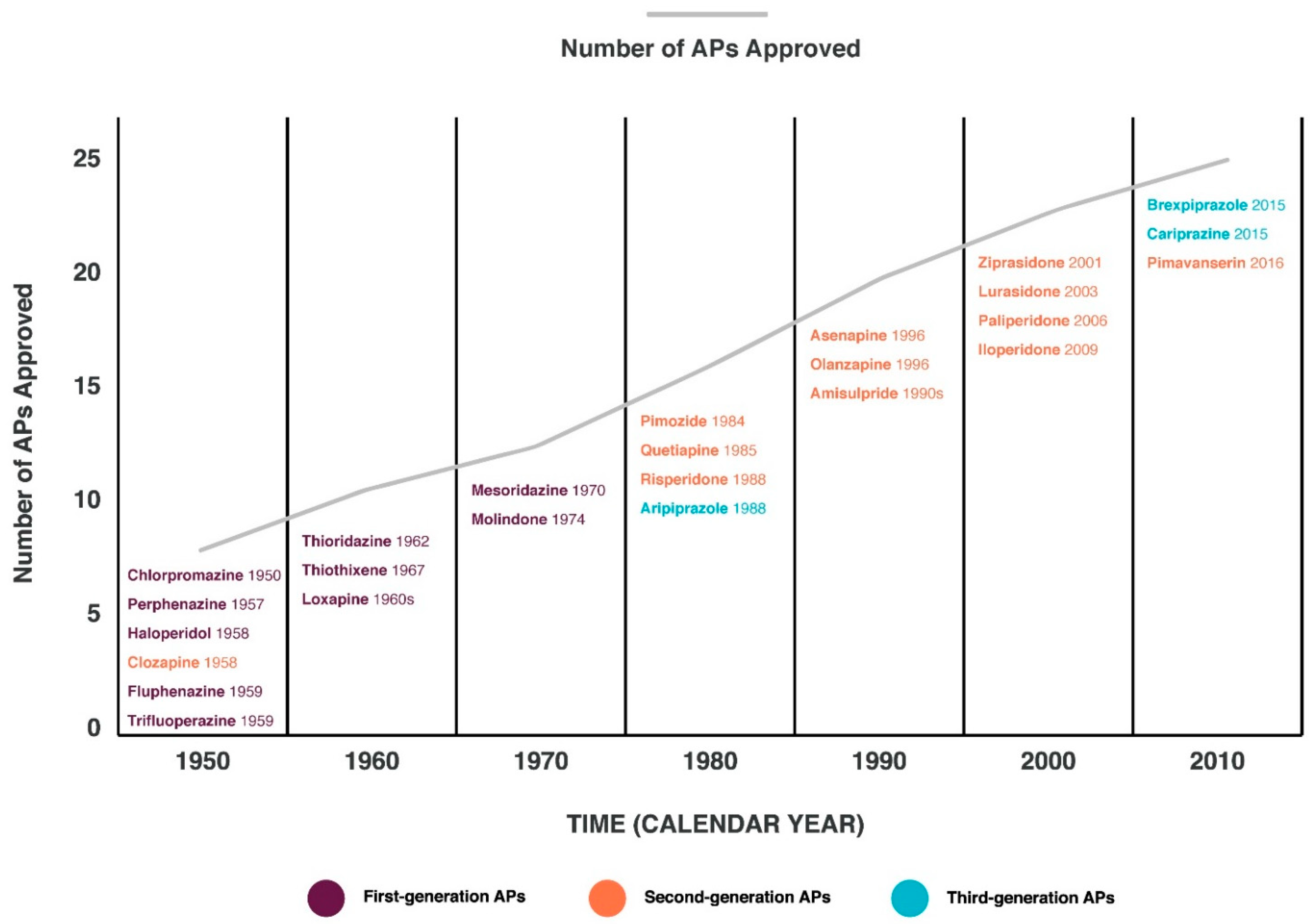{kind=link}
{kind=link}
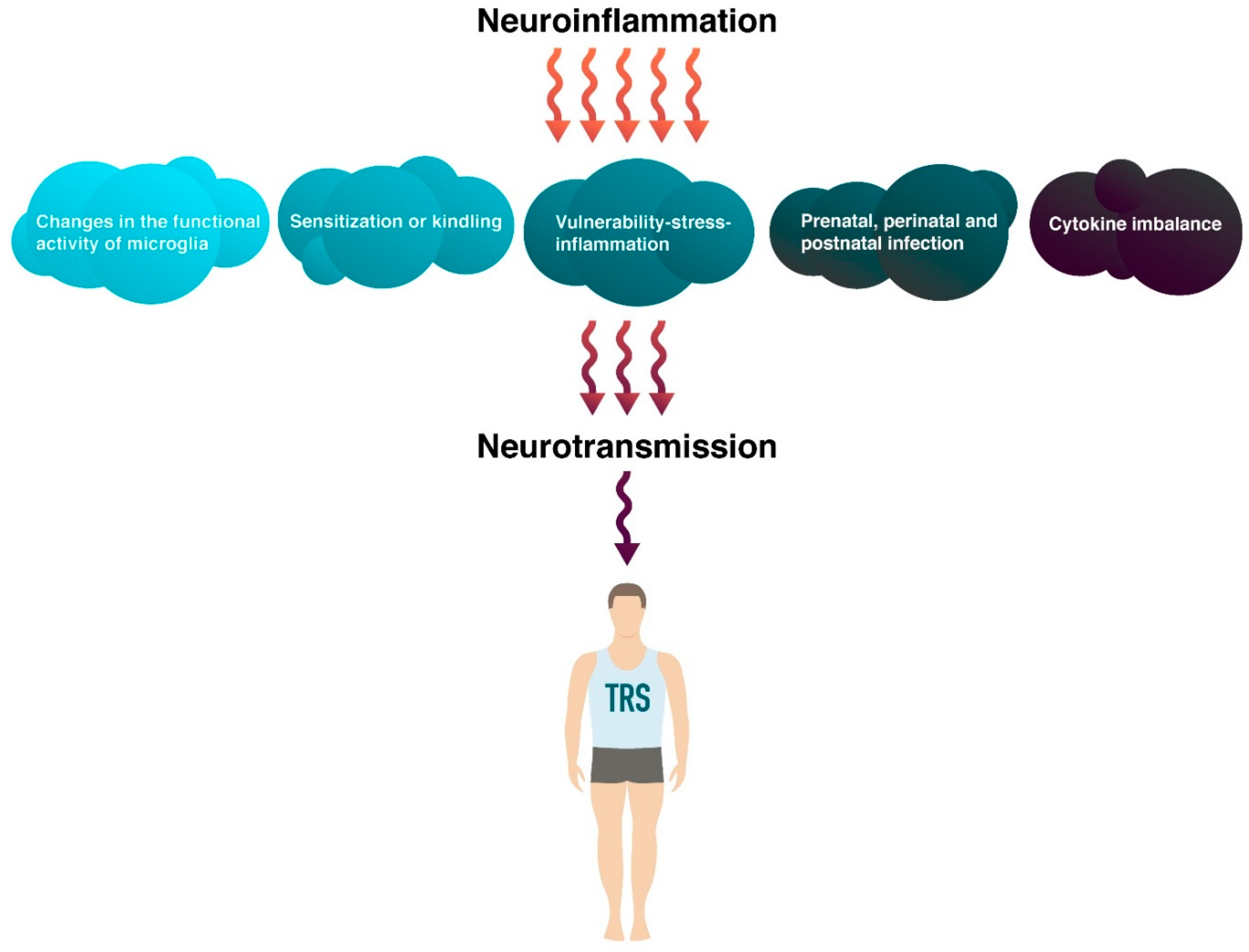{kind=link}
{kind=link}
| Hypothesis | Mechanism | References |
|---|---|---|
| Genetic | Genetic predisposition to low affinity of targets (dopaminergic receptors) to APs of the first and new generations. | [22,23] |
| Neurodevelopmental | Congenital minor anomalies of brain development (microdysgenesis) in brain regions critical for Sch development. | [24,25] |
| Neurotransmitter | Violation of the synthesis, release, or breakdown of dopamine and other neurotransmitters (serotonin, melatonin, etc.). | [19,26,27,28] |
| Metabolic | Primary (genetically determined) and secondary metabolic disorders of APs of the first and new generation in the liver. | [29,30,31] |
| Transport | Primary (genetically determined) and secondary impairment of expression and/or functional activity of APs transporter proteins of the first and new generations across the blood–brain barrier. | [32,33,34] |
| Oxidative stress | Violation of the prooxidant-antioxidant balance in favor of the former, which leads to oxidative damage to cellular lipids, proteins, enzymes, carbohydrates and DNA, which contributes to a worsening of the course and an unfavorable outcome of Sch. | [35,36,37] |
| Inflammatory | Primary (genetically determined) and secondary violation of the cytokine status (absolute or relative hyperproduction of pro-inflammatory cytokines). | [38,39,40] |
| Microbiome | Microbiota through the gut–brain axis is associated with the development and severity of Sch, intestinal microbiota is associated with the response to APs. | [41,42,43] |
| Nutritional | Deficiency or excess of nutrients (vitamins, minerals, amino acids) necessary for the functioning of the dopaminergic system of the brain. | [44,45,46] |
| Author, Year | Mechanism | Pathogenesis | Reference |
|---|---|---|---|
| Meehan et al., 2017 | Prenatal, perinatal and postnatal infection | Immune activation. Violation of neurogenesis processes, including dopaminergic and glutamatergic neurotransmission. | [75] |
| Frank et al., 2018 | Sensitization or kindling | Stimulation of the immune response. Activation of cell proliferation, increased production and release of pro-inflammatory cytokines. | [57] |
| Momtazmanesh et al., 2019 | Cytokine imbalance | Increased serum levels of pro-inflammatory cytokines IL-1β, IL-6 and TGF-β. | [79] |
| Wang et al., 2020 | Cytokine imbalance | Interactions between cytokines and neurotransmitters in certain areas of the brain, as well as during brain development. Induction of IL-1β conversion of mesencephalic progenitor cells into a dopaminergic phenotype. Reduced survival of serotonergic neurons through IL-6. | [83] |
| Kumar et al., 2020 | Sensitization or kindling | Stimulation of the glutamatergic system, ionotropic and metabotropic glutamate receptors that excite amino acid transporters. Increased levels of glutamate in the anterior cingulate cortex. | [61] |
| Woodburn et al., 2021 | Changes in the functional activity of microglia | Priming of microglia causes an exaggerated immune response. Proliferation and increased production of pro-inflammatory cytokines. | [52] |
| Müller et al., 2021 | Prenatal, perinatal and postnatal infection | Increased levels of CRP and pro-inflammatory cytokines in childhood. | [76] |
| Dziurkowska et al., 2021 | Cytokine imbalance | Increased plasma levels of IL-2 and IL-6, activation of IRS. Positive correlation of IL-2, IL-6 and cortisol, hypercortisolemia. | [89] |
| Woodburn et al., 2021 | Sensitization or kindling | Pro-inflammatory immune response in the CNS. Activation and proliferation of microglia. Mediated neurotransmitter disorders. | [52] |
| Rovira et al., 2022 | Prenatal, perinatal and postnatal infection | Violation of the structure, exposure to inflammatory factors, neurochemical changes. Increased dopamine levels, impaired COMT activity. | [66] |
| Pro-Inflammatory Cytokines | Anti-Inflammatory Cytokines |
|---|---|
| Ciliary neurotrophic factor (CNTF) Granulocytic-macrophage colony-stimulating factor (GM-CSF) Interferon gamma (IFN-γ) Interleukin 20 (IL-20) Interleukin 1 alpha (IL1-α) Interleukin 1 β (IL1-β) Interleukin 11 (IL-11) Interleukin 12 (IL-12) Interleukin 17 (IL-17) Interleukin 18 (IL-18) Interleukin 18 (IL-8) Interleukin 33 (IL-33) Interleukin 6 (IL-6) Leukemia inhibitory factor (LIF) Oncostatin M (OSM) Transforming growth factor beta (TGF-β) Tumor necrosis factor alpha (TNF-α) | Interleukin 1 receptor antagonist (IL-1Ra) Interleukin 10 (IL-10) Interleukin 11 (IL-11) Interleukin 13 (IL-13) Interleukin 4 (IL-4) Interleukin 6 (IL-6) Interleukin-18-binding protein (IL-18BP) Transforming growth factor beta (TGF-β) |
| Cytokine | Gene: OMIM | Role in Neuroinflammation | Role in TRS | References |
|---|---|---|---|---|
| IL-1β | IL1B: 147720 | Stimulation of the synthesis of other pro-inflammatory and chemotactic mediators in the CNS. Stimulation of aberrant release and accumulation of glutamate, which subsequently leads to neuronal death in most neurodegenerative diseases. | +/− or + | [99,100,101,102] |
| TNF-α | TNFA: 191160 | Regulation of several processes including sleep, learning and memory, synaptic plasticity and astrocytic-induced synaptic strengthening. Initiation of inflammatory, apoptotic and neurodegenerative cascades, while TNF-α signaling via TNFR2 is anti-inflammatory and cytoprotective, resulting in induction of proliferation, differentiation, angiogenesis and tissue repair. | +++ | [38,104,105,106,107,108] |
| IFN-γ | IFNG: 147570 | Priming of microglia, which is associated with various cellular adaptations, including changes in morphology, upregulation of receptors and an increase in pro-inflammatory cytokines. | +/− | [102,109,110] |
| IL-12A | IL12A: 161560 | Stimulation of proliferation. Activation and increase in the cytotoxicity of NK cells and T cells. Stimulation of differentiation in Th1. Induction of IFN-γ and TNF-α secretion, synergism with pro-inflammatory cytokines with IL-18. | +++ | [38,92] |
| IL-18 | IL18: 600953 | Potentiation of the development of the relationship between the immune and nervous systems, since IL-18 and its receptors in the CNS mediate neuroinflammation of the brain, modulating homeostasis and behavior. | ++ | [111,112] |
| IL-8 | CXCL8: 146930 | Increased migration of neutrophils, T cells and monocytes, whose enzymes produce free oxygen radicals Indirect increase in oxidative stress, which can lead to neuronal death. | +++ | [102,113,114] |
| IL-17 | IL17A: 603149 | Stimulation of macrophages and microglia to secrete pro-inflammatory cytokines in the CNS. | +++ | [38,109,115] |
| Cytokine | Gene: OMIM | Role in Neuroinflammation | Role in TRS | References |
|---|---|---|---|---|
| IL-4 | IL4: 147780 | Initiation of T-helper differentiation into T-helper 2 lymphocytes. Increased Th2 cytotoxicity. Modulation of the function of macrophages and microglial cells. Decreased cytotoxicity. | +/− | [118,119] |
| IL-6 | IL6: 147620 | A key role in the processes associated with immunity and neuroinflammation. Modulation of the sensitivity of neurons to neurotransmitters. | +++ | [120,121,122,123,124] |
| IL-10 | IL10: 124092 | Initiation of cellular effects through canonical JAK/ STAT, which includes JAK1 and STAT3. Induction of expression of genes associated with immunosuppression. | +++ | [38,116,125,126,127] |
| Drug | Mechanism | Results | References |
|---|---|---|---|
| Non-steroidal anti-inflammatory drugs | |||
| Celecoxib | Selective inhibition of COX-2. | Significant reduction in PANSS positive TRS symptom scores and overall PANSS score, but no significant change in negative TRS symptoms. Improvement in conceptual disorganization and abstract thinking by PANSS in patients with TRS. | [133,134] |
| Acetylsalicylic acid | Inhibition of COX-1 and c COX-2. | Improvement in PANSS symptoms. | [135,136] |
| Statins | |||
| Simvastatin | Inhibition of HMG-CoA reductase, anti-inflammatory effect, reduction of pro-inflammatory cytokines (IL-1β, IL-6, TNF-α) and CRP. | Decrease in negative symptom scores on the PANSS scale in patients with TRS, decrease in the total score on the PANSS scale. | [137] |
| Pravastatin | Inhibition of HMG-CoA reductase, anti-inflammatory effect, reduction of pro-inflammatory cytokines (IL-1β, IL-6, TNF-α) and CRP. | Marked decrease in scores positive symptoms on the PANSS scale. | [138] |
| Corticosteroids | |||
| Cortisone | Influence on carbohydrate and electrolyte metabolism, anti-inflammatory (inhibition of phospholipase A2), desensitizing and anti-allergic, immunosuppressive effects. | Most patients with Sch did not show significant changes in Sch symptoms. | [139] |
| Prednisolone | Suppression of the function of leukocytes and tissue macrophages. Limitation of migration of leukocytes to the area of inflammation, impairment of the ability of macrophages to phagocytosis, as well as to the formation of IL-1, inhibition of the activity of phospholipase A2, suppression of the release of COX-1 and COX-2, etc. | There was no significant difference in improvement in the severity of Sch symptoms with the placebo group in patients with Sch. | [140] |
| Monoclonal antibody | |||
| Tocilizumab | Selective binding and suppression of expression and functional activity of IL-6 receptors. | No significant change in scores for positive and negative TRS symptoms, but improvement in BACS cognition. | [141,142] |
| Cytokines | |||
| - IFN-γ-1b | Activation of macrophages and induction of expression of the class II major histocompatibility complex molecule, inhibition of virus replication. | A pronounced decrease in the total PANSS score in patients with TRS. | [143] |
| Intravenous immunoglobulins | |||
| - IgG | Increasing the content of antibodies in the blood to a physiological level, creating passive immunity. | A pronounced decrease in the total PANSS score in patients with antibody positive psychosis. Most patients gave a clinical response to therapy. | [144,145] |
| Other groups of drugs | |||
| Mucolytics/antioxidants: - N-acetylcysteine | Precursor of the biological antioxidant glutathione, anti-inflammatory and antioxidant effect. | A decrease in scores on all three PANSS scales, an improvement on the CGI-S, CGI-I scales in patients with TRS. The reduction in negative symptom scores on the PANSS scale was more significant in patients with TRS. | [146,147] |
| Antibiotics: - Minocycline | Bacteriostatic action due to the suppression of protein synthesis by reversible binding to the 30S ribosomal subunit of sensitive microorganisms. | Decrease in scores on all three PANSS scales, improvement in BPRS scores, no changes in cognitive function in patients with TRS. | [148] |
| Polyunsaturated fatty acids: - Omega-3 fatty acids | Antioxidant, anti-inflammatory and neuroprotective effect. | Significant improvement on the three PANSS scales, as well as improvement in cognitive functions, was not revealed. | [149,150] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shnayder, N.A.; Khasanova, A.K.; Strelnik, A.I.; Al-Zamil, M.; Otmakhov, A.P.; Neznanov, N.G.; Shipulin, G.A.; Petrova, M.M.; Garganeeva, N.P.; Nasyrova, R.F. Cytokine Imbalance as a Biomarker of Treatment-Resistant Schizophrenia. Int. J. Mol. Sci. 2022, 23, 11324. https://doi.org/10.3390/ijms231911324
Shnayder NA, Khasanova AK, Strelnik AI, Al-Zamil M, Otmakhov AP, Neznanov NG, Shipulin GA, Petrova MM, Garganeeva NP, Nasyrova RF. Cytokine Imbalance as a Biomarker of Treatment-Resistant Schizophrenia. International Journal of Molecular Sciences. 2022; 23(19):11324. https://doi.org/10.3390/ijms231911324
Chicago/Turabian StyleShnayder, Natalia A., Aiperi K. Khasanova, Anna I. Strelnik, Mustafa Al-Zamil, Andrey P. Otmakhov, Nikolay G. Neznanov, German A. Shipulin, Marina M. Petrova, Natalia P. Garganeeva, and Regina F. Nasyrova. 2022. "Cytokine Imbalance as a Biomarker of Treatment-Resistant Schizophrenia" International Journal of Molecular Sciences 23, no. 19: 11324. https://doi.org/10.3390/ijms231911324
APA StyleShnayder, N. A., Khasanova, A. K., Strelnik, A. I., Al-Zamil, M., Otmakhov, A. P., Neznanov, N. G., Shipulin, G. A., Petrova, M. M., Garganeeva, N. P., & Nasyrova, R. F. (2022). Cytokine Imbalance as a Biomarker of Treatment-Resistant Schizophrenia. International Journal of Molecular Sciences, 23(19), 11324. https://doi.org/10.3390/ijms231911324