
Genome-Wide Identification and Functional Characterization Reveals the Pivotal Roles of BnaA8.ATG8F in Salt Stress Tolerance and Nitrogen Limitation Adaptation in Allotetraploid Rapeseed

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Genome-Wide Identification of ATG8s in Brassica Species
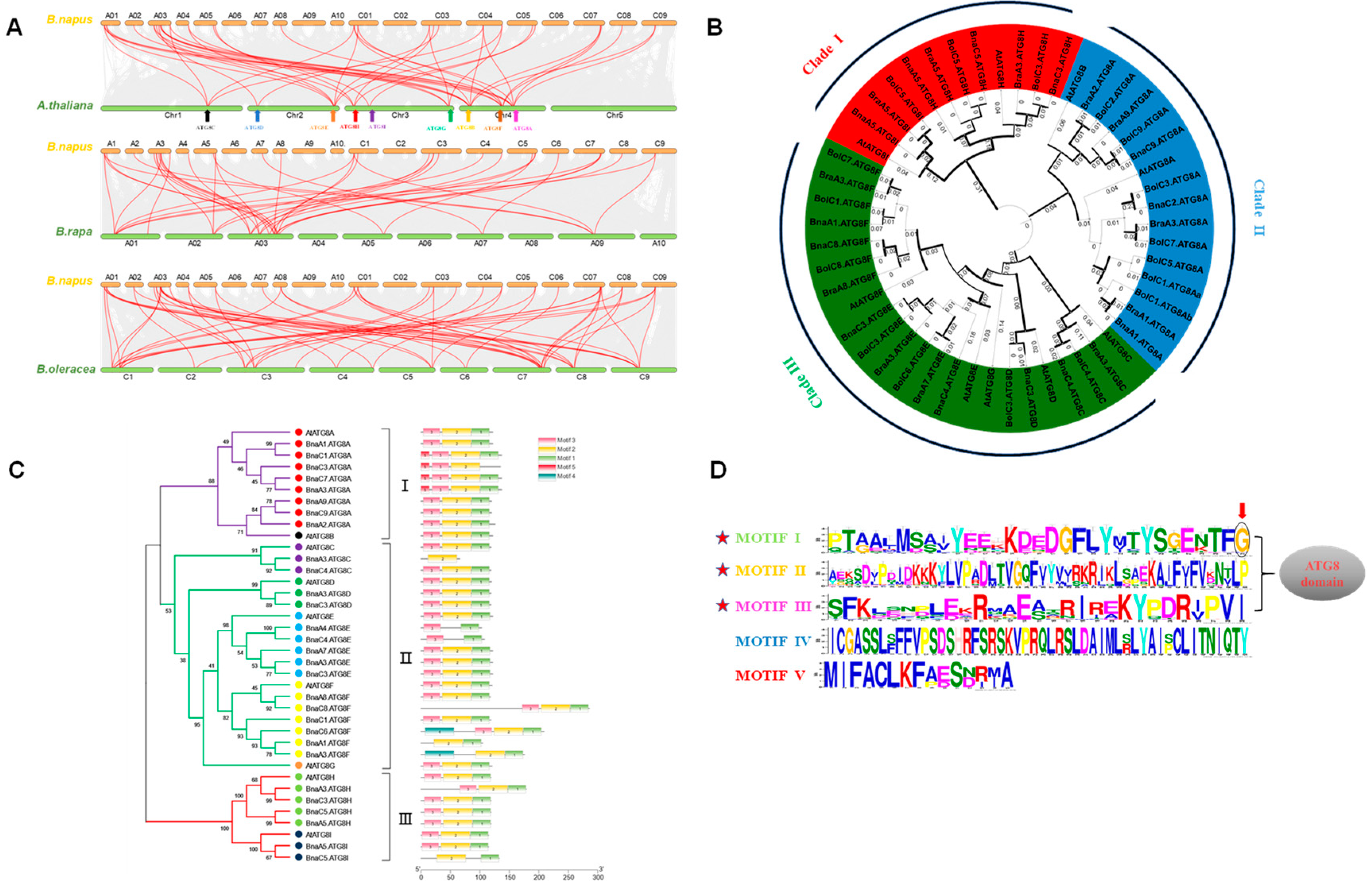
2.2. Genomic Distribution, Expansion, and Syntenic Analysis of BnaATG8s
2.3. Phylogenetic Relationships and Evolutionary Selection Pressure of the ATG8 Proteins
2.4. Molecular Characterization of BnaATG8s
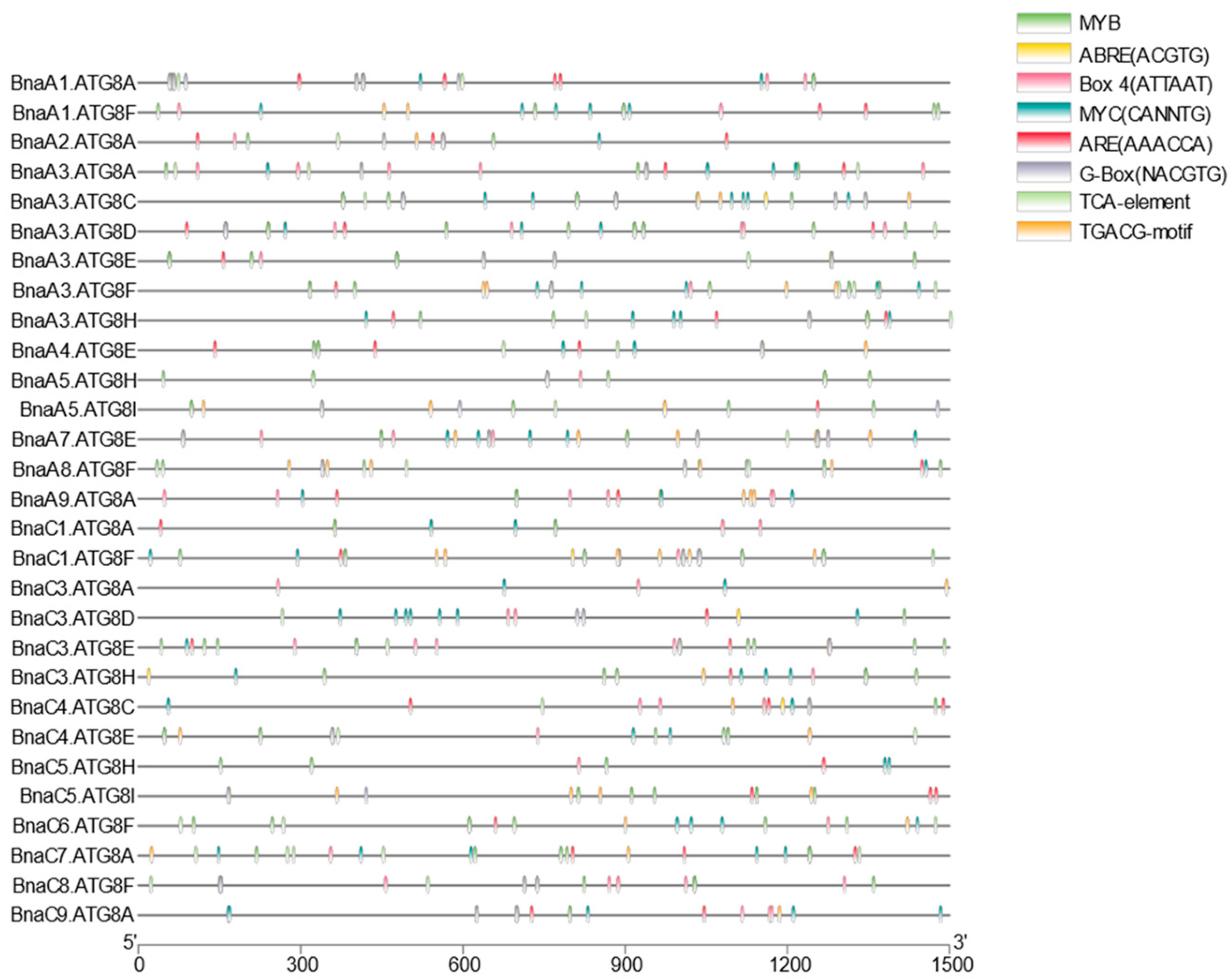
2.5. Conserved Domains, Gene Structure, Protein Interaction, and Transcriptional Regulatory Analysis
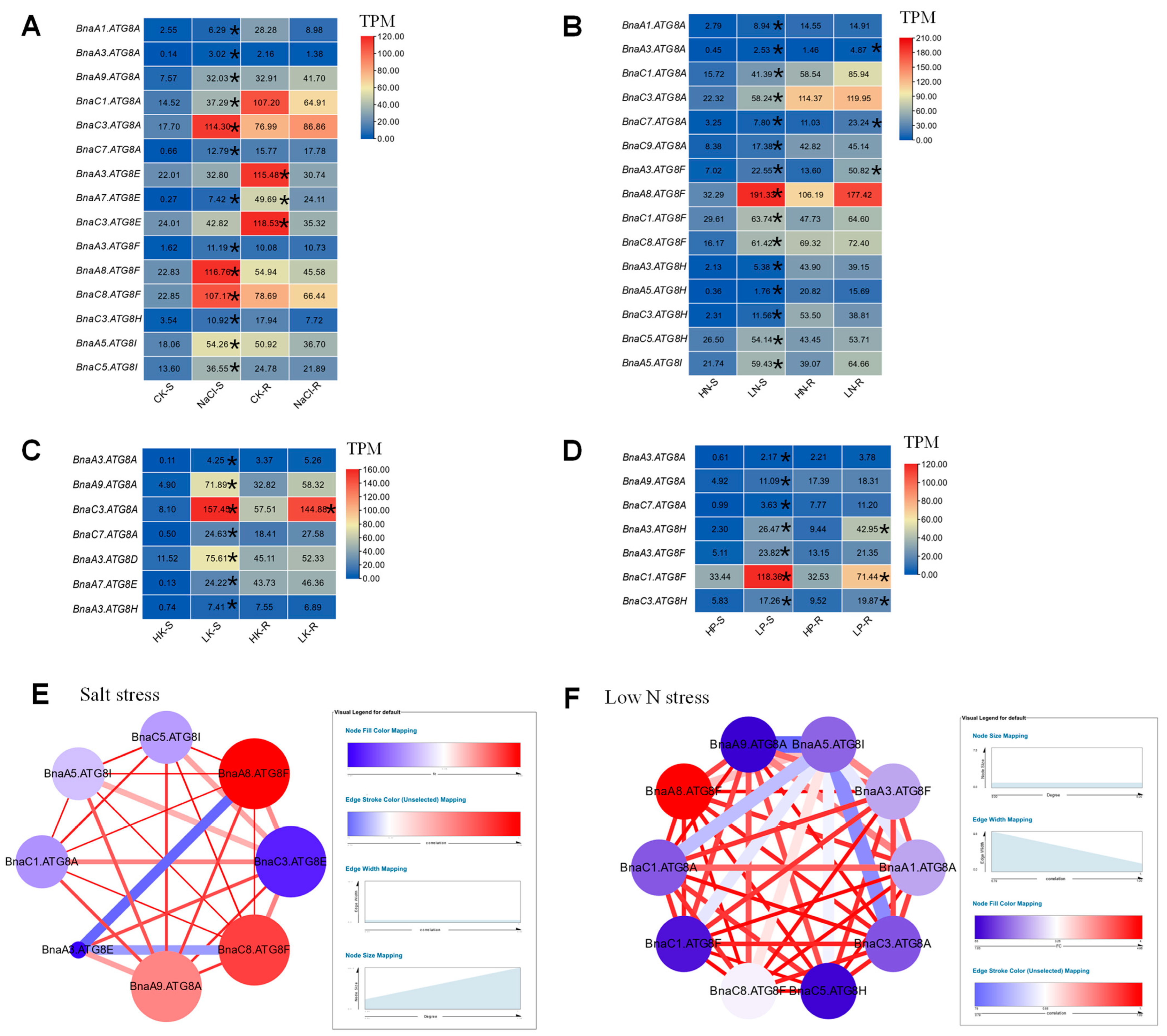
2.6. Transcriptional Analysis of BnaATG8s under Diverse Nutrient Stresses
2.7. Gene Co-Expression Network Analysis of the Core BnaATG8 Members
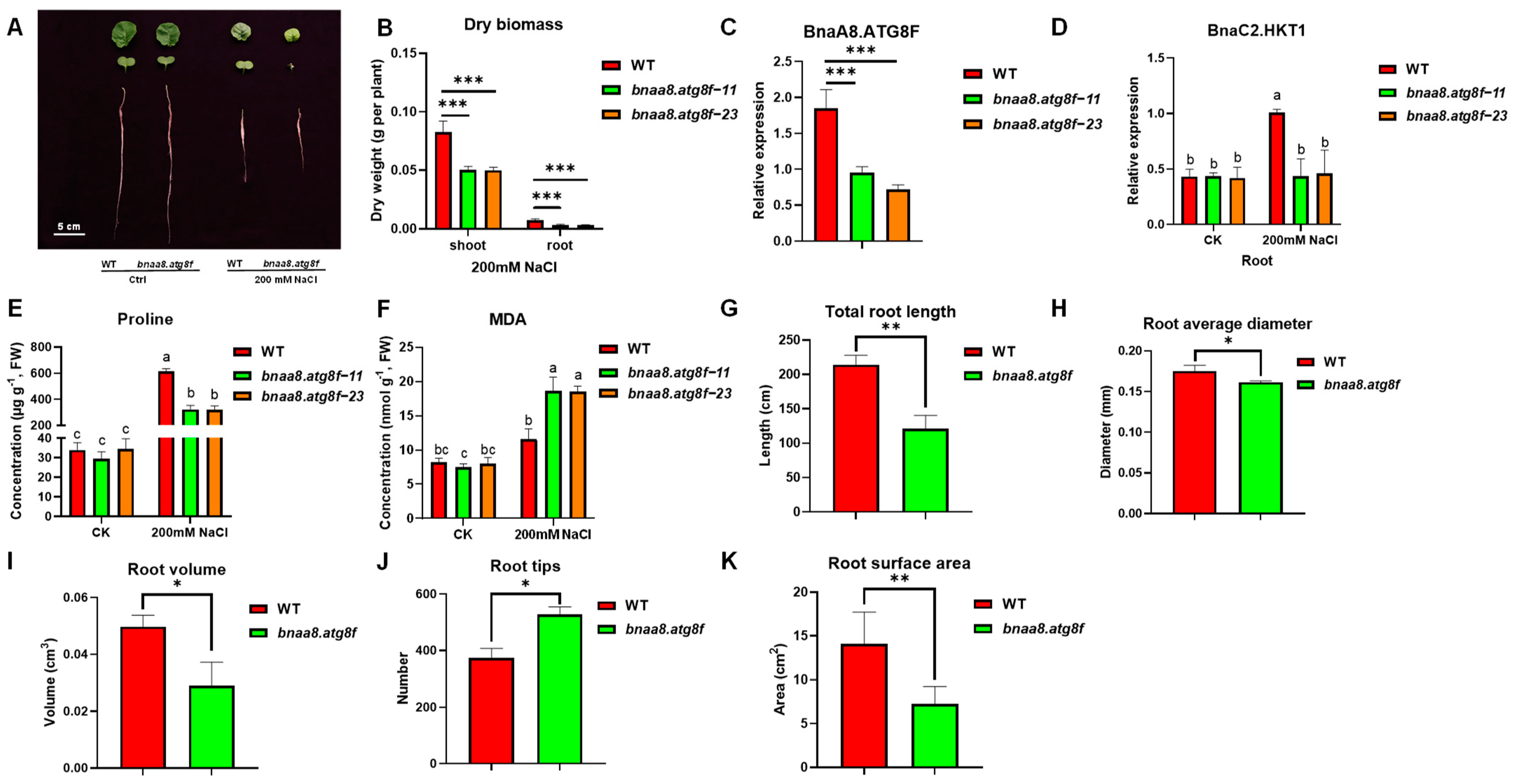
2.8. BnaA8.ATG8F May Be Indispensable for The Resistance to Salt Stress and Low Nitrogen Stress
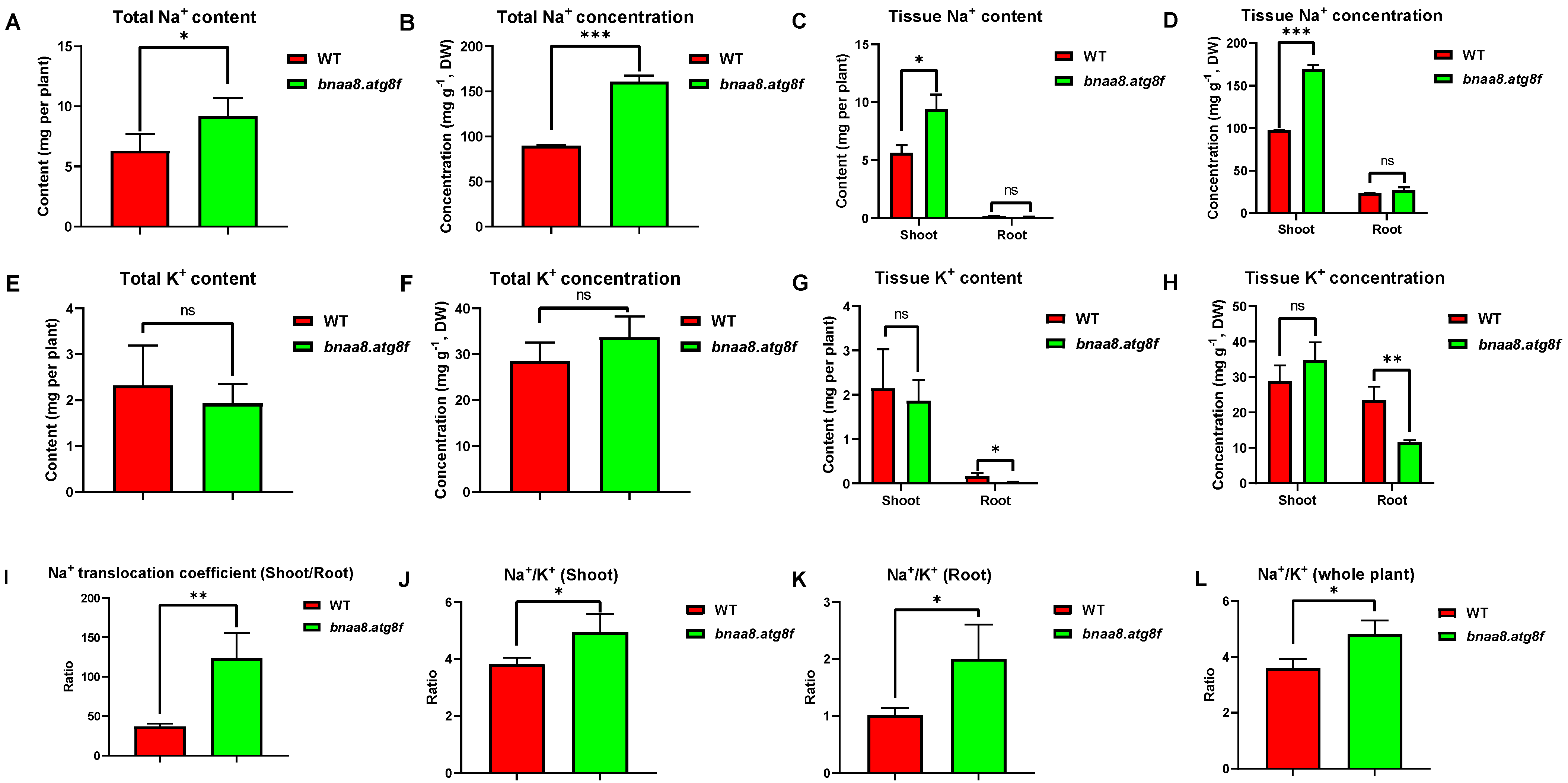
2.9. Effects of BnaA8.ATG8F on Morpho-Physiological and Mineral Nutrients in Rapeseed under Salt Stress
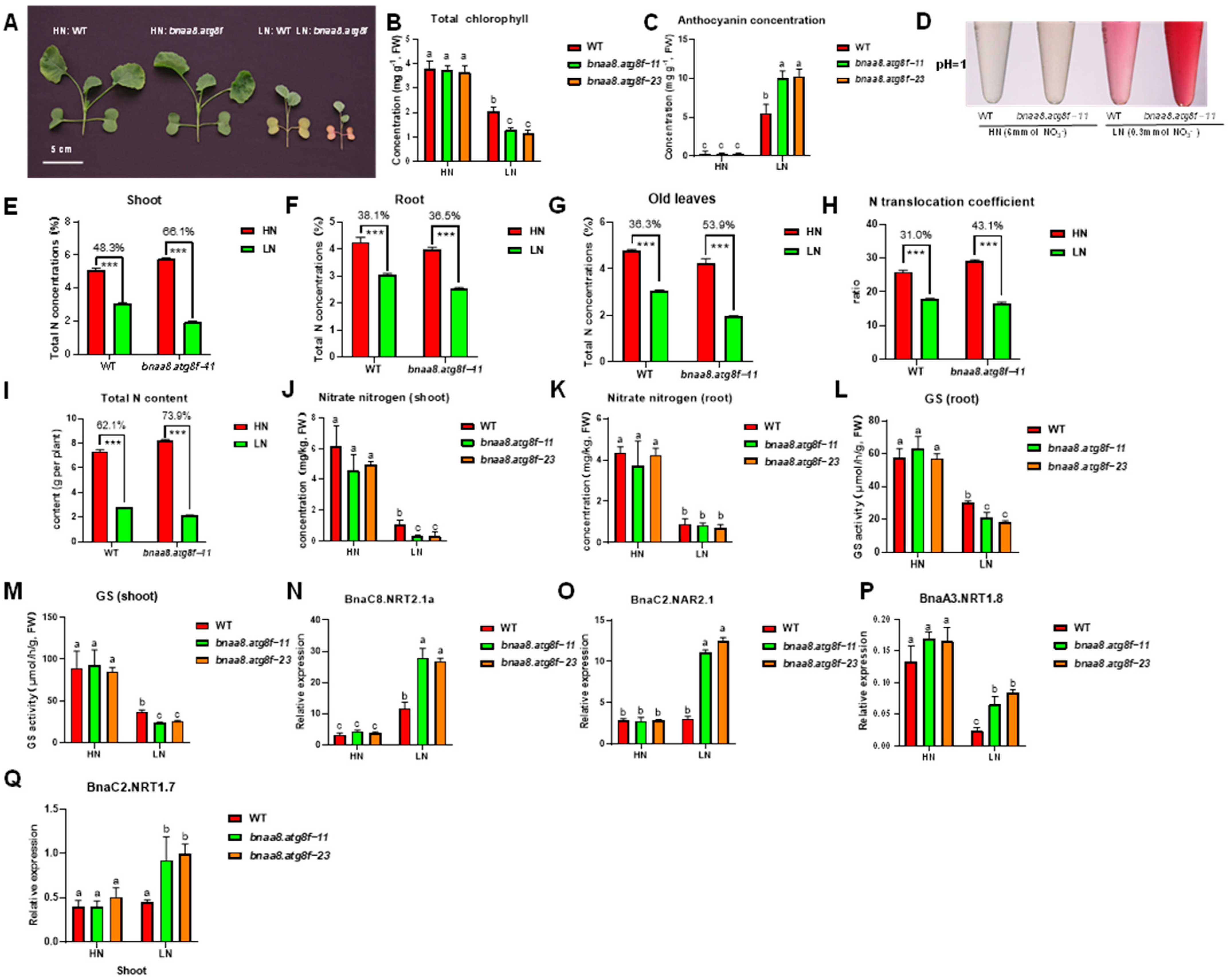
2.10. BnaA8.ATG8F Plays a Key Role in Rapeseed Resistance to Low Nitrate Stress
3. Discussion
3.1. Integrated Bioinformatics Analysis Provided a Comprehensive Insight into the Molecular Characteristics of BnaATG8s
3.2. Transcriptional Analysis Revealed the Response of the BnaATG8 Family to Various Nutrient Stresses and Identified the Core Member BnaA8.ATG8F
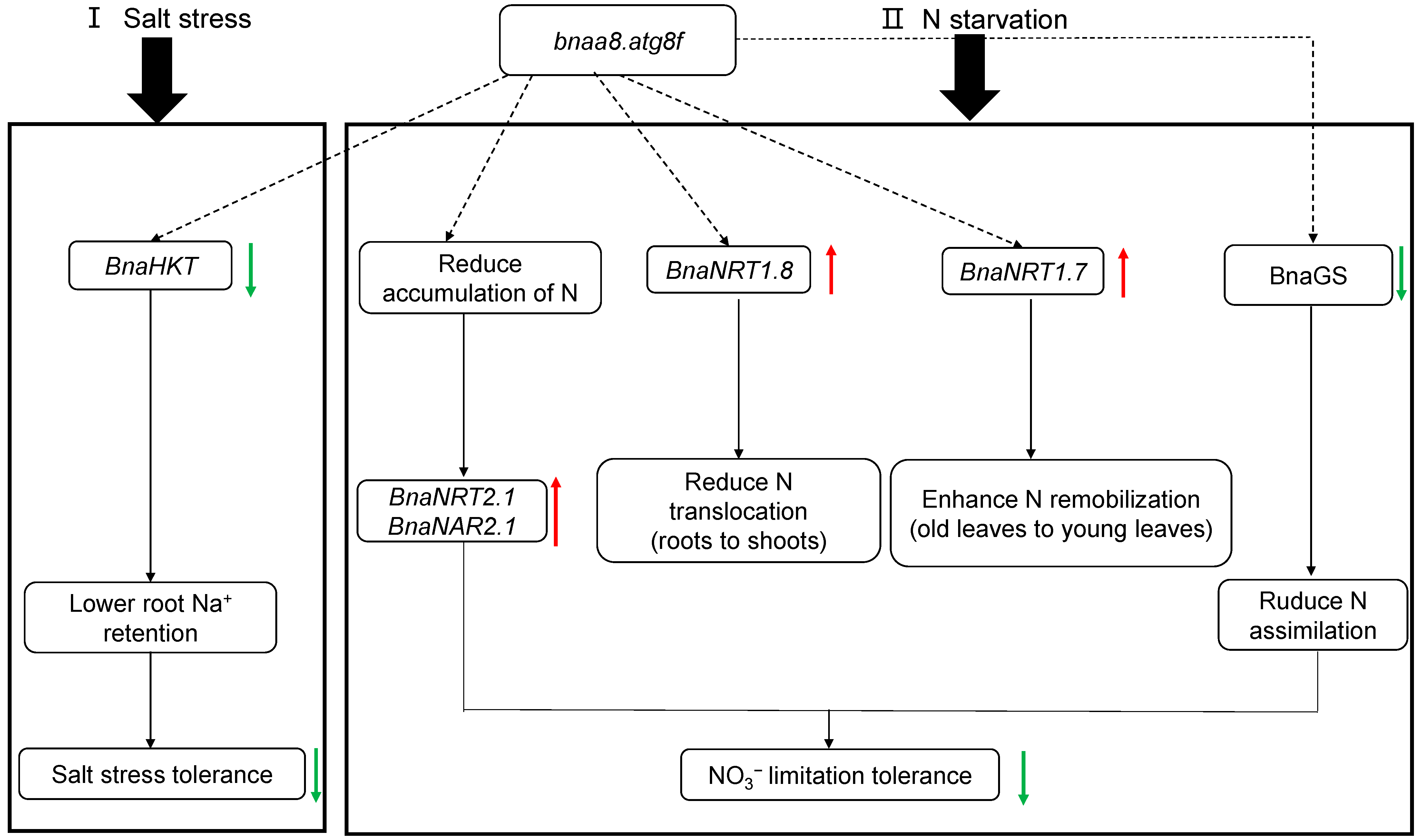
3.3. Molecular Strategy Model of BnaA8.ATG8F Regulating Salt Stress Resistance and Nitrogen Limitation Adaptation in Allotetraploid Rapeseed
4. Materials and Methods
4.1. Identification and Gene Nomenclature of Autophagy Gene 8 (ATG8) Family in Brassica
4.2. Chromosomal Localization and Syntenic Analysis of ATG8s in Brassica
4.3. Analysis of Evolutionary Selection Pressure and Phylogeny Relationships
4.4. Identification of Exon-Intron Structures, Conserved Motifs, and Putative Cis-Acting Regulatory Elements (CREs)
4.5. Molecular Characterization of the ATG8 Proteins
4.6. Transcriptional Responses of BnaATG8s and Functionality Analysis under Different Nutrient Stresses
4.7. Differential Expression and Gene Co-Expression Network Analysis
4.8. Construction of Recombinant Vector and Acquisition of Transgenic Rapeseed
4.9. Determination of Osmoregulatory Substances, Chlorophyll, Anthocyanin, Nitrate Nitrogen, Total Nitrogen Concentration, and Enzyme Activities
4.10. Ionomic Analysis
4.11. Quantitative Reverse-Transcription PCR (RT-qPCR) Assays
4.12. Statistical Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Avin-Wittenberg, T. Autophagy and its role in plant abiotic stress management. Plant Cell Environ. 2019, 42, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Hayward, A.; Dinesh-Kumar, S. What can plant autophagy do for an innate immune response? Annu. Rev. Phytopathol. 2011, 49, 557–576. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Vierstra, R. Autophagic recycling: Lessons from yeast help define the process in plants. Curr. Opin. Plant Biol. 2005, 8, 165–173. [Google Scholar] [CrossRef]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bassham, D. Autophagy: Pathways for self-eating in plant cells. Annu. Rev. Plant Biol. 2012, 63, 215–237. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Contento, A.; Nguyen, P.; Bassham, D. Degradation of oxidized proteins by autophagy during oxidative stress in Arabidopsis. Plant Physiol. 2007, 143, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Dong, J.; Wang, T. Autophagy in Plant Abiotic Stress Management. Int. J. Mol. Sci. 2021, 22, 4075. [Google Scholar] [CrossRef]
- Signorelli, S.; Tarkowski, Ł.; Van den Ende, W.; Bassham, D. Linking Autophagy to Abiotic and Biotic Stress Responses. Trends Plant Sci. 2019, 24, 413–430. [Google Scholar] [CrossRef]
- Su, W.; Bao, Y.; Yu, X.; Xia, X.; Liu, C.; Yin, W. Autophagy and Its Regulators in Response to Stress in Plants. Int. J. Mol. Sci. 2020, 21, 8889. [Google Scholar] [CrossRef]
- Avila-Ospina, L.; Moison, M.; Yoshimoto, K.; Masclaux-Daubresse, C. Autophagy, plant senescence, and nutrient recycling. J. Exp. Bot. 2014, 65, 3799–3811. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Chung, K.; Luo, M.; Jiang, L. Autophagosome Biogenesis and the Endoplasmic Reticulum: A Plant Perspective. Trends Plant Sci. 2018, 23, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.; Vierstra, R. Autophagy: The Master of Bulk and Selective Recycling. Annu. Rev. Plant Biol. 2018, 69, 173–208. [Google Scholar] [CrossRef]
- Chen, Q.; Soulay, F.; Saudemont, B.; Elmayan, T.; Marmagne, A.; Masclaux-Daubresse, C. Overexpression of ATG8 in Arabidopsis Stimulates Autophagic Activity and Increases Nitrogen Remobilization Efficiency and Grain Filling. Plant Cell Physiol. 2019, 60, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Kajikawa, M.; Yamauchi, M.; Shinkawa, H.; Tanaka, M.; Hatano, K.; Nishimura, Y.; Kato, M.; Fukuzawa, H. Isolation and Characterization of Chlamydomonas Autophagy-Related Mutants in Nutrient-Deficient Conditions. Plant Cell Physiol. 2019, 60, 126–138. [Google Scholar] [CrossRef]
- Honig, A.; Avin-Wittenberg, T.; Galili, G. Selective autophagy in the aid of plant germination and response to nutrient starvation. Autophagy 2012, 8, 838–839. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 3930. [Google Scholar] [CrossRef]
- An, H.; Qi, X.; Gaynor, M.; Hao, Y.; Gebken, S.; Mabry, M.; McAlvay, A.; Teakle, G.; Conant, G.; Barker, M.; et al. Transcriptome and organellar sequencing highlights the complex origin and diversification of allotetraploid Brassica napus. Nat. Commun. 2019, 10, 2878. [Google Scholar] [CrossRef]
- Song, J.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Tang, H.; Bowers, J.; Wang, X.; Paterson, A. Angiosperm genome comparisons reveal early polyploidy in the monocot lineage. Proc. Natl. Acad. Sci. USA 2010, 107, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Schranz, M.; Lysak, M.; Mitchell-Olds, T. The ABC’s of comparative genomics in the Brassicaceae: Building blocks of crucifer genomes. Trends Plant Sci. 2006, 11, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Parkin, I.; Gulden, S.; Sharpe, A.; Lukens, L.; Trick, M.; Osborn, T.; Lydiate, D. Segmental structure of the Brassica napus genome based on comparative analysis with Arabidopsis thaliana. Genetics 2005, 171, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Nekrutenko, A.; Makova, K.D.; Li, W.H. The KA/KS Ratio Test for Assessing the Protein-Coding Potential of Genomic Regions: An Empirical and Simulation Study. Genome Res. 2002, 12, 198–202. [Google Scholar] [CrossRef]
- Blanc, G.; Wolfe, K. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef]
- Town, C.; Cheung, F.; Maiti, R.; Crabtree, J.; Haas, B.; Wortman, J.; Hine, E.; Althoff, R.; Arbogast, T.; Tallon, L.; et al. Comparative genomics of Brassica oleracea and Arabidopsis thaliana reveal gene loss, fragmentation, and dispersal after polyploidy. Plant Cell 2006, 18, 1348–1359. [Google Scholar] [CrossRef]
- Mijakovic, I.; Grangeasse, C.; Turgay, K. Exploring the diversity of protein modifications: Special bacterial phosphorylation systems. FEMS Microbiol. Rev. 2016, 40, 398–417. [Google Scholar] [CrossRef]
- Shpilka, T.; Weidberg, H.; Pietrokovski, S.; Elazar, Z. Atg8: An autophagy-related ubiquitin-like protein family. Genome Biol. 2011, 12, 226. [Google Scholar] [CrossRef]
- Su, T.; Li, X.; Yang, M.; Shao, Q.; Zhao, Y.; Ma, C.; Wang, P. Autophagy: An Intracellular Degradation Pathway Regulating Plant Survival and Stress Response. Front. Plant Sci. 2020, 11, 164. [Google Scholar] [CrossRef]
- Cheng, X.; Wang, Z. Overexpression of COL9, a CONSTANS-LIKE gene, delays flowering by reducing expression of CO and FT in Arabidopsis thaliana. Plant J. Cell Mol. Biol. 2005, 43, 758–768. [Google Scholar] [CrossRef]
- Amin, I.; Rasool, S.; Mir, M.; Wani, W.; Masoodi, K.; Ahmad, P. Ion homeostasis for salinity tolerance in plants: A molecular approach. Physiol. Plant. 2021, 171, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Berthomieu, P.; Conéjéro, G.; Nublat, A.; Brackenbury, W.; Lambert, C.; Savio, C.; Uozumi, N.; Oiki, S.; Yamada, K.; Cellier, F.; et al. Functional analysis of AtHKT1 in Arabidopsis shows that Na+ recirculation by the phloem is crucial for salt tolerance. EMBO J. 2003, 22, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Yue, C.; Liu, Y.; Zhang, T.; Huang, J.; Hua, Y. Multiomics reveal pivotal roles of sodium translocation and compartmentation in regulating salinity resistance in allotetraploid rapeseed. J. Exp. Bot. 2021, 72, 5687–5708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, T.; Liao, Q.; Yao, J.; Liang, G.; Song, H.; Guan, C.; Hua, Y. Integrated physiologic, genomic and transcriptomic strategies involving the adaptation of allotetraploid rapeseed to nitrogen limitation. BMC Plant Biol. 2018, 18, 322. [Google Scholar] [CrossRef]
- Tang, J.; Bassham, D. Autophagy during drought: Function, regulation, and potential application. Plant J. Cell Mol. Biol. 2022, 109, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Slavikova, S.; Ufaz, S.; Avin-Wittenberg, T.; Levanony, H.; Galili, G. An autophagy-associated Atg8 protein is involved in the responses of Arabidopsis seedlings to hormonal controls and abiotic stresses. J. Exp. Bot. 2008, 59, 4029–4043. [Google Scholar] [CrossRef]
- Bu, F.; Yang, M.; Guo, X.; Huang, W.; Chen, L. Multiple Functions of ATG8 Family Proteins in Plant Autophagy. Front. Cell Dev. Biol. 2020, 8, 466. [Google Scholar] [CrossRef]
- Yoshimoto, K.; Hanaoka, H.; Sato, S.; Kato, T.; Tabata, S.; Noda, T.; Ohsumi, Y. Processing of ATG8s, ubiquitin-like proteins, and their deconjugation by ATG4s are essential for plant autophagy. Plant Cell 2004, 16, 2967–2983. [Google Scholar] [CrossRef]
- Seo, E.; Woo, J.; Park, E.; Bertolani, S.; Siegel, J.; Choi, D.; Dinesh-Kumar, S. Comparative analyses of ubiquitin-like ATG8 and cysteine protease ATG4 autophagy genes in the plant lineage and cross-kingdom processing of ATG8 by ATG4. Autophagy 2016, 12, 2054–2068. [Google Scholar] [CrossRef]
- Wang, P.; Nolan, T.; Yin, Y.; Bassham, D. ATG8Identification of transcription factors that regulate expression and autophagy in. Autophagy 2020, 16, 123–139. [Google Scholar] [CrossRef]
- Chung, T.; Suttangkakul, A.; Vierstra, R. The ATG autophagic conjugation system in maize: ATG transcripts and abundance of the ATG8-lipid adduct are regulated by development and nutrient availability. Plant Physiol. 2009, 149, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Lemieux, C.; Jorgensen, R. Introduction of a Chimeric Chalcone Synthase Gene into Petunia Results in Reversible Co-Suppression of Homologous Genes in trans. Plant Cell 1990, 2, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Liu, D.; Xie, W.; Yang, Z.; Guo, L.; Liu, K.; Yang, Q.; Chen, L. BnPIR: Brassica napus pan-genome information resource for 1689 accessions. Plant Biotechnol. J. 2021, 19, 412–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, J.; Liang, J.; Cheng, F.; Wang, X. Brassica database (BRAD) version 2.0: Integrating and mining Brassicaceae species genomic resources. Database J. Biol. Databases Curation 2015, 2015, bav093. [Google Scholar] [CrossRef]
- Ostergaard, L.; King, G. Standardized gene nomenclature for the Brassica genus. Plant Methods 2008, 4, 10. [Google Scholar] [CrossRef]
- Smith, T.; Waterman, M. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Lee, T.; Tang, H.; Wang, X.; Paterson, A. PGDD: A database of gene and genome duplication in plants. Nucleic Acids Res. 2013, 41, D1152–D1158. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.; Frank, M.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Larkin, M.; Blackshields, G.; Brown, N.; Chenna, R.; McGettigan, P.; McWilliam, H.; Valentin, F.; Wallace, I.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Bailey, T.; Boden, M.; Buske, F.; Frith, M.; Grant, C.; Clementi, L.; Ren, J.; Li, W.; Noble, W. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Horton, P.; Park, K.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed]
- Möller, S.; Croning, M.; Apweiler, R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 2001, 17, 646–653. [Google Scholar] [CrossRef]
- Blom, N.; Sicheritz-Pontén, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.; Morris, J.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Hua, Y.-P.; Zhou, T.; Song, H.-X.; Guan, C.-Y.; Zhang, Z.-H. Integrated genomic and transcriptomic insights into the two-component high-affinity nitrate transporters in allotetraploid rapeseed. Plant Soil 2018, 427, 245–268. [Google Scholar] [CrossRef]
- Han, Y.; Song, H.; Liao, Q.; Yu, Y.; Jian, S.; Lepo, J.; Liu, Q.; Rong, X.; Tian, C.; Zeng, J.; et al. Nitrogen Use Efficiency Is Mediated by Vacuolar Nitrate Sequestration Capacity in Roots of Brassica napus. Plant Physiol. 2016, 170, 1684–1698. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, D.; Tang, Y.; Wu, F.; Wang, J. CytoCluster: A Cytoscape Plugin for Cluster Analysis and Visualization of Biological Networks. Int. J. Mol. Sci. 2017, 18, 1880. [Google Scholar] [CrossRef] [PubMed]
- De Block, M.; De Brouwer, D.; Tenning, P. Transformation of Brassica napus and Brassica oleracea Using Agrobacterium tumefaciens and the Expression of the bar and neo Genes in the Transgenic Plants. Plant physiology. 1989, 91, 694–701. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, Z.; Chu, G.; Huang, C.; Tian, S.; Zhao, Z.; Chen, G. Anthocyanin accumulation and molecular analysis of anthocyanin biosynthesis-associated genes in eggplant (Solanum melongena L.). J. Agric. Food Chem. 2014, 62, 2906–2912. [Google Scholar] [CrossRef]
- Patterson, K.; Cakmak, T.; Cooper, A.; Lager, I.; Rasmusson, A.; Escobar, M. Distinct signalling pathways and transcriptome response signatures differentiate ammonium- and nitrate-supplied plants. Plant Cell Environ. 2010, 33, 1486–1501. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, Q.; Ding, L.; Sun, Y. Effect of cadmium toxicity on nitrogen metabolism in leaves of Solanum nigrum L. as a newly found cadmium hyperaccumulator. J. Hazard. Mater. 2008, 154, 818–825. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Zhou, T.; Zhang, Y.; Chen, J.; Song, H.; Wu, P.; Yue, C.; Huang, J.; Zhang, Z.; Hua, Y. Genome-Wide Identification and Functional Characterization Reveals the Pivotal Roles of BnaA8.ATG8F in Salt Stress Tolerance and Nitrogen Limitation Adaptation in Allotetraploid Rapeseed. Int. J. Mol. Sci. 2022, 23, 11318. https://doi.org/10.3390/ijms231911318
Zhang T, Zhou T, Zhang Y, Chen J, Song H, Wu P, Yue C, Huang J, Zhang Z, Hua Y. Genome-Wide Identification and Functional Characterization Reveals the Pivotal Roles of BnaA8.ATG8F in Salt Stress Tolerance and Nitrogen Limitation Adaptation in Allotetraploid Rapeseed. International Journal of Molecular Sciences. 2022; 23(19):11318. https://doi.org/10.3390/ijms231911318
Chicago/Turabian StyleZhang, Tianyu, Ting Zhou, Yifan Zhang, Junfan Chen, Haili Song, Pengjia Wu, Caipeng Yue, Jinyong Huang, Zhenhua Zhang, and Yingpeng Hua. 2022. "Genome-Wide Identification and Functional Characterization Reveals the Pivotal Roles of BnaA8.ATG8F in Salt Stress Tolerance and Nitrogen Limitation Adaptation in Allotetraploid Rapeseed" International Journal of Molecular Sciences 23, no. 19: 11318. https://doi.org/10.3390/ijms231911318
APA StyleZhang, T., Zhou, T., Zhang, Y., Chen, J., Song, H., Wu, P., Yue, C., Huang, J., Zhang, Z., & Hua, Y. (2022). Genome-Wide Identification and Functional Characterization Reveals the Pivotal Roles of BnaA8.ATG8F in Salt Stress Tolerance and Nitrogen Limitation Adaptation in Allotetraploid Rapeseed. International Journal of Molecular Sciences, 23(19), 11318. https://doi.org/10.3390/ijms231911318