
Antagonist of Growth Hormone-Releasing Hormone Potentiates the Antitumor Effect of Pemetrexed and Cisplatin in Pleural Mesothelioma

 , , , ,
, , , ,  ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
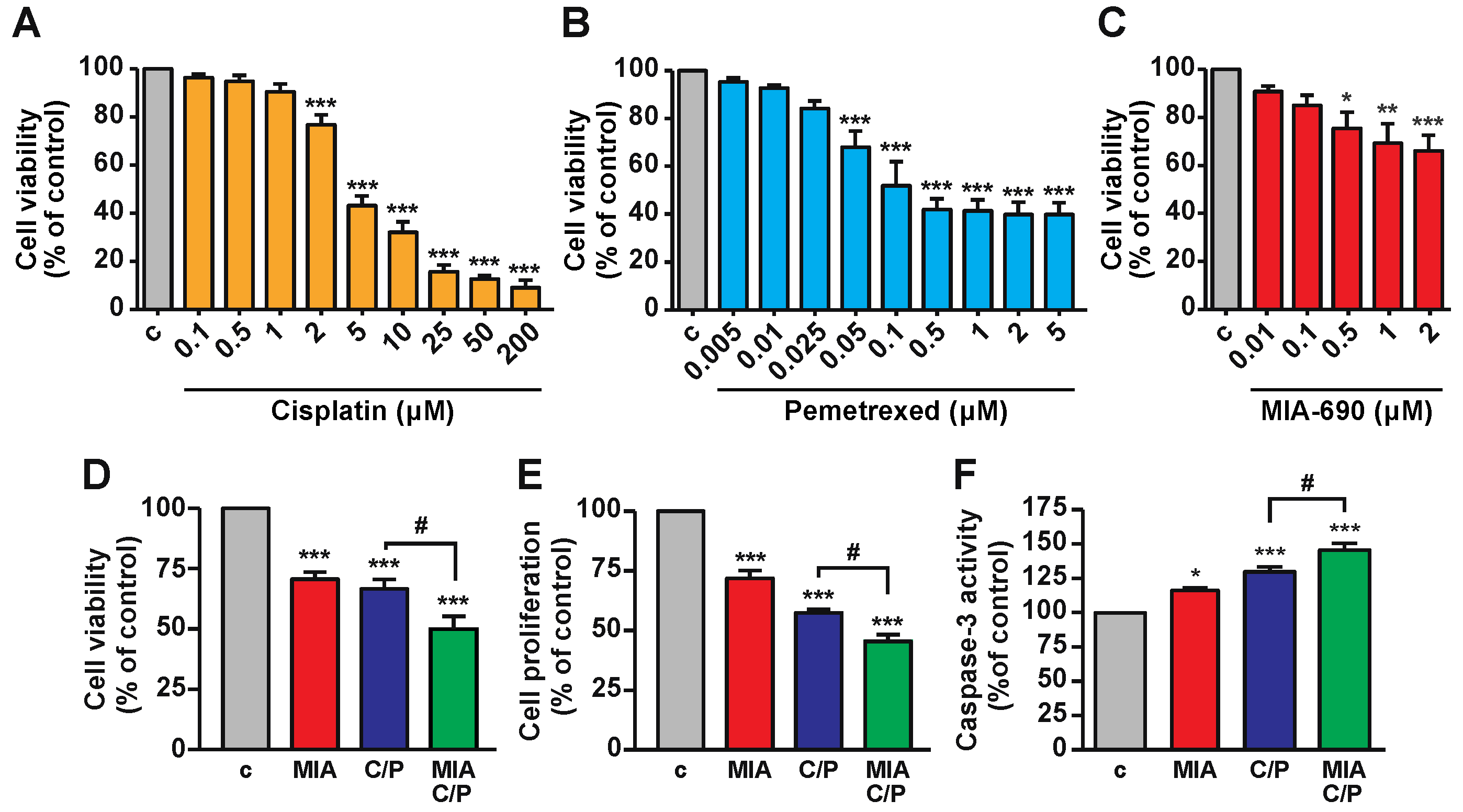
2.1. MIA-690 Increases the Inhibitory Effects of Cisplatin/Pemetrexed on Cell Viability of PM Cells
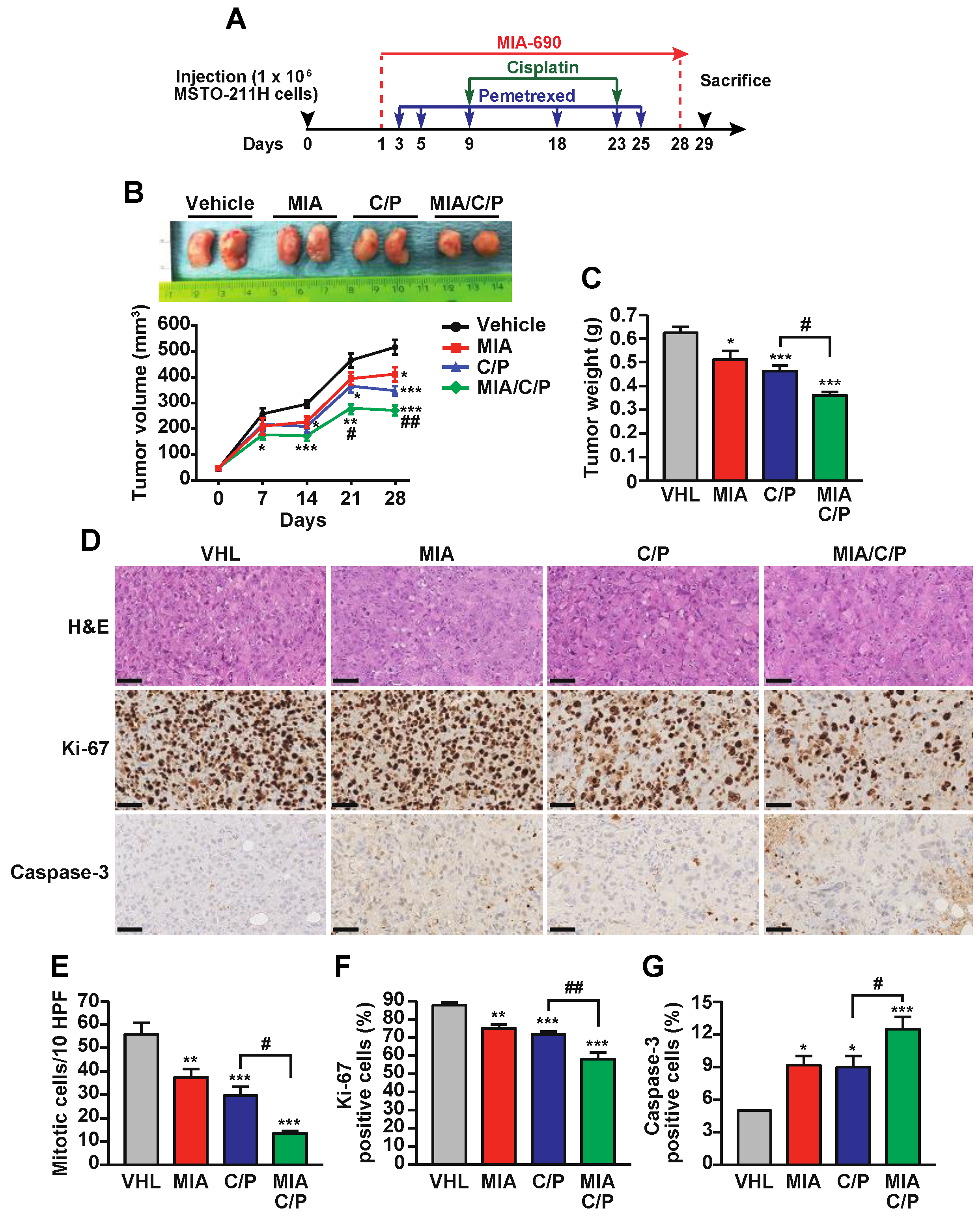
2.2. MIA-690 Potentiates the In Vivo Antitumor Effect of Cisplatin/Pemetrexed in PM Xenografts
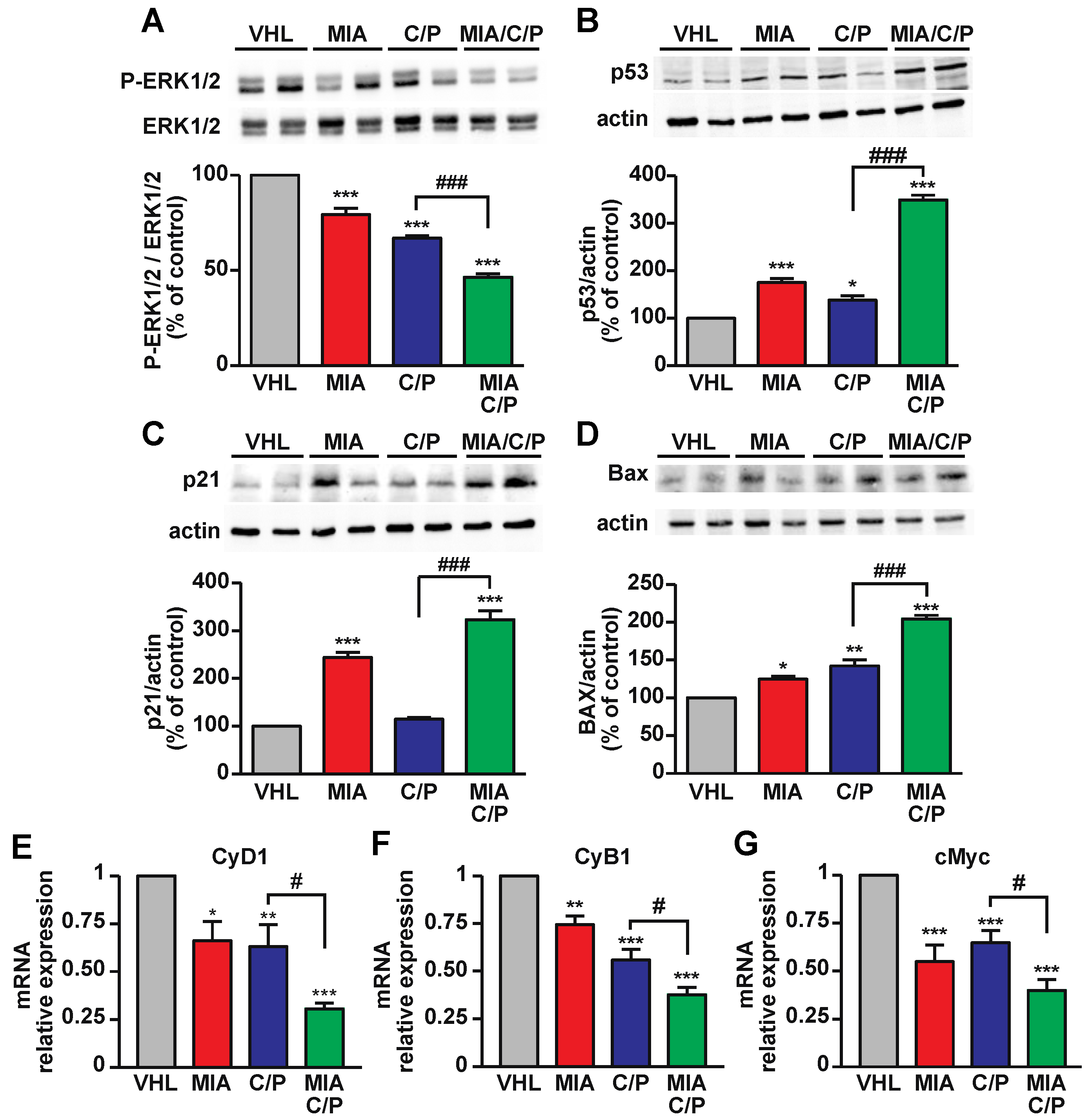
2.3. The Combination of MIA-690 with Cisplatin and Pemetrexed Is More Effective than Chemotherapeutic Drugs Alone on the Inhibition of Proliferative and Survival Pathways
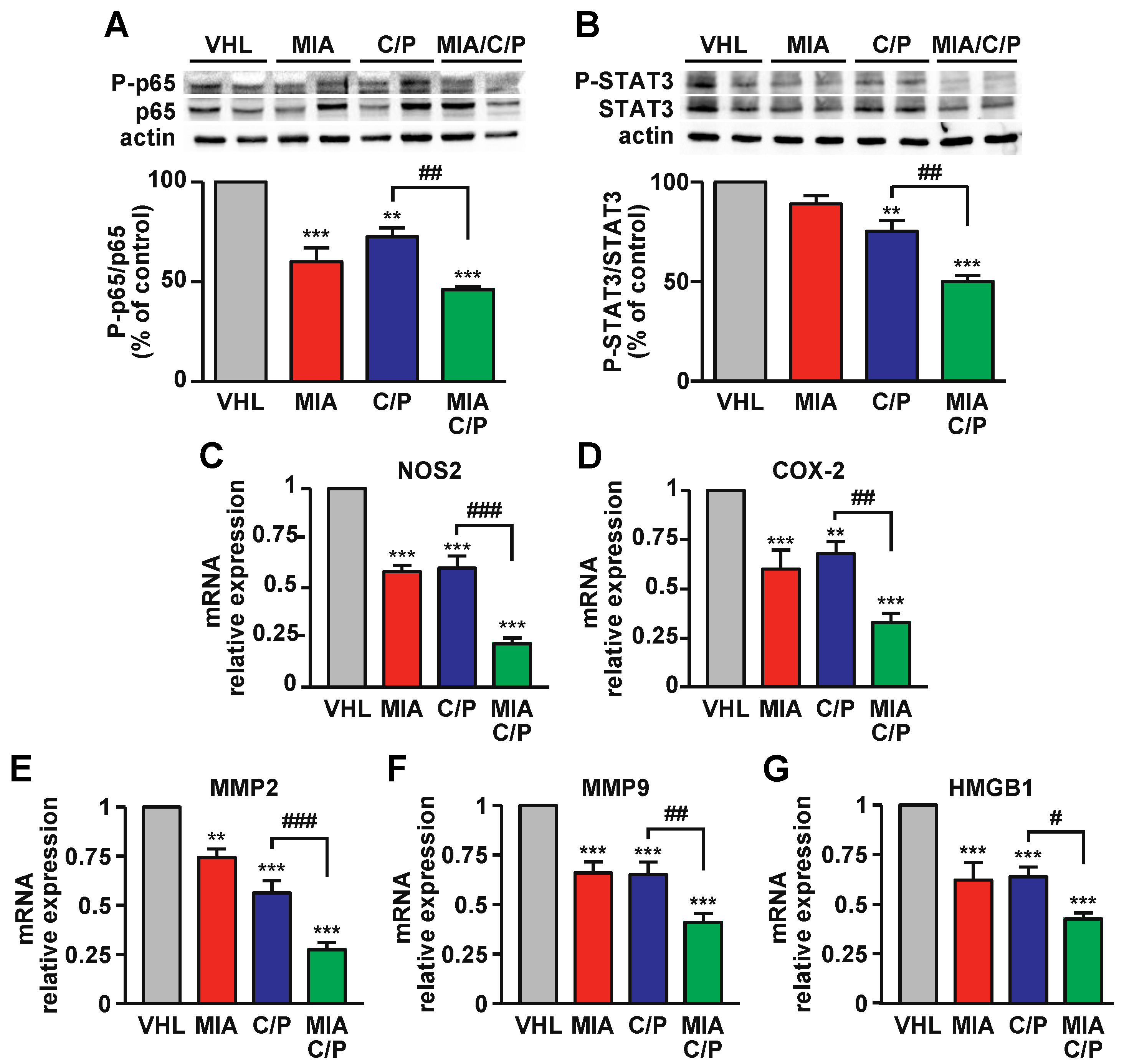
2.4. MIA-690 Increases the Anti-inflammatory Action of Cisplatin and Pemetrexed in PM Tumors
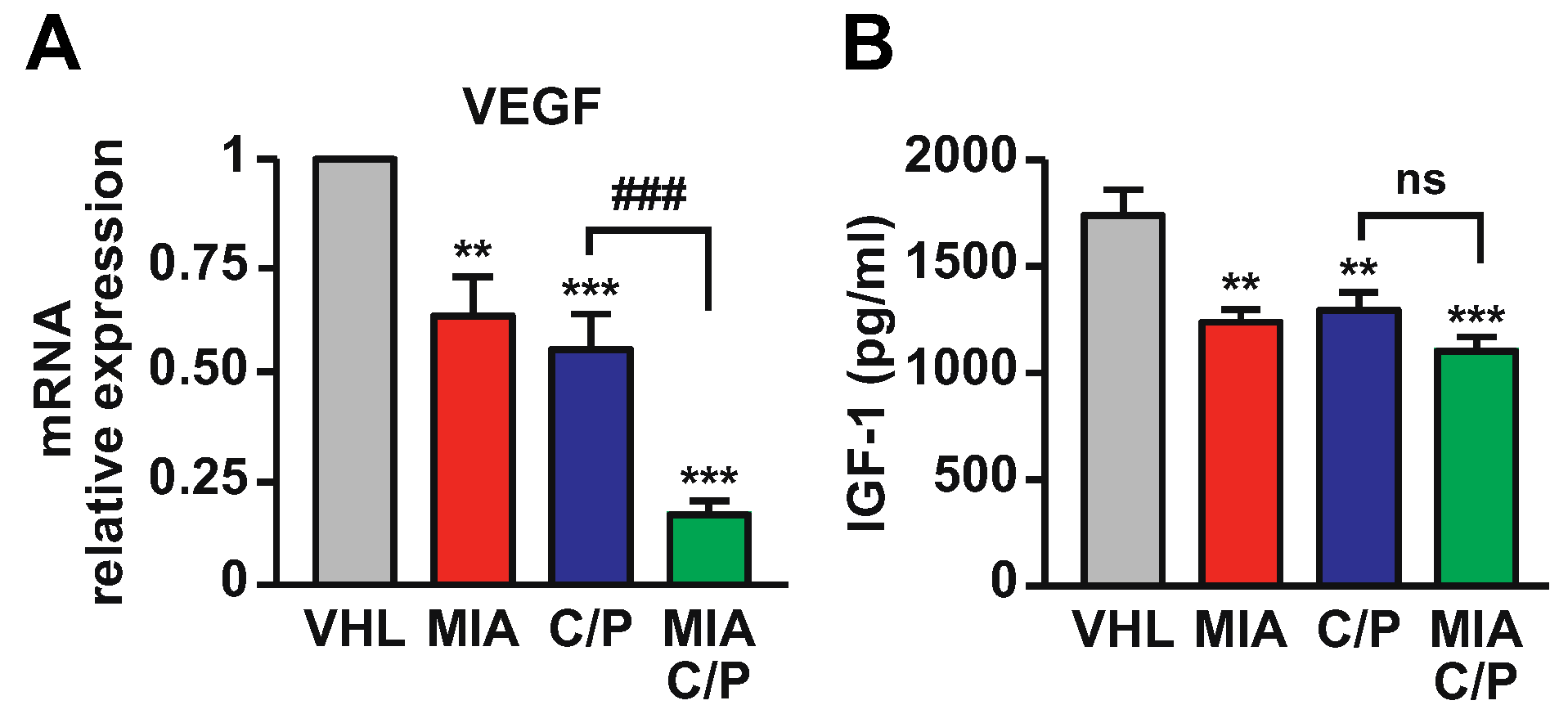
2.5. MIA-690 Potentiates the Effect of Cisplatin/Pemetrexed on Inhibition of VEGF and IGF-1 Expression in PM Tumors
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Survival and Proliferation
4.4. Caspase-3 Activity
4.5. Combination Studies
4.6. In Vivo Tumor Growth
4.7. Histochemistry
4.8. Western Blot Analysis
4.9. Real-Time PCR
4.10. IGF-1 Analysis
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Yap, T.A.; Aerts, J.G.; Popat, S.; Fennell, D.A. Novel insights into mesothelioma biology and implications for therapy. Nat. Rev. Cancer 2017, 17, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Rodella, G.; Staniszewska, M.; Pancer, K.W.; Wieczorek, M.; Salmaso, S.; Caliceti, P.; Garofalo, M. Novel Insights Into Mesothelioma Therapy: Emerging Avenues and Future Prospects. Front. Oncol. 2022, 12, 916839. [Google Scholar] [CrossRef]
- Sauter, J.L.; Dacic, S.; Galateau-Salle, F.; Attanoos, R.L.; Butnor, K.J.; Churg, A.; Husain, A.N.; Kadota, K.; Khoor, A.; Nicholson, A.G.; et al. The 2021 WHO Classification of Tumors of the Pleura: Advances Since the 2015 Classification. J. Thorac. Oncol. 2022, 17, 608–622. [Google Scholar] [CrossRef]
- Obacz, J.; Yung, H.; Shamseddin, M.; Linnane, E.; Liu, X.; Azad, A.A.; Rassl, D.M.; Fairen-Jimenez, D.; Rintoul, R.C.; Nikolic, M.Z.; et al. Biological basis for novel mesothelioma therapies. Br. J. Cancer 2021, 125, 1039–1055. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef]
- Tsao, A.S.; Pass, H.I.; Rimner, A.; Mansfield, A.S. New Era for Malignant Pleural Mesothelioma: Updates on Therapeutic Options. J. Clin. Oncol. 2022, 40, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Mujoomdar, A.A.; Tilleman, T.R.; Richards, W.G.; Bueno, R.; Sugarbaker, D.J. Prevalence of in vitro chemotherapeutic drug resistance in primary malignant pleural mesothelioma: Result in a cohort of 203 resection specimens. J. Thorac. Cardiovasc. Surg. 2010, 140, 352–355. [Google Scholar] [CrossRef]
- Zalcman, G.; Mazieres, J.; Margery, J.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Molinier, O.; Corre, R.; Monnet, I.; Gounant, V.; et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): A randomised, controlled, open-label, phase 3 trial. Lancet 2016, 387, 1405–1414. [Google Scholar] [CrossRef]
- Lapidot, M.; Saladi, S.V.; Salgia, R.; Sattler, M. Novel Therapeutic Targets and Immune Dysfunction in Malignant Pleural Mesothelioma. Front. Pharmacol. 2021, 12, 806570. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, R.; Brazeau, P.; Bohlen, P.; Esch, F.; Ling, N.; Wehrenberg, W.B. Growth hormone-releasing factor from a human pancreatic tumor that caused acromegaly. Science 1982, 218, 585–587. [Google Scholar] [CrossRef] [PubMed]
- Granata, R. Peripheral activities of growth hormone-releasing hormone. J. Endocrinol. Investig. 2016, 39, 721–727. [Google Scholar] [CrossRef]
- Schally, A.V.; Zhang, X.; Cai, R.; Hare, J.M.; Granata, R.; Bartoli, M. Actions and Potential Therapeutic Applications of Growth Hormone-Releasing Hormone Agonists. Endocrinology 2019, 160, 1600–1612. [Google Scholar] [CrossRef]
- Cai, R.; Schally, A.V.; Cui, T.; Szalontay, L.; Halmos, G.; Sha, W.; Kovacs, M.; Jaszberenyi, M.; He, J.; Rick, F.G.; et al. Synthesis of new potent agonistic analogs of growth hormone-releasing hormone (GHRH) and evaluation of their endocrine and cardiac activities. Peptides 2014, 52, 104–112. [Google Scholar] [CrossRef]
- Granata, R.; Trovato, L.; Gallo, M.P.; Destefanis, S.; Settanni, F.; Scarlatti, F.; Brero, A.; Ramella, R.; Volante, M.; Isgaard, J.; et al. Growth hormone-releasing hormone promotes survival of cardiac myocytes in vitro and protects against ischaemia-reperfusion injury in rat heart. Cardiovasc. Res. 2009, 83, 303–312. [Google Scholar] [CrossRef]
- Kanashiro-Takeuchi, R.M.; Tziomalos, K.; Takeuchi, L.M.; Treuer, A.V.; Lamirault, G.; Dulce, R.; Hurtado, M.; Song, Y.; Block, N.L.; Rick, F.; et al. Cardioprotective effects of growth hormone-releasing hormone agonist after myocardial infarction. Proc. Natl. Acad. Sci. USA 2010, 107, 2604–2609. [Google Scholar] [CrossRef]
- Recinella, L.; Chiavaroli, A.; Orlando, G.; Ferrante, C.; Marconi, G.D.; Gesmundo, I.; Granata, R.; Cai, R.; Sha, W.; Schally, A.V.; et al. Antinflammatory, antioxidant, and behavioral effects induced by administration of growth hormone-releasing hormone analogs in mice. Sci. Rep. 2020, 10, 732. [Google Scholar] [CrossRef]
- Zhang, X.; Cui, T.; He, J.; Wang, H.; Cai, R.; Popovics, P.; Vidaurre, I.; Sha, W.; Schmid, J.; Ludwig, B.; et al. Beneficial effects of growth hormone-releasing hormone agonists on rat INS-1 cells and on streptozotocin-induced NOD/SCID mice. Proc. Natl. Acad. Sci. USA 2015, 112, 13651–13656. [Google Scholar] [CrossRef]
- Kiaris, H.; Chatzistamou, I.; Papavassiliou, A.G.; Schally, A.V. Growth hormone-releasing hormone: Not only a neurohormone. Trends Endocrinol. Metab. TEM 2011, 22, 311–317. [Google Scholar] [CrossRef]
- Schally, A.V.; Varga, J.L.; Engel, J.B. Antagonists of growth-hormone-releasing hormone: An emerging new therapy for cancer. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 33–43. [Google Scholar] [CrossRef]
- Sato, K.; Hotta, M.; Kageyama, J.; Hu, H.Y.; Dong, M.H.; Ling, N. Synthetic analogs of growth hormone-releasing factor with antagonistic activity in vitro. Biochem. Biophys. Res. Commun. 1990, 167, 360–366. [Google Scholar] [CrossRef]
- Zarandi, M.; Cai, R.; Kovacs, M.; Popovics, P.; Szalontay, L.; Cui, T.; Sha, W.; Jaszberenyi, M.; Varga, J.; Zhang, X.; et al. Synthesis and structure-activity studies on novel analogs of human growth hormone releasing hormone (GHRH) with enhanced inhibitory activities on tumor growth. Peptides 2017, 89, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Zhang, X.; Wang, H.; Cui, T.; Halmos, G.; Sha, W.; He, J.; Popovics, P.; Vidaurre, I.; Zhang, C.; et al. Synthesis of potent antagonists of receptors for growth hormone-releasing hormone with antitumor and anti-inflammatory activity. Peptides 2022, 150, 170716. [Google Scholar] [CrossRef] [PubMed]
- Recinella, L.; Chiavaroli, A.; Veschi, S.; Di Valerio, V.; Lattanzio, R.; Orlando, G.; Ferrante, C.; Gesmundo, I.; Granata, R.; Cai, R.; et al. Antagonist of growth hormone-releasing hormone MIA-690 attenuates the progression and inhibits growth of colorectal cancer in mice. Biomed. Pharmacother. 2022, 146, 112554. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, X.; Vidaurre, I.; Cai, R.; Sha, W.; Schally, A.V. Inhibition of experimental small-cell and non-small-cell lung cancers by novel antagonists of growth hormone-releasing hormone. Int. J. Cancer 2018, 142, 2394–2404. [Google Scholar] [CrossRef] [PubMed]
- Gan, J.; Ke, X.; Jiang, J.; Dong, H.; Yao, Z.; Lin, Y.; Lin, W.; Wu, X.; Yan, S.; Zhuang, Y.; et al. Growth hormone-releasing hormone receptor antagonists inhibit human gastric cancer through downregulation of PAK1-STAT3/NF-kappaB signaling. Proc. Natl. Acad. Sci. USA 2016, 113, 14745–14750. [Google Scholar] [CrossRef]
- Zhang, C.; Cui, T.; Cai, R.; Wangpaichitr, M.; Mirsaeidi, M.; Schally, A.V.; Jackson, R.M. Growth Hormone-Releasing Hormone in Lung Physiology and Pulmonary Disease. Cells 2020, 9, 2331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cai, R.; Lazerson, A.; Delcroix, G.; Wangpaichitr, M.; Mirsaeidi, M.; Griswold, A.J.; Schally, A.V.; Jackson, R.M. Growth Hormone-Releasing Hormone Receptor Antagonist Modulates Lung Inflammation and Fibrosis due to Bleomycin. Lung 2019, 197, 541–549. [Google Scholar] [CrossRef]
- Zhang, C.; Tian, R.; Dreifus, E.M.; Hashemi Shahraki, A.; Holt, G.; Cai, R.; Griswold, A.; Bejarano, P.; Jackson, R.; A, V.S.; et al. Activity of the growth hormone-releasing hormone antagonist MIA602 and its underlying mechanisms of action in sarcoidosis-like granuloma. Clin. Transl. Immunol. 2021, 10, e1310. [Google Scholar] [CrossRef]
- Villanova, T.; Gesmundo, I.; Audrito, V.; Vitale, N.; Silvagno, F.; Musuraca, C.; Righi, L.; Libener, R.; Riganti, C.; Bironzo, P.; et al. Antagonists of growth hormone-releasing hormone (GHRH) inhibit the growth of human malignant pleural mesothelioma. Proc. Natl. Acad. Sci. USA 2019, 116, 2226–2231. [Google Scholar] [CrossRef] [PubMed]
- Arulananda, S.; O’Brien, M.; Evangelista, M.; Jenkins, L.J.; Poh, A.R.; Walkiewicz, M.; Leong, T.; Mariadason, J.M.; Cebon, J.; Balachander, S.B.; et al. A novel BH3-mimetic, AZD0466, targeting BCL-XL and BCL-2 is effective in pre-clinical models of malignant pleural mesothelioma. Cell Death Discov. 2021, 7, 122. [Google Scholar] [CrossRef]
- Cho, H.; Matsumoto, S.; Fujita, Y.; Kuroda, A.; Menju, T.; Sonobe, M.; Kondo, N.; Torii, I.; Nakano, T.; Lara, P.N.; et al. Trametinib plus 4-Methylumbelliferone Exhibits Antitumor Effects by ERK Blockade and CD44 Downregulation and Affects PD-1 and PD-L1 in Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2017, 12, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.S.; Klein, M.A. Targeting cyclin D1 in non-small cell lung cancer and mesothelioma cells by antisense oligonucleotides. Anticancer. Res. 2011, 31, 3683–3690. [Google Scholar] [PubMed]
- Romagnoli, S.; Fasoli, E.; Vaira, V.; Falleni, M.; Pellegrini, C.; Catania, A.; Roncalli, M.; Marchetti, A.; Santambrogio, L.; Coggi, G.; et al. Identification of potential therapeutic targets in malignant mesothelioma using cell-cycle gene expression analysis. Am. J. Pathol. 2009, 174, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Canino, C.; Luo, Y.; Marcato, P.; Blandino, G.; Pass, H.I.; Cioce, M. A STAT3-NFkB/DDIT3/CEBPbeta axis modulates ALDH1A3 expression in chemoresistant cell subpopulations. Oncotarget 2015, 6, 12637–12653. [Google Scholar] [CrossRef]
- Rick, F.G.; Seitz, S.; Schally, A.V.; Szalontay, L.; Krishan, A.; Datz, C.; Stadlmayr, A.; Buchholz, S.; Block, N.L.; Hohla, F. GHRH antagonist when combined with cytotoxic agents induces S-phase arrest and additive growth inhibition of human colon cancer. Cell Cycle 2012, 11, 4203–4210. [Google Scholar] [CrossRef]
- Seitz, S.; Rick, F.G.; Schally, A.V.; Treszl, A.; Hohla, F.; Szalontay, L.; Zarandi, M.; Ortmann, O.; Engel, J.B.; Buchholz, S. Combination of GHRH antagonists and docetaxel shows experimental effectiveness for the treatment of triple-negative breast cancers. Oncol. Rep. 2013, 30, 413–418. [Google Scholar] [CrossRef][Green Version]
- Schally, A.V.; Perez, R.; Block, N.L.; Rick, F.G. Potentiating effects of GHRH analogs on the response to chemotherapy. Cell Cycle 2015, 14, 699–704. [Google Scholar] [CrossRef]
- Hohla, F.; Schally, A.V.; Szepeshazi, K.; Varga, J.L.; Buchholz, S.; Koster, F.; Heinrich, E.; Halmos, G.; Rick, F.G.; Kannadka, C.; et al. Synergistic inhibition of growth of lung carcinomas by antagonists of growth hormone-releasing hormone in combination with docetaxel. Proc. Natl. Acad. Sci. USA 2006, 103, 14513–14518. [Google Scholar] [CrossRef]
- Malakoti, F.; Targhazeh, N.; Abadifard, E.; Zarezadeh, R.; Samemaleki, S.; Asemi, Z.; Younesi, S.; Mohammadnejad, R.; Hadi Hossini, S.; Karimian, A.; et al. DNA repair and damage pathways in mesothelioma development and therapy. Cancer Cell Int. 2022, 22, 176. [Google Scholar] [CrossRef] [PubMed]
- Rick, F.G.; Schally, A.V.; Szalontay, L.; Block, N.L.; Szepeshazi, K.; Nadji, M.; Zarandi, M.; Hohla, F.; Buchholz, S.; Seitz, S. Antagonists of growth hormone-releasing hormone inhibit growth of androgen-independent prostate cancer through inactivation of ERK and Akt kinases. Proc. Natl. Acad. Sci. USA 2012, 109, 1655–1660. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.; Tranchant, R.; Risa-Ebri, B.; Suarez-Solis, M.L.; Fernandez, L.C.; Carrillo-de-Santa-Pau, E.; Del Pozo, N.; Martinez de Villarreal, J.; Meiller, C.; Allory, Y.; et al. Combined MEK and PI3K/p110beta Inhibition as a Novel Targeted Therapy for Malignant Mesothelioma Displaying Sarcomatoid Features. Cancer Res. 2020, 80, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Gesmundo, I.; Granato, G.; Fuentes-Fayos, A.C.; Alvarez, C.V.; Dieguez, C.; Zatelli, M.C.; Congiusta, N.; Banfi, D.; Prencipe, N.; Leone, S.; et al. Antagonists of Growth Hormone-Releasing Hormone Inhibit the Growth of Pituitary Adenoma Cells by Hampering Oncogenic Pathways and Promoting Apoptotic Signaling. Cancers 2021, 13, 3950. [Google Scholar] [CrossRef] [PubMed]
- Barabutis, N.; Siejka, A.; Schally, A.V. Growth hormone releasing hormone induces the expression of nitric oxide synthase. J. Cell Mol. Med. 2011, 15, 1148–1155. [Google Scholar] [CrossRef]
- Barabutis, N.; Siejka, A. The highly interrelated GHRH, p53, and Hsp90 universe. Cell Biol. Int. 2020, 44, 1558–1563. [Google Scholar] [CrossRef]
- Volakaki, A.A.; Lafkas, D.; Kassi, E.; Schally, A.V.; Papavassiliou, A.G.; Kiaris, H. Essential role of p21/waf1 in the mediation of the anti-proliferative effects of GHRH antagonist JMR-132. J. Mol. Endocrinol. 2008, 41, 389–392. [Google Scholar] [CrossRef]
- Hohla, F.; Buchholz, S.; Schally, A.V.; Seitz, S.; Rick, F.G.; Szalontay, L.; Varga, J.L.; Zarandi, M.; Halmos, G.; Vidaurre, I.; et al. GHRH antagonist causes DNA damage leading to p21 mediated cell cycle arrest and apoptosis in human colon cancer cells. Cell Cycle 2009, 8, 3149–3156. [Google Scholar] [CrossRef]
- Uxa, S.; Castillo-Binder, P.; Kohler, R.; Stangner, K.; Muller, G.A.; Engeland, K. Ki-67 gene expression. Cell Death Differ. 2021, 28, 3357–3370. [Google Scholar] [CrossRef]
- Perez-Roger, I.; Kim, S.H.; Griffiths, B.; Sewing, A.; Land, H. Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1). EMBO J. 1999, 18, 5310–5320. [Google Scholar] [CrossRef]
- Choi, Y.J.; Anders, L. Signaling through cyclin D-dependent kinases. Oncogene 2014, 33, 1890–1903. [Google Scholar] [CrossRef] [PubMed]
- Ramael, M.; Van den Bossche, J.; Buysse, C.; Deblier, I.; Segers, K.; Van Marck, E. Immunoreactivity for c-fos and c-myc protein with the monoclonal antibodies 14E10 and 6E10 in malignant mesothelioma and non-neoplastic mesothelium of the pleura. Histol. Histopathol. 1995, 10, 639–643. [Google Scholar] [PubMed]
- Riquelme, E.; Suraokar, M.B.; Rodriguez, J.; Mino, B.; Lin, H.Y.; Rice, D.C.; Tsao, A.; Wistuba, I.I. Frequent coamplification and cooperation between C-MYC and PVT1 oncogenes promote malignant pleural mesothelioma. J. Thorac. Oncol. 2014, 9, 998–1007. [Google Scholar] [CrossRef]
- Tan, Y.; Sementino, E.; Chernoff, J.; Testa, J.R. Targeting MYC sensitizes malignant mesothelioma cells to PAK blockage-induced cytotoxicity. Am. J. Cancer Res. 2017, 7, 1724–1737. [Google Scholar] [PubMed]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene-the grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Cioce, M.; Canino, C.; Pulito, C.; Muti, P.; Strano, S.; Blandino, G. Butein impairs the protumorigenic activity of malignant pleural mesothelioma cells. Cell Cycle 2012, 11, 132–140. [Google Scholar] [CrossRef]
- Rick, F.G.; Schally, A.V.; Block, N.L.; Nadji, M.; Szepeshazi, K.; Zarandi, M.; Vidaurre, I.; Perez, R.; Halmos, G.; Szalontay, L. Antagonists of growth hormone-releasing hormone (GHRH) reduce prostate size in experimental benign prostatic hyperplasia. Proc. Natl. Acad. Sci. USA 2011, 108, 3755–3760. [Google Scholar] [CrossRef]
- Barabutis, N.; Schally, A.V.; Siejka, A. P53, GHRH, inflammation and cancer. EBioMedicine 2018, 37, 557–562. [Google Scholar] [CrossRef]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef]
- Nuvoli, B.; Galati, R. Cyclooxygenase-2, epidermal growth factor receptor, and aromatase signaling in inflammation and mesothelioma. Mol. Cancer Ther. 2013, 12, 844–852. [Google Scholar] [CrossRef]
- Soini, Y.; Kahlos, K.; Puhakka, A.; Lakari, E.; Saily, M.; Paakko, P.; Kinnula, V. Expression of inducible nitric oxide synthase in healthy pleura and in malignant mesothelioma. Br. J. Cancer 2000, 83, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Recinella, L.; Chiavaroli, A.; Di Valerio, V.; Veschi, S.; Orlando, G.; Ferrante, C.; Gesmundo, I.; Granata, R.; Cai, R.; Sha, W.; et al. Protective effects of growth hormone-releasing hormone analogs in DSS-induced colitis in mice. Sci. Rep. 2021, 11, 2530. [Google Scholar] [CrossRef] [PubMed]
- Strbac, D.; Dolzan, V. Matrix Metalloproteinases as Biomarkers and Treatment Targets in Mesothelioma: A Systematic Review. Biomolecules 2021, 11, 1272. [Google Scholar] [CrossRef]
- Yang, H.; Rivera, Z.; Jube, S.; Nasu, M.; Bertino, P.; Goparaju, C.; Franzoso, G.; Lotze, M.T.; Krausz, T.; Pass, H.I.; et al. Programmed necrosis induced by asbestos in human mesothelial cells causes high-mobility group box 1 protein release and resultant inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 12611–12616. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Patergnani, S.; Giorgi, C.; Suarez, J.; Goto, K.; Bononi, A.; Tanji, M.; Novelli, F.; Pastorino, S.; Xu, R.; et al. Asbestos induces mesothelial cell transformation via HMGB1-driven autophagy. Proc. Natl. Acad. Sci. USA 2020, 117, 25543–25552. [Google Scholar] [CrossRef]
- Munoz-Moreno, L.; Schally, A.V.; Prieto, J.C.; Carmena, M.J.; Bajo, A.M. Growth hormone-releasing hormone receptor antagonists modify molecular machinery in the progression of prostate cancer. Prostate 2018, 78, 915–926. [Google Scholar] [CrossRef]
- Siejka, A.; Barabutis, N.; Schally, A.V. GHRH antagonist inhibits focal adhesion kinase (FAK) and decreases expression of vascular endothelial growth factor (VEGF) in human lung cancer cells in vitro. Peptides 2012, 37, 63–68. [Google Scholar] [CrossRef]
- Hoang, C.D.; Zhang, X.; Scott, P.D.; Guillaume, T.J.; Maddaus, M.A.; Yee, D.; Kratzke, R.A. Selective activation of insulin receptor substrate-1 and -2 in pleural mesothelioma cells: Association with distinct malignant phenotypes. Cancer Res. 2004, 64, 7479–7485. [Google Scholar] [CrossRef][Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gesmundo, I.; Pedrolli, F.; Vitale, N.; Bertoldo, A.; Orlando, G.; Banfi, D.; Granato, G.; Kasarla, R.; Balzola, F.; Deaglio, S.; et al. Antagonist of Growth Hormone-Releasing Hormone Potentiates the Antitumor Effect of Pemetrexed and Cisplatin in Pleural Mesothelioma. Int. J. Mol. Sci. 2022, 23, 11248. https://doi.org/10.3390/ijms231911248
Gesmundo I, Pedrolli F, Vitale N, Bertoldo A, Orlando G, Banfi D, Granato G, Kasarla R, Balzola F, Deaglio S, et al. Antagonist of Growth Hormone-Releasing Hormone Potentiates the Antitumor Effect of Pemetrexed and Cisplatin in Pleural Mesothelioma. International Journal of Molecular Sciences. 2022; 23(19):11248. https://doi.org/10.3390/ijms231911248
Chicago/Turabian StyleGesmundo, Iacopo, Francesca Pedrolli, Nicoletta Vitale, Alessia Bertoldo, Giulia Orlando, Dana Banfi, Giuseppina Granato, Ramesh Kasarla, Federico Balzola, Silvia Deaglio, and et al. 2022. "Antagonist of Growth Hormone-Releasing Hormone Potentiates the Antitumor Effect of Pemetrexed and Cisplatin in Pleural Mesothelioma" International Journal of Molecular Sciences 23, no. 19: 11248. https://doi.org/10.3390/ijms231911248
APA StyleGesmundo, I., Pedrolli, F., Vitale, N., Bertoldo, A., Orlando, G., Banfi, D., Granato, G., Kasarla, R., Balzola, F., Deaglio, S., Cai, R., Sha, W., Papotti, M., Ghigo, E., Schally, A. V., & Granata, R. (2022). Antagonist of Growth Hormone-Releasing Hormone Potentiates the Antitumor Effect of Pemetrexed and Cisplatin in Pleural Mesothelioma. International Journal of Molecular Sciences, 23(19), 11248. https://doi.org/10.3390/ijms231911248