
Plasma Proteomics Enable Differentiation of Lung Adenocarcinoma from Chronic Obstructive Pulmonary Disease (COPD)
 , , , , , ,
, , , , , ,  , , and
, , and
Abstract
1. Introduction
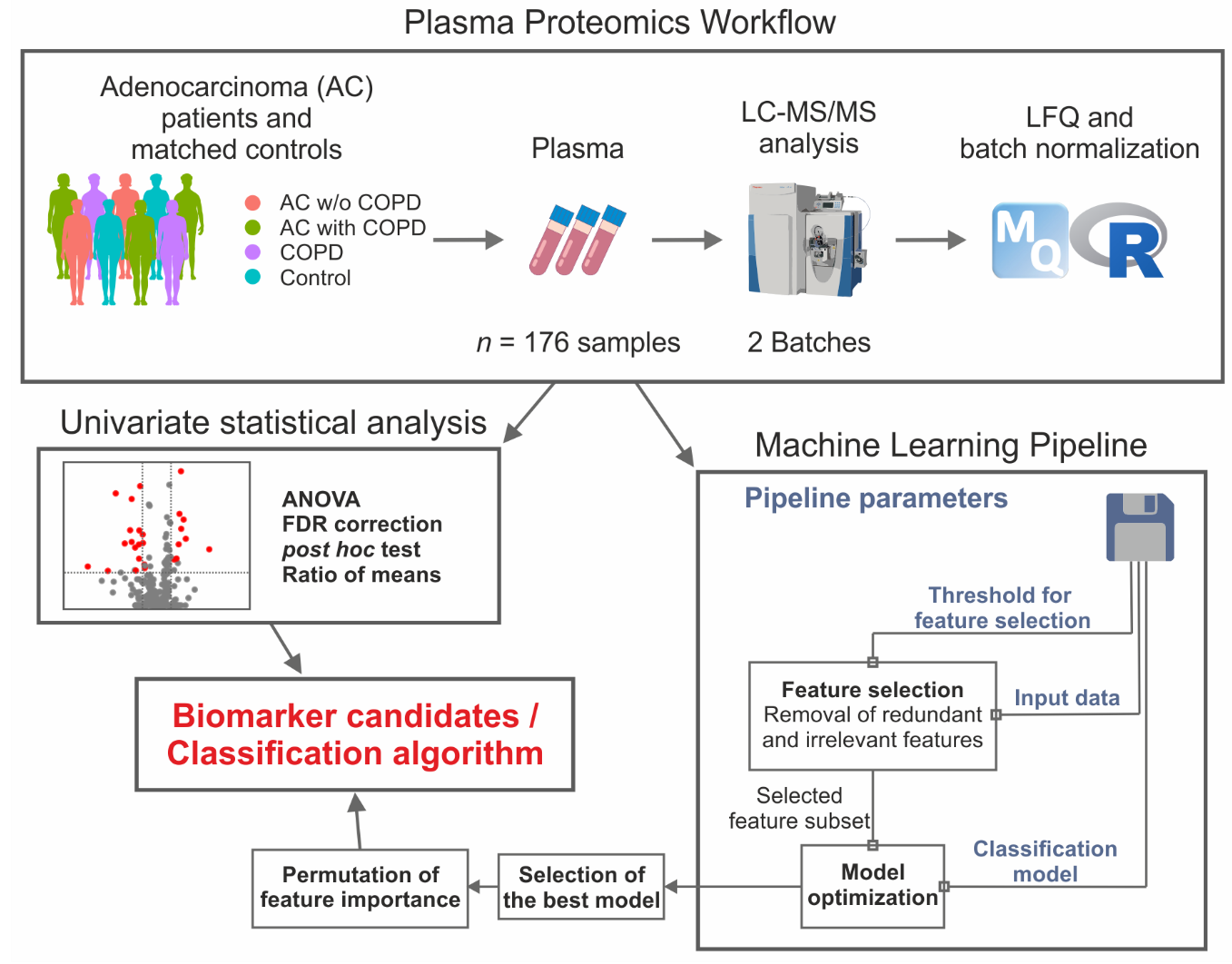
2. Results
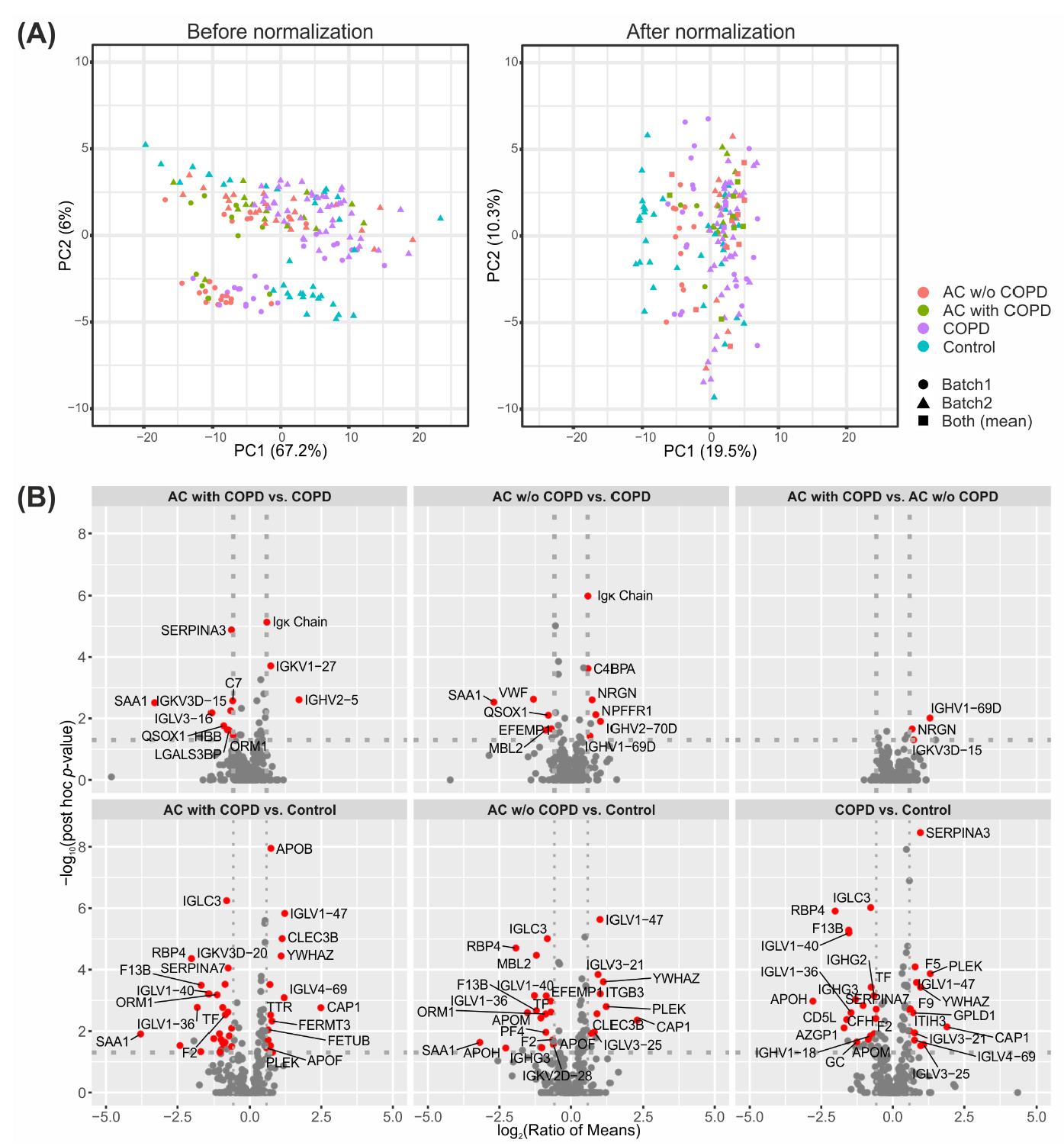
2.1. Successful Normalization of Label-Free Proteomics Data
2.2. Univariate Statistics Reveal Proteins Discriminating AC and COPD
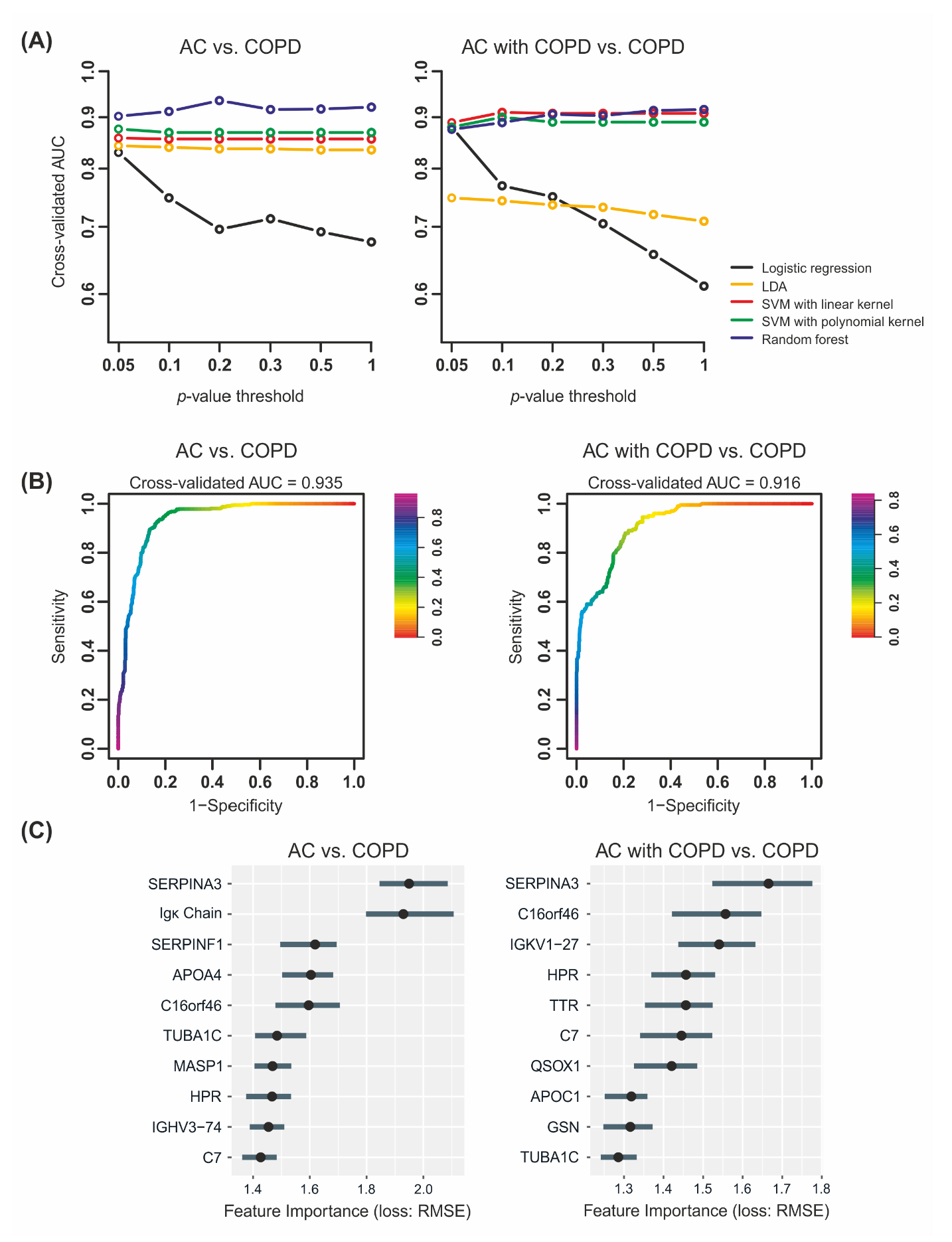
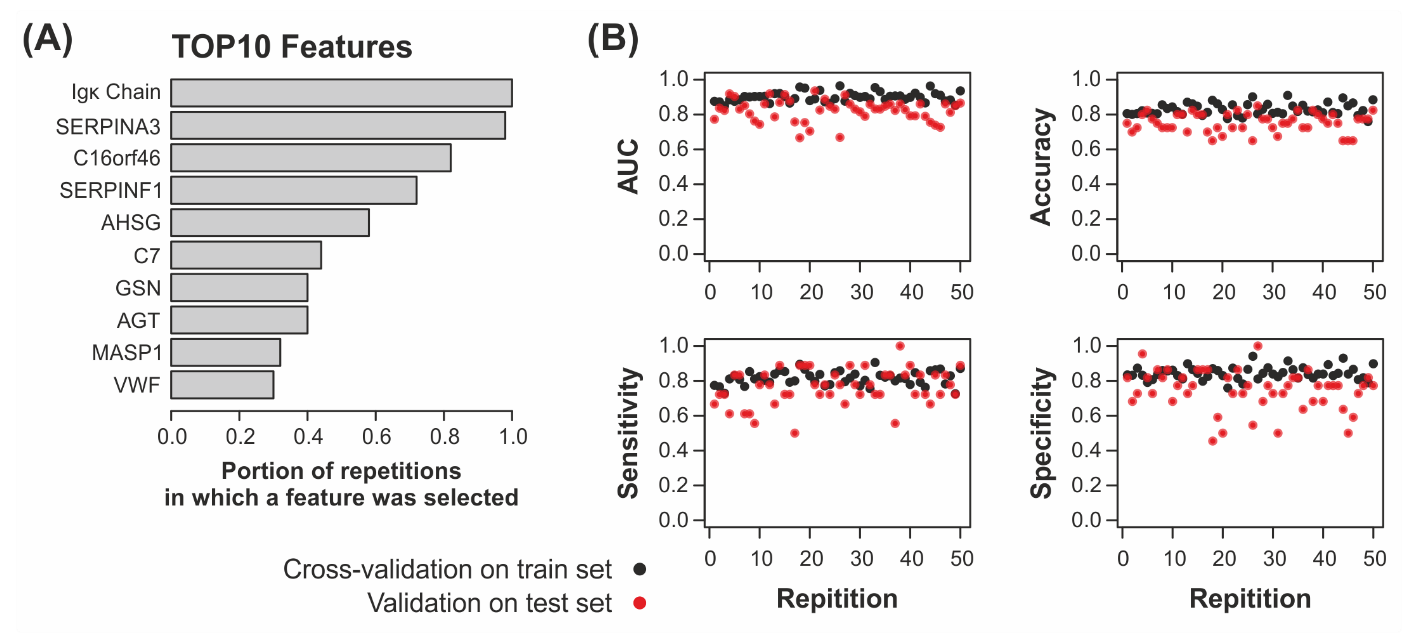
2.3. Machine Learning Yields Highly Predictive Classification Models
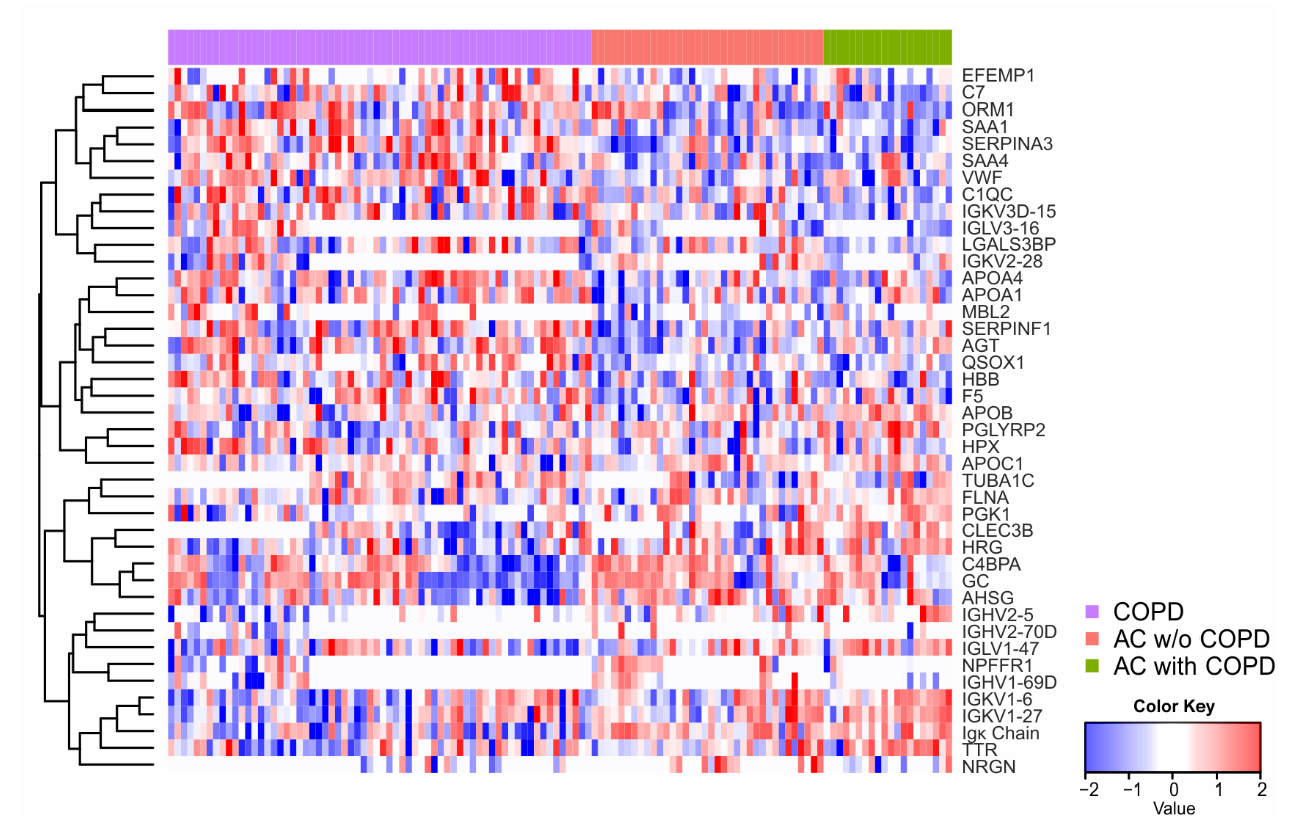
2.4. Univariate Statistics and Machine Learning Reveal Candidates for a Biomarker Panel
3. Discussion
4. Materials and Methods
4.1. Patient Plasma Samples and Clinical Data
4.2. Sample Preparation for LC–MS/MS Analysis
4.3. LC–MS/MS Analysis
4.4. Protein Identification and Quantification
4.5. Batch Normalization
4.6. Statistical Analysis
4.7. Machine Learning
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Adcock, I.M.; Caramori, G.; Barnes, P.J. Chronic obstructive pulmonary disease and lung cancer: New molecular insights. Respir. Int. Rev. Thorac. Dis. 2011, 81, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Parris, B.A.; O’Farrell, H.E.; Fong, K.M.; Yang, I.A. Chronic obstructive pulmonary disease (copd) and lung cancer: Common pathways for pathogenesis. J. Thorac. Dis. 2019, 11, S2155–S2172. [Google Scholar] [CrossRef] [PubMed]
- Lundback, B.; Lindberg, A.; Lindstrom, M.; Ronmark, E.; Jonsson, A.C.; Jonsson, E.; Larsson, L.G.; Andersson, S.; Sandstrom, T.; Larsson, K. Not 15 but 50% of smokers develop copd?—Report from the obstructive lung disease in northern sweden studies. Respir. Med. 2003, 97, 115–122. [Google Scholar] [CrossRef]
- Burney, P.G.; Patel, J.; Newson, R.; Minelli, C.; Naghavi, M. Global and regional trends in copd mortality, 1990–2010. Eur. Respir. J. 2015, 45, 1239–1247. [Google Scholar] [CrossRef]
- Young, R.P.; Duan, F.; Chiles, C.; Hopkins, R.J.; Gamble, G.D.; Greco, E.M.; Gatsonis, C.; Aberle, D. Airflow limitation and histology shift in the national lung screening trial. The nlst-acrin cohort substudy. Am. J. Respir. Crit. Care Med. 2015, 192, 1060–1067. [Google Scholar] [CrossRef]
- Young, R.P.; Hopkins, R.J.; Christmas, T.; Black, P.N.; Metcalf, P.; Gamble, G.D. Copd prevalence is increased in lung cancer, independent of age, sex and smoking history. Eur. Respir. J. 2009, 34, 380–386. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Conrads, T.P.; Hood, B.L.; Veenstra, T.D. Sampling and analytical strategies for biomarker discovery using mass spectrometry. BioTechniques 2006, 40, 799–805. [Google Scholar] [CrossRef]
- Szabo, M.; Hajba, L.; Kun, R.; Guttman, A.; Csanky, E. Proteomic and glycomic markers to differentiate lung adenocarcinoma from copd. Curr. Med. Chem. 2020, 27, 3302–3313. [Google Scholar] [CrossRef]
- Zamay, T.N.; Zamay, G.S.; Kolovskaya, O.S.; Zukov, R.A.; Petrova, M.M.; Gargaun, A.; Berezovski, M.V.; Kichkailo, A.S. Current and prospective protein biomarkers of lung cancer. Cancers 2017, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.; Nishiyama, N.; Yamano, S.; Komatsu, H.; Hanada, S.; Wei, M.; Wanibuchi, H.; Suehiro, S.; Kakehashi, A. Serum agr2 as an early diagnostic and postoperative prognostic biomarker of human lung adenocarcinoma. Cancer Biomark. Sect. A Dis. Markers 2011, 10, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Sholl, L.M. Biomarkers in lung adenocarcinoma: A decade of progress. Arch. Pathol. Lab. Med. 2015, 139, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Bittner, N.; Ostoros, G.; Geczi, L. New treatment options for lung adenocarcinoma—In view of molecular background. Pathol. Oncol. Res. POR 2014, 20, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gao, Y.; Hao, F.; Lou, X.; Zhang, X.; Li, Y.; Wu, D.; Xiao, T.; Yang, L.; Li, Q.; et al. Secretomes are a potential source of molecular targets for cancer therapies and indicate that apoe is a candidate biomarker for lung adenocarcinoma metastasis. Mol. Biol. Rep. 2014, 41, 7507–7523. [Google Scholar] [CrossRef]
- Li, W.; Zheng, H.; Qin, H.; Liu, G.; Ke, L.; Li, Y.; Li, N.; Zhong, X. Exploration of differentially expressed plasma proteins in patients with lung adenocarcinoma using itraq-coupled 2d lc-ms/ms. Clin. Respir. J. 2018, 12, 2036–2045. [Google Scholar] [CrossRef]
- Lai, T.; Wu, D.; Chen, M.; Cao, C.; Jing, Z.; Huang, L.; Lv, Y.; Zhao, X.; Lv, Q.; Wang, Y.; et al. Ykl-40 expression in chronic obstructive pulmonary disease: Relation to acute exacerbations and airway remodeling. Respir. Res. 2016, 17, 31. [Google Scholar] [CrossRef]
- Johansson, S.L.; Roberts, N.B.; Schlosser, A.; Andersen, C.B.; Carlsen, J.; Wulf-Johansson, H.; Saekmose, S.G.; Titlestad, I.L.; Tornoe, I.; Miller, B.; et al. Microfibrillar-associated protein 4: A potential biomarker of chronic obstructive pulmonary disease. Respir. Med. 2014, 108, 1336–1344. [Google Scholar] [CrossRef]
- Angata, T.; Fujinawa, R.; Kurimoto, A.; Nakajima, K.; Kato, M.; Takamatsu, S.; Korekane, H.; Gao, C.X.; Ohtsubo, K.; Kitazume, S.; et al. Integrated approach toward the discovery of glyco-biomarkers of inflammation-related diseases. Ann. N. Y. Acad. Sci. 2012, 1253, 159–169. [Google Scholar] [CrossRef]
- Camacho, D.M.; Collins, K.M.; Powers, R.K.; Costello, J.C.; Collins, J.J. Next-generation machine learning for biological networks. Cell 2018, 173, 1581–1592. [Google Scholar] [CrossRef]
- Byvatov, E.; Schneider, G. Support vector machine applications in bioinformatics. Appl. Bioinform. 2003, 2, 67–77. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef] [PubMed]
- Megger, D.A.; Bracht, T.; Meyer, H.E.; Sitek, B. Label-free quantification in clinical proteomics. Biochim. Biophys. Acta 2013, 1834, 1581–1590. [Google Scholar] [CrossRef]
- Witzke, K.E.; Grosserueschkamp, F.; Jutte, H.; Horn, M.; Roghmann, F.; von Landenberg, N.; Bracht, T.; Kallenbach-Thieltges, A.; Kafferlein, H.; Bruning, T.; et al. Integrated fourier transform infrared imaging and proteomics for identification of a candidate histochemical biomarker in bladder cancer. Am. J. Pathol. 2019, 189, 619–631. [Google Scholar] [CrossRef]
- Bracht, T.; Schweinsberg, V.; Trippler, M.; Kohl, M.; Ahrens, M.; Padden, J.; Naboulsi, W.; Barkovits, K.; Megger, D.A.; Eisenacher, M.; et al. Analysis of disease-associated protein expression using quantitative proteomics-fibulin-5 is expressed in association with hepatic fibrosis. J. Proteome Res. 2015, 14, 2278–2286. [Google Scholar] [CrossRef]
- Naboulsi, W.; Megger, D.A.; Bracht, T.; Kohl, M.; Turewicz, M.; Eisenacher, M.; Voss, D.M.; Schlaak, J.F.; Hoffmann, A.C.; Weber, F.; et al. Quantitative tissue proteomics analysis reveals versican as potential biomarker for early-stage hepatocellular carcinoma. J. Proteome Res. 2016, 15, 38–47. [Google Scholar] [CrossRef]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. MCP 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Niu, L.; Geyer, P.E.; Wewer Albrechtsen, N.J.; Gluud, L.L.; Santos, A.; Doll, S.; Treit, P.V.; Holst, J.J.; Knop, F.K.; Vilsboll, T.; et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol. Syst. Biol. 2019, 15, e8793. [Google Scholar] [CrossRef]
- Captur, G.; Heywood, W.E.; Coats, C.; Rosmini, S.; Patel, V.; Lopes, L.R.; Collis, R.; Patel, N.; Syrris, P.; Bassett, P.; et al. Identification of a multiplex biomarker panel for hypertrophic cardiomyopathy using quantitative proteomics and machine learning. Mol. Cell. Proteom. MCP 2020, 19, 114–127. [Google Scholar] [CrossRef]
- Durham, A.L.; Adcock, I.M. The relationship between copd and lung cancer. Lung Cancer 2015, 90, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Koshiol, J.; Rotunno, M.; Consonni, D.; Pesatori, A.C.; De Matteis, S.; Goldstein, A.M.; Chaturvedi, A.K.; Wacholder, S.; Landi, M.T.; Lubin, J.H.; et al. Chronic obstructive pulmonary disease and altered risk of lung cancer in a population-based case-control study. PLoS ONE 2009, 4, e7380. [Google Scholar] [CrossRef] [PubMed]
- Mouronte-Roibas, C.; Leiro-Fernandez, V.; Ruano-Ravina, A.; Ramos-Hernandez, C.; Casado-Rey, P.; Botana-Rial, M.; Garcia-Rodriguez, E.; Fernandez-Villar, A. Predictive value of a series of inflammatory markers in copd for lung cancer diagnosis: A case-control study. Respir. Res. 2019, 20, 198. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.C.; Kwan, C.K.; Yau, S.; So, P.P.; Poon, P.C.; Au, J.S. The role of inflammation in the pathogenesis of lung cancer. Expert Opin. Ther. Targets 2011, 15, 1127–1137. [Google Scholar] [CrossRef]
- Basile, U.; Gulli, F.; Gragnani, L.; Napodano, C.; Pocino, K.; Rapaccini, G.L.; Mussap, M.; Zignego, A.L. Free light chains: Eclectic multipurpose biomarker. J. Immunol. Methods 2017, 451, 11–19. [Google Scholar] [CrossRef]
- Braber, S.; Thio, M.; Blokhuis, B.R.; Henricks, P.A.; Koelink, P.J.; Groot Kormelink, T.; Bezemer, G.F.; Kerstjens, H.A.; Postma, D.S.; Garssen, J.; et al. An association between neutrophils and immunoglobulin free light chains in the pathogenesis of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2012, 185, 817–824. [Google Scholar] [CrossRef]
- Hu, J.; Boeri, M.; Sozzi, G.; Liu, D.; Marchiano, A.; Roz, L.; Pelosi, G.; Gatter, K.; Pastorino, U.; Pezzella, F. Gene signatures stratify computed tomography screening detected lung cancer in high-risk populations. EBioMedicine 2015, 2, 831–840. [Google Scholar] [CrossRef][Green Version]
- Jung, Y.J.; Oh, I.J.; Kim, Y.; Jung, J.H.; Seok, M.; Lee, W.; Park, C.K.; Lim, J.H.; Kim, Y.C.; Kim, W.S.; et al. Clinical validation of a protein biomarker panel for non-small cell lung cancer. J. Korean Med. Sci. 2018, 33, e342. [Google Scholar] [CrossRef]
- Sanchez-Navarro, A.; Murillo-de-Ozores, A.R.; Perez-Villalva, R.; Linares, N.; Carbajal-Contreras, H.; Flores, M.E.; Gamba, G.; Castaneda-Bueno, M.; Bobadilla, N.A. Transient response of serpina3 during cellular stress. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2022, 36, e22190. [Google Scholar] [CrossRef]
- Sanchez-Navarro, A.; Gonzalez-Soria, I.; Caldino-Bohn, R.; Bobadilla, N.A. An integrative view of serpins in health and disease: The contribution of serpina3. Am. J. Physiol. Cell Physiol. 2021, 320, C106–C118. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, J.; Qu, C.; Peng, Y.; Lei, J.; Li, K.; Zong, B.; Sun, L.; Liu, S. Overexpression of serpina3 promotes tumor invasion and migration, epithelial-mesenchymal-transition in triple-negative breast cancer cells. Breast Cancer 2021, 28, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Katilius, E.; Ostroff, R.M.; Kim, Y.; Seok, M.; Lee, S.; Jang, S.; Kim, W.S.; Choi, C.M. Development of a protein biomarker panel to detect non-small-cell lung cancer in korea. Clin. Lung Cancer 2017, 18, e99–e107. [Google Scholar] [CrossRef] [PubMed]
- Borlak, J.; Langer, F.; Chatterji, B. Serum proteome mapping of egf transgenic mice reveal mechanistic biomarkers of lung cancer precursor lesions with clinical significance for human adenocarcinomas. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 3122–3144. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Gallien, S.; El-Khoury, V.; Goswami, P.; Sertamo, K.; Schlesser, M.; Berchem, G.; Domon, B. Quantification of saa1 and saa2 in lung cancer plasma using the isotype-specific prm assays. Proteomics 2015, 15, 3116–3125. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.J.; Ahn, J.M.; Yoon, Y.H.; Rhim, T.Y.; Park, C.S.; Park, J.Y.; Lee, S.Y.; Kim, J.W.; Cho, J.Y. Identification and validation of saa as a potential lung cancer biomarker and its involvement in metastatic pathogenesis of lung cancer. J. Proteome Res. 2011, 10, 1383–1395. [Google Scholar] [CrossRef]
- Sung, H.J.; Jeon, S.A.; Ahn, J.M.; Seul, K.J.; Kim, J.Y.; Lee, J.Y.; Yoo, J.S.; Lee, S.Y.; Kim, H.; Cho, J.Y. Large-scale isotype-specific quantification of serum amyloid a 1/2 by multiple reaction monitoring in crude sera. J. Proteom. 2012, 75, 2170–2180. [Google Scholar] [CrossRef]
- Hughes, C.S.; Moggridge, S.; Muller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Valikangas, T.; Suomi, T.; Elo, L.L. A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief. Bioinform. 2018, 19, 1–11. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison (Condition A vs. Condition B) | Protein Groups Considered for Statistical Testing 1 | Significantly Differentially Abundant Protein Groups 2 | Higher Abundance in Condition A | Higher Abundance in Condition B |
|---|---|---|---|---|
| AC with COPD vs. COPD | 325 | 11 | 3 | 8 |
| AC w/o COPD vs. COPD | 349 | 11 | 6 | 5 |
| AC w/o COPD vs. AC with COPD | 324 | 3 | 3 | 0 |
| AC with COPD vs. Control | 271 | 39 | 14 | 25 |
| AC w/o COPD vs. Control | 278 | 26 | 9 | 17 |
| COPD vs. Control | 283 | 31 | 14 | 18 |
| (1) AC vs. COPD 1 | (2) AC with COPD vs. COPD 2 | ||
|---|---|---|---|
| AUC | 0.935 | AUC | 0.916 |
| PRAUC | 0.928 | PRAUC | 0.882 |
| Accuracy | 0.865 | Accuracy | 0.873 |
| Sensitivity 3 | 0.848 | Sensitivity 5 | 0.570 |
| Specificity 4 | 0.879 | Specificity 4 | 0.965 |
| Minimum 1 | Mean 1 | Maximum 1 | ||
|---|---|---|---|---|
| AUC | Train set 2 | 0.85 | 0.901 | 0.965 |
| Test set 3 | 0.667 | 0.823 | 0.936 | |
| PRAUC | Train set | 0.763 | 0.864 | 0.968 |
| Test set | 0.554 | 0.766 | 0.931 | |
| Accuracy | Train set | 0.76 | 0.831 | 0.91 |
| Test set | 0.65 | 0.753 | 0.85 | |
| Sensitivity | Train set | 0.726 | 0.815 | 0.905 |
| Test set | 0.5 | 0.763 | 1 | |
| Specificity | Train set | 0.759 | 0.844 | 0.941 |
| Test set | 0.455 | 0.745 | 1 |
| Group | Description | Mean Age (Years) | Sex | Smoking Behavior | |
|---|---|---|---|---|---|
| AC * (n = 64) | AC w/o COPD (n = 43) | AC-patients without diagnosed COPD | 67.17 ± 9.43, min. 41, max. 85 | 25 female, 18 male | 20 smokers, 10 ex-smokers, 13 never-smokers |
| AC with COPD (n = 21) | AC-patients with diagnosed COPD | 64.48 ± 8.89, min. 52, max. 84 | 12 female, 9 male | 11 smokers, 8 ex-smokers, 2 never-smokers | |
| COPD § (n = 77) | COPD-patients without AC | 68.61 ± 10.43, min. 38, max. 87 | 36 female, 41 male | 36 smokers, 33 ex-smokers, 6 never-smokers, 2 NA | |
| HC (n = 35) | Hospital controls | 65.34 ± 12.40 min. 41, max. 82 | 16 female, 19 male | 14 smokers, 13 ex-smokers, 8 never-smokers | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bracht, T.; Kleefisch, D.; Schork, K.; Witzke, K.E.; Chen, W.; Bayer, M.; Hovanec, J.; Johnen, G.; Meier, S.; Ko, Y.-D.; et al. Plasma Proteomics Enable Differentiation of Lung Adenocarcinoma from Chronic Obstructive Pulmonary Disease (COPD). Int. J. Mol. Sci. 2022, 23, 11242. https://doi.org/10.3390/ijms231911242
Bracht T, Kleefisch D, Schork K, Witzke KE, Chen W, Bayer M, Hovanec J, Johnen G, Meier S, Ko Y-D, et al. Plasma Proteomics Enable Differentiation of Lung Adenocarcinoma from Chronic Obstructive Pulmonary Disease (COPD). International Journal of Molecular Sciences. 2022; 23(19):11242. https://doi.org/10.3390/ijms231911242
Chicago/Turabian StyleBracht, Thilo, Daniel Kleefisch, Karin Schork, Kathrin E. Witzke, Weiqiang Chen, Malte Bayer, Jan Hovanec, Georg Johnen, Swetlana Meier, Yon-Dschun Ko, and et al. 2022. "Plasma Proteomics Enable Differentiation of Lung Adenocarcinoma from Chronic Obstructive Pulmonary Disease (COPD)" International Journal of Molecular Sciences 23, no. 19: 11242. https://doi.org/10.3390/ijms231911242
APA StyleBracht, T., Kleefisch, D., Schork, K., Witzke, K. E., Chen, W., Bayer, M., Hovanec, J., Johnen, G., Meier, S., Ko, Y.-D., Behrens, T., Brüning, T., Fassunke, J., Buettner, R., Uszkoreit, J., Adamzik, M., Eisenacher, M., & Sitek, B. (2022). Plasma Proteomics Enable Differentiation of Lung Adenocarcinoma from Chronic Obstructive Pulmonary Disease (COPD). International Journal of Molecular Sciences, 23(19), 11242. https://doi.org/10.3390/ijms231911242