
AMBRA1 p.Gln30Arg Mutation, Identified in a Cowden Syndrome Family, Exhibits Hyperproliferative Potential in hTERT-RPE1 Cells

 , ,
, ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
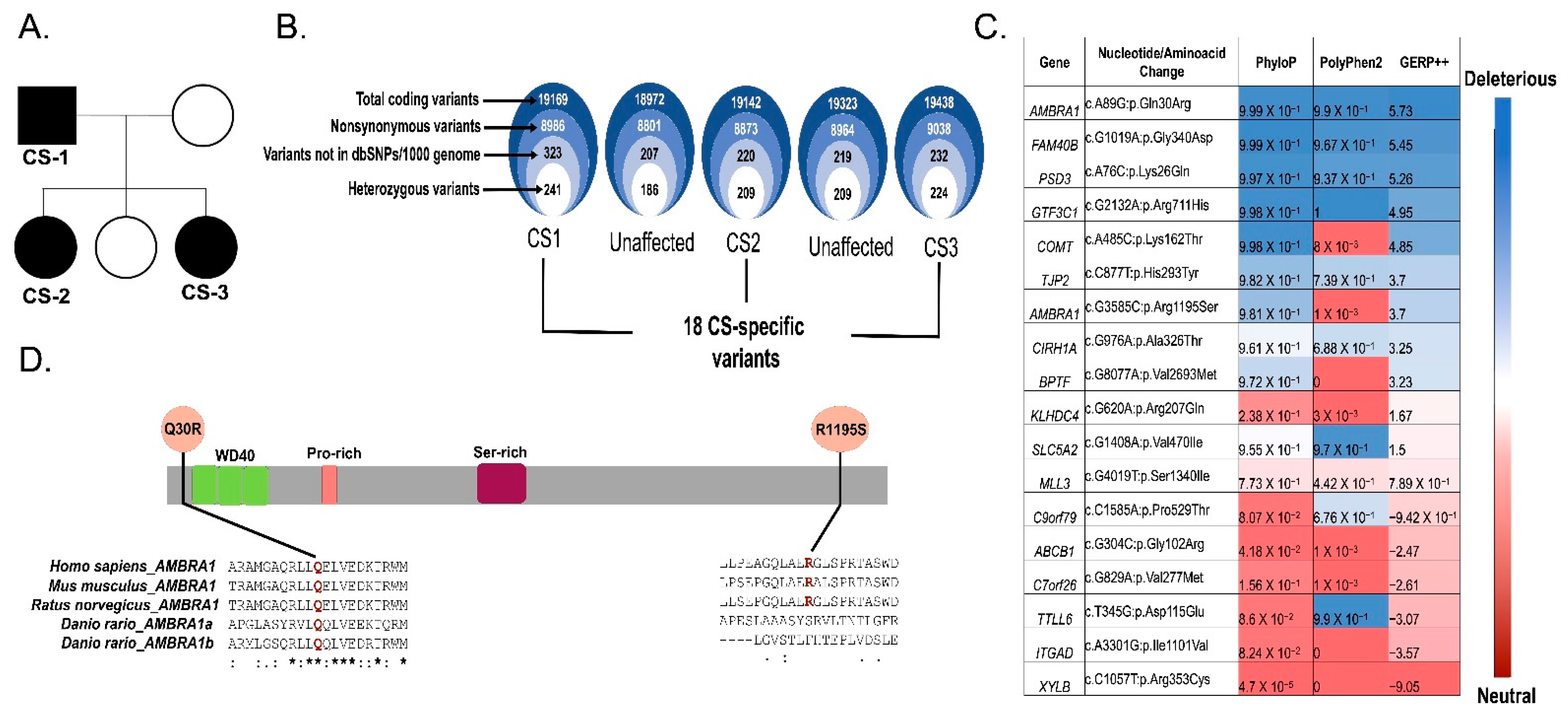
2.1. Study Participants
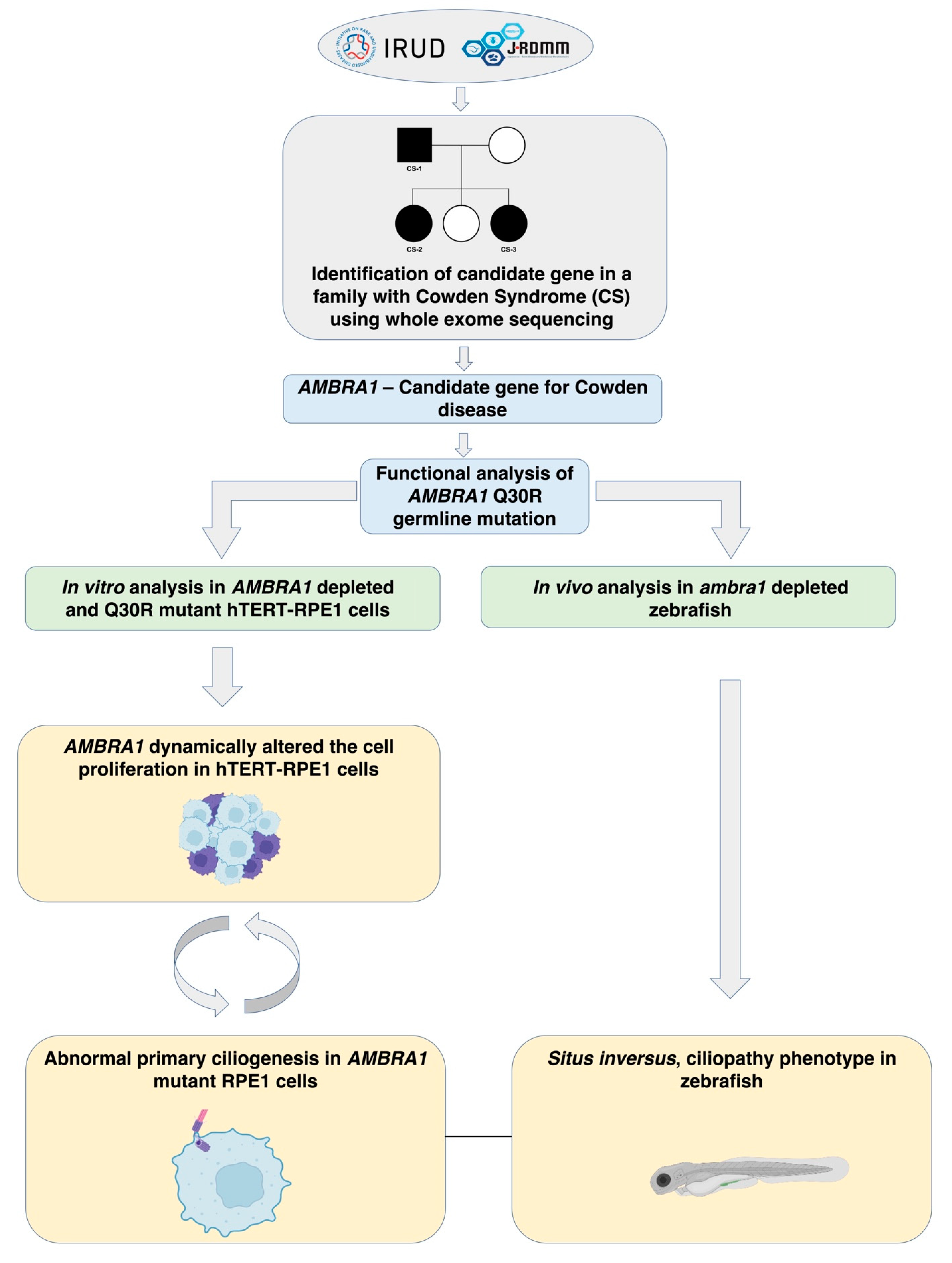
2.2. AMBRA1 Is a Candidate Gene of Cowden Syndrome
2.3. AMBRA1 Dynamically Altered the Cell Cycle in hTERT-RPE1 Cells
2.3.1. AMBRA1 Depletion Caused Hyperproliferation
2.3.2. AMBRA1 Q30R Mutation Disturbed the G1/S Transition of Cells
2.3.3. AMBRA1 Knockouts and Q30R Mutants Exhibited Different Cell Cycle Behaviors
2.4. AMBRA1 Depletion Affected Primary Ciliogenesis, the Structural Checkpoint of the Cell Cycle
2.4.1. AMBRA1 Q30R Mutant hTERT-RPE1 Cells Exhibited Abnormal Primary Cilia
2.4.2. In Vivo Knockout Model of ambra1 in Zebrafish Showed Situs Inversus Phenotype
3. Discussion
4. Materials and Methods
4.1. Identification of Candidate Gene of PTEN Wildtype CS
4.2. Functional Analysis of AMBRA1 Q30R Using hTERT-RPE1 Cells
4.3. Manipulation of ambra1 in Zebrafish
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gammon, A.; Jasperson, K.; Champine, M. Genetic basis of Cowden syndrome and its implications for clinical practice and risk management. Appl. Clin. Genet. 2016, 9, 83–92. [Google Scholar] [CrossRef]
- Garofola, C.; Jamal, Z.; Gross, G.P. Cowden Disease; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Yehia, L.; Ni, Y.; Sesock, K.; Niazi, F.; Fletcher, B.; Chen, H.J.L.; LaFramboise, T.; Eng, C. Unexpected cancer-predisposition gene variants in Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome patients without underlying germline PTEN mutations. PLoS Genet. 2018, 14, e1007352. [Google Scholar] [CrossRef] [PubMed]
- Mester, J.; Eng, C. Cowden syndrome: Recognizing and managing a not-so-rare hereditary cancer syndrome. J. Surg. Oncol. 2015, 111, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Marsh, D.J.; Coulon, V.; Lunetta, K.L.; Rocca-Serra, P.; Dahia, P.L.; Zheng, Z.; Liaw, D.; Caron, S.; Duboué, B.; Lin, A.Y.; et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum. Mol. Genet. 1998, 7, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Cianfanelli, V.; De Zio, D.; Di Bartolomeo, S.; Nazio, F.; Strappazzon, F.; Cecconi, F. Ambra1 at a glance. J. Cell Sci. 2015, 128, 2003–2008. [Google Scholar] [CrossRef]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Maiani, E.; Milletti, G.; Nazio, F.; Holdgaard, S.G.; Bartkova, J.; Rizza, S.; Cianfanelli, V.; Lorente, M.; Simoneschi, D.; Di Marco, M.; et al. AMBRA1 regulates cyclin D to guard S-phase entry and genomic integrity. Nature 2021, 592, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Tong, Y.; Lv, J.; Peng, R.; Chen, S.; Kuang, L.; Su, K.; Zheng, Y.; Zhang, T.; Zhang, F.; et al. Rare mutations in the autophagy-regulating gene AMBRA1 contribute to human neural tube defects. Hum. Mutat. 2020, 41, 1383–1393. [Google Scholar] [CrossRef]
- Plotnikova, O.V.; Pugacheva, E.N.; Golemis, E.A. Primary cilia and the cell cycle. Methods Cell Biol. 2009, 94, 137–160. [Google Scholar]
- Lai, Y.; Jiang, Y. Reciprocal regulation between primary cilia and mTORC1. Genes 2020, 11, 711. [Google Scholar] [CrossRef]
- Di Rita, A.; Strappazzon, F. AMBRA1, a Novel BH3-Like Protein: New Insights Into the AMBRA1–BCL2-Family Proteins Relationship. Int. Rev. Cell Mol. Biol. 2017, 330, 85–113. [Google Scholar] [PubMed]
- Nigg, E.A.; Raff, J.W. Centrioles, centrosomes, and cilia in health and disease. Cell 2009, 139, 663–678. [Google Scholar] [CrossRef]
- Cianfanelli, V.; Nazio, F.; Cecconi, F. Connecting autophagy: AMBRA1 and its network of regulation. Mol. Cell. Oncol. 2015, 2, e970059. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Jang, G.M.; Hüttenhain, R.; Gordon, D.E.; Du, D.; Newton, B.W.; Johnson, J.R.; Hiatt, J.; Hultquist, J.F.; Johnson, T.L.; et al. CRL4AMBRA1 targets Elongin C for ubiquitination and degradation to modulate CRL5 signaling. EMBO J. 2018, 37, e97508. [Google Scholar] [CrossRef]
- Tse, G.M.K.; Law, B.K.B.; Ma, T.K.F.; Chan, A.B.W.; Pang, L.M.; Chu, W.C.W.; Cheung, H.S. Hamartoma of the breast: A clinicopathological review. J. Clin. Pathol. 2002, 55, 951–954. [Google Scholar] [CrossRef]
- Leiter Herrán, F.; Restrepo, C.S.; Alvarez Gómez, D.I.; Suby-Long, T.; Ocazionez, D.; Vargas, D. Hamartomas from head to toe: An imaging overview. Br. J. Radiol. 2017, 90, 20160607. [Google Scholar] [CrossRef]
- Chaikovsky, A.C.; Li, C.; Jeng, E.E.; Loebell, S.; Lee, M.C.; Murray, C.W.; Cheng, R.; Demeter, J.; Swaney, D.L.; Chen, S.H.; et al. The AMBRA1 E3 ligase adaptor regulates the stability of cyclin D. Nature 2021, 592, 794–798. [Google Scholar] [CrossRef]
- Simoneschi, D.; Rona, G.; Zhou, N.; Jeong, Y.T.; Jiang, S.; Milletti, G.; Arbini, A.A.; O’Sullivan, A.; Wang, A.A.; Nithikasem, S.; et al. CRL4 AMBRA1 is a master regulator of D-type cyclins. Nature 2021, 592, 789–793. [Google Scholar] [CrossRef]
- Phua, S.C.; Chiba, S.; Suzuki, M.; Su, E.; Roberson, E.C.; Pusapati, G.V.; Schurmans, S.; Setou, M.; Rohatgi, R.; Reiter, J.F.; et al. Dynamic remodeling of membrane composition drives cell cycle through primary cilia excision. Cell 2017, 168, 264–279. [Google Scholar] [CrossRef]
- Chebib, J.; Guillaume, F. The relative impact of evolving pleiotropy and mutational correlation on trait divergence. bioRxiv 2021. [Google Scholar] [CrossRef]
- Griffiths AJ, F.; Miller, J.H.; Suzuki, D.T.; Lewontin, R.C.; Gelbart, W.M. An Introduction to Genetic Analysis; WH Freeman: New York, NY, USA, 2000; p. 960. [Google Scholar]
- Mayes, J.; Heaton, N. Angiomyolipoma in a Patient with Situs inversus Totalis: Managing Two Rare Diseases. Case Rep. Hepatol. 2016, 2016, 5060284. [Google Scholar] [CrossRef]
- Rallis, E.; Giannopoulos, C.; Korfitis, C.; Kyriakos, N.; Liatsos, C. Tuberous sclerosis in a patient with situs inversus totalis and polysplenia. Indian J. Dermatol. Venereol. Leprol. 2012, 78, 520. [Google Scholar] [CrossRef]
- El-Asmar, J.; Hirmas, N.; Faraj, W.; Akel, A.; Taslakian, B. Undifferentiated Embryonic Liver Sarcoma with Situs inversus Totalis, Polysplenia, and Patent Ductus Venosus. J. Mol. Biomark. Diagn. 2016, 7, 2. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Kabir, A.; Inan, T.; Shehu, A. Analysis of AlphaFold2 for Modeling Structures of Wildtype and Variant Protein Sequences. In Proceedings of the 14th International Conference on Bioinformatics and Computational Biology, Online, 21–23 March 2022; Volume 83, pp. 53–65. [Google Scholar]
- Wu, R.S.; Lam, I.I.; Clay, H.; Duong, D.N.; Deo, R.C.; Coughlin, S.R. A rapid method for directed gene knockout for screening in G0 zebrafish. Dev. Cell 2018, 46, 112–129. [Google Scholar] [CrossRef] [PubMed]
- Ansai, S.; Kinoshita, M. Targeted mutagenesis using CRISPR/Cas system in medaka. Biol. Open 2014, 3, 362–371. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Revathidevi, S.; Hosomichi, K.; Natsume, T.; Nakaoka, H.; Fujito, N.T.; Akatsuka, H.; Sato, T.; Munirajan, A.K.; Inoue, I. AMBRA1 p.Gln30Arg Mutation, Identified in a Cowden Syndrome Family, Exhibits Hyperproliferative Potential in hTERT-RPE1 Cells. Int. J. Mol. Sci. 2022, 23, 11124. https://doi.org/10.3390/ijms231911124
Revathidevi S, Hosomichi K, Natsume T, Nakaoka H, Fujito NT, Akatsuka H, Sato T, Munirajan AK, Inoue I. AMBRA1 p.Gln30Arg Mutation, Identified in a Cowden Syndrome Family, Exhibits Hyperproliferative Potential in hTERT-RPE1 Cells. International Journal of Molecular Sciences. 2022; 23(19):11124. https://doi.org/10.3390/ijms231911124
Chicago/Turabian StyleRevathidevi, Sundaramoorthy, Kazuyoshi Hosomichi, Toyoaki Natsume, Hirofumi Nakaoka, Naoko T. Fujito, Hisako Akatsuka, Takehito Sato, Arasambattu Kannan Munirajan, and Ituro Inoue. 2022. "AMBRA1 p.Gln30Arg Mutation, Identified in a Cowden Syndrome Family, Exhibits Hyperproliferative Potential in hTERT-RPE1 Cells" International Journal of Molecular Sciences 23, no. 19: 11124. https://doi.org/10.3390/ijms231911124
APA StyleRevathidevi, S., Hosomichi, K., Natsume, T., Nakaoka, H., Fujito, N. T., Akatsuka, H., Sato, T., Munirajan, A. K., & Inoue, I. (2022). AMBRA1 p.Gln30Arg Mutation, Identified in a Cowden Syndrome Family, Exhibits Hyperproliferative Potential in hTERT-RPE1 Cells. International Journal of Molecular Sciences, 23(19), 11124. https://doi.org/10.3390/ijms231911124