
The Roles of Mitophagy and Autophagy in Ineffective Erythropoiesis in β-Thalassemia

Abstract
1. Introduction
2. Clinical Manifestation of β-Thalassemia and β-Thalassemia/HbE Diseases
2.1. Etiology and Epidemiology of β-Thalassemia and β-Thalassemia/HbE Diseases
2.2. Pathophysiology of β-Thalassemia and β-Thalassemia/HbE Diseases
2.3. Current Treatment for β-Thalassemia and β-Thalassemia/HbE Diseases
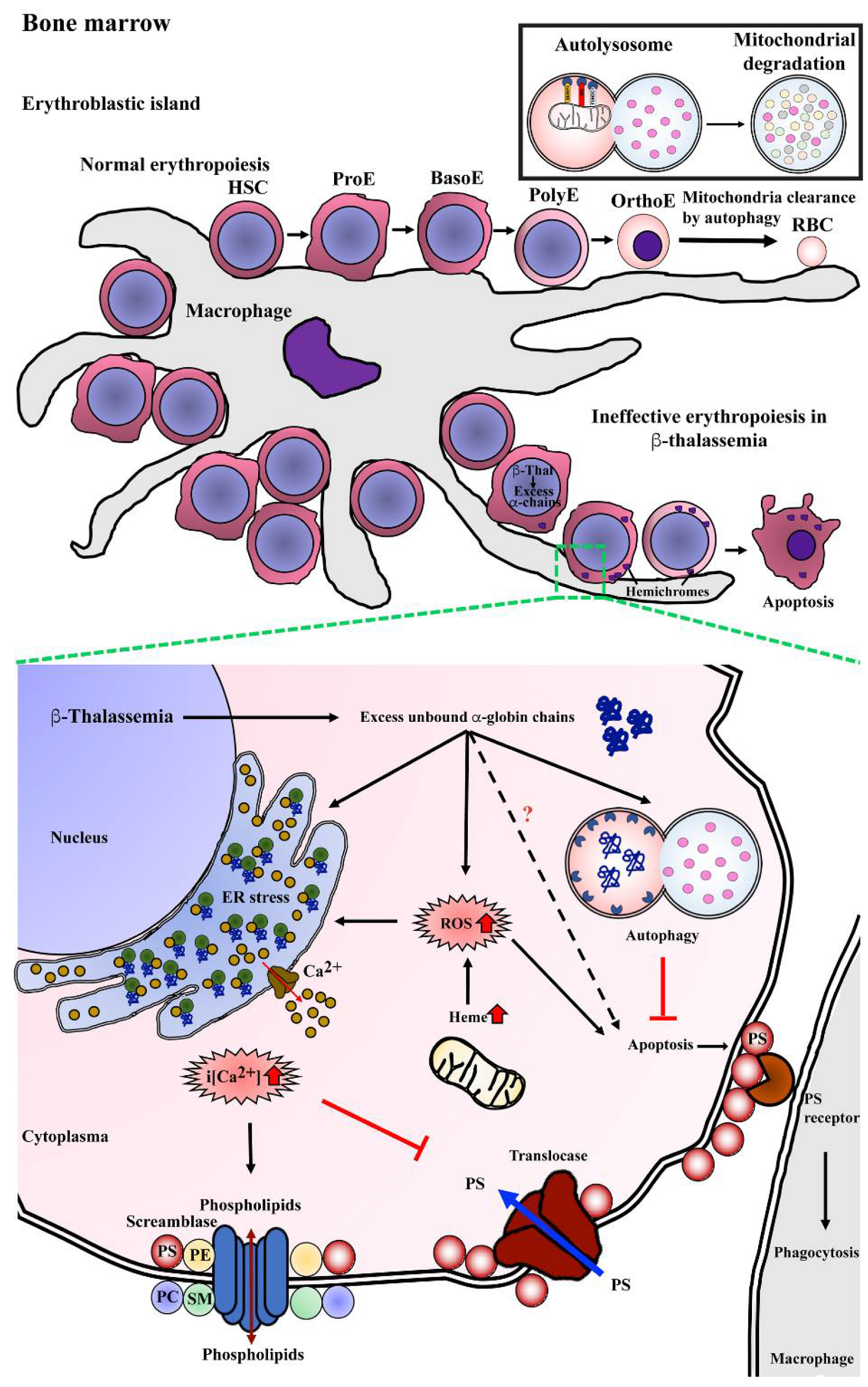
3. Molecular Mechanism of Ineffective Erythropoiesis in β-Thalassemia
4. Autophagy
5. Autophagy during Normal Erythropoiesis
6. Autophagy and Ineffective Erythropoiesis in Thalassemia
7. Novel Therapeutic Drugs Targeting Ineffective Erythropoiesis
7.1. Janus-Associated Kinase (JAK) 2 Inhibitors
7.2. Pyruvate Kinase Activator
7.3. Activin II Receptor Ligand Traps

{kind=link}
{kind=link}
{kind=link}
| Drug | Mechanism of Action | Clinical Trial Identifier | Phase/Status * | Results | Reference |
|---|---|---|---|---|---|
| Ruxolitinib | Janus-associated kinase (JAK) inhibitors | NCT02049450 | Phase 2 completed | No change in transfusion requirement, Reduction in spleen volume | [81] |
| Mitapivat (AG-348) | Pyruvate kinase activator | NCT03692052 | Phase 2 completed | Increase Hb concentration | [83] |
| Mitapivat (AG-348) | Pyruvate kinase activator | NCT04770779, NCT04770753 | Phase 3 recruiting | ||
| Sotatercept (ACE-011) | Activin II receptor ligand traps | NCT01571635 | Phase 2 completed | Increased Hb concentration, Reduced transfusion burden | [87] |
| Luspatercept (ACE-536) | Activin II receptor ligand traps | NCT03342404 | Phase 2 active, not recruiting | Improves Hb concentration, Reduces transfusion burden | [89] |
| Luspatercept (ACE-536) | Activin II receptor ligand traps | NCT02604433 | Phase 3 completed | Reduces transfusion burden | [88] |
8. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rund, D.; Rachmilewitz, E. Beta-thalassemia. N. Engl. J. Med. 2005, 353, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D. beta-Thalassemias. N. Engl. J. Med. 2021, 384, 727–743. [Google Scholar] [CrossRef] [PubMed]
- Pootrakul, P.; Sirankapracha, P.; Hemsorach, S.; Moungsub, W.; Kumbunlue, R.; Piangitjagum, A.; Wasi, P.; Ma, L.; Schrier, S.L. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in thai patients with thalassemia. Blood 2000, 96, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy. Curr. Biol. 2005, 15, R282–R283. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell. Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol 2006, 26, 9220–9231. [Google Scholar] [CrossRef]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxid. Redox. Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef]
- Ashley-Koch, A.; Yang, Q.; Olney, R.S. Sickle hemoglobin (HbS) allele and sickle cell disease: A HuGE review. Am. J. Epidemiol. 2000, 151, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Fucharoen, S.; Weatherall, D.J. The hemoglobin E thalassemias. Cold Spring Harb. Perspect. Med. 2012, 2, a011734. [Google Scholar] [CrossRef] [PubMed]
- Modell, B.; Darlison, M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ. 2008, 86, 480–487. [Google Scholar] [CrossRef]
- Rees, D.C.; Styles, L.; Vichinsky, E.P.; Clegg, J.B.; Weatherall, D.J. The hemoglobin E syndromes. Ann. N. Y. Acad. Sci. 1998, 850, 334–343. [Google Scholar] [CrossRef]
- Finotti, A.; Breda, L.; Lederer, C.W.; Bianchi, N.; Zuccato, C.; Kleanthous, M.; Rivella, S.; Gambari, R. Recent trends in the gene therapy of beta-thalassemia. J. Blood Med. 2015, 6, 69–85. [Google Scholar]
- Thein, S.L. The molecular basis of beta-thalassemia. Cold Spring Harb. Perspect. Med. 2013, 3, a011700. [Google Scholar] [CrossRef] [PubMed]
- Fucharoen, S.; Ketvichit, P.; Pootrakul, P.; Siritanaratkul, N.; Piankijagum, A.; Wasi, P. Clinical manifestation of beta-thalassemia/hemoglobin E disease. J. Pediatr. Hematol. Oncol. 2000, 22, 552–557. [Google Scholar] [CrossRef]
- Fibach, E.; Rachmilewitz, E.A. Pathophysiology and treatment of patients with beta-thalassemia—An update. F1000Res 2017, 6, 2156. [Google Scholar] [CrossRef]
- Atichartakarn, V.; Chuncharunee, S.; Archararit, N.; Udomsubpayakul, U.; Lee, R.; Tunhasiriwet, A.; Aryurachai, K. Prevalence and risk factors for pulmonary hypertension in patients with hemoglobin E/beta-thalassemia disease. Eur. J. Haematol. 2014, 92, 346–353. [Google Scholar] [CrossRef]
- Chuncharunee, S.; Teawtrakul, N.; Siritanaratkul, N.; Chueamuangphan, N. Review of disease-related complications and management in adult patients with thalassemia: A multi-center study in Thailand. PLoS ONE 2019, 14, e0214148. [Google Scholar] [CrossRef]
- Teawtrakul, N.; Jetsrisuparb, A.; Sirijerachai, C.; Chansung, K.; Wanitpongpun, C. Severe bacterial infections in patients with non-transfusion-dependent thalassemia: Prevalence and clinical risk factors. Int. J. Infect. Dis. 2015, 39, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.S.; Lin, C.L.; Lin, C.L.; Kao, C.H. Thalassaemia and risk of cancer: A population-based cohort study. J. Epidemiol. Community Health 2015, 69, 1066–1070. [Google Scholar] [CrossRef]
- Taher, A.T.; Musallam, K.M.; Karimi, M.; El-Beshlawy, A.; Belhoul, K.; Daar, S.; Saned, M.; Cesaretti, C.; Cappellini, M.D. Splenectomy and thrombosis: The case of thalassemia intermedia. J. Thromb. Haemost. 2010, 8, 2152–2158. [Google Scholar] [CrossRef] [PubMed]
- Borgna-Pignatti, C.; Cappellini, M.D.; De Stefano, P.; Del Vecchio, G.C.; Forni, G.L.; Gamberini, M.R.; Ghilardi, R.; Origa, R.; Piga, A.; Romeo, M.A.; et al. Survival and complications in thalassemia. Ann. N. Y. Acad. Sci. 2005, 1054, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Rachmilewitz, E.A.; Giardina, P.J. How I treat thalassemia. Blood 2011, 118, 3479–3488. [Google Scholar] [CrossRef]
- Aydinok, Y. Iron Chelation Therapy as a Modality of Management. Hematol. Oncol. Clin. North Am. 2018, 32, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Italia, K.Y.; Jijina, F.F.; Merchant, R.; Panjwani, S.; Nadkarni, A.H.; Sawant, P.M.; Nair, S.B.; Ghosh, K.; Colah, R.B. Effect of hydroxyurea on the transfusion requirements in patients with severe HbE-beta-thalassaemia: A genotypic and phenotypic study. J. Clin. Pathol. 2010, 63, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Strocchio, L.; Locatelli, F. Hematopoietic Stem Cell Transplantation in Thalassemia. Hematol. Oncol. Clin. North Am. 2018, 32, 317–328. [Google Scholar] [CrossRef]
- Boulad, F.; Mansilla-Soto, J.; Cabriolu, A.; Riviere, I.; Sadelain, M. Gene Therapy and Genome Editing. Hematol. Oncol. Clin. North Am. 2018, 32, 329–342. [Google Scholar] [CrossRef]
- Mathias, L.A.; Fisher, T.C.; Zeng, L.; Meiselman, H.J.; Weinberg, K.I.; Hiti, A.L.; Malik, P. Ineffective erythropoiesis in beta-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp. Hematol. 2000, 28, 1343–1353. [Google Scholar] [CrossRef]
- Yuan, J.; Angelucci, E.; Lucarelli, G.; Aljurf, M.; Snyder, L.M.; Kiefer, C.R.; Ma, L.; Schrier, S.L. Accelerated programmed cell death (apoptosis) in erythroid precursors of patients with severe beta-thalassemia (Cooley’s anemia). Blood 1993, 82, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, S.N.; Lee, M.J. Observations on the relationship between gamma-globin chain content and globin chain precipitation in thalassaemic erythroblasts and on the composition of erythroblastic inclusions in HbE/beta-thalassaemia. Eur. J. Haematol. 1997, 59, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Srinoun, K.; Svasti, S.; Chumworathayee, W.; Vadolas, J.; Vattanaviboon, P.; Fucharoen, S.; Winichagoon, P. Imbalanced globin chain synthesis determines erythroid cell pathology in thalassemic mice. Haematologica 2009, 94, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Leecharoenkiat, A.; Wannatung, T.; Lithanatudom, P.; Svasti, S.; Fucharoen, S.; Chokchaichamnankit, D.; Srisomsap, C.; Smith, D.R. Increased oxidative metabolism is associated with erythroid precursor expansion in beta0-thalassaemia/Hb E disease. Blood Cells Mol. Dis 2011, 47, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Malvezzi, M.; Andra, K.K.; Pandey, K.; Lee, B.C.; Falzone, M.E.; Brown, A.; Iqbal, R.; Menon, A.K.; Accardi, A. Out-of-the-groove transport of lipids by TMEM16 and GPCR scramblases. Proc. Natl. Acad. Sci. USA 2018, 115, E7033–E7042. [Google Scholar] [CrossRef]
- Kean, L.S.; Brown, L.E.; Nichols, J.W.; Mohandas, N.; Archer, D.R.; Hsu, L.L. Comparison of mechanisms of anemia in mice with sickle cell disease and b-thalassemia: Peripheral destruction, ineffective erythropoiesis, and phospholipid scramblase-mediated phosphatidylserine exposure. Exp. Hematol. 2002, 30, 394–402. [Google Scholar] [CrossRef]
- Wannatung, T.; Lithanatudom, P.; Leecharoenkiat, A.; Svasti, S.; Fucharoen, S.; Smith, D.R. Increased erythropoiesis of b-thalassaemia/Hb E proerythroblasts is mediated by high basal levels of ERK1/2 activation. Br. J. Haematol 2009, 146, 557–568. [Google Scholar] [CrossRef]
- Lithanatudom, P.; Leecharoenkiat, A.; Wannatung, T.; Svasti, S.; Fucharoen, S.; Smith, D.R. A mechanism of ineffective erythropoiesis in beta-thalassemia/Hb E disease. Haematologica 2010, 95, 716–723. [Google Scholar] [CrossRef]
- Lithanatudom, P.; Wannatung, T.; Leecharoenkiat, A.; Svasti, S.; Fucharoen, S.; Smith, D.R. Enhanced activation of autophagy in beta-thalassemia/Hb E erythroblasts during erythropoiesis. Ann. Hematol. 2011, 90, 747–758. [Google Scholar] [CrossRef]
- Wickramasinghe, S.N.; Hughes, M. Some features of bone marrow macrophages in patients with homozygous beta-thalassaemia. Br. J. Haematol. 1978, 38, 23–28. [Google Scholar] [CrossRef]
- Angelucci, E.; Bai, H.; Centis, F.; Bafti, M.S.; Lucarelli, G.; Ma, L.; Schrier, S. Enhanced macrophagic attack on beta-thalassemia major erythroid precursors. Haematologica 2002, 87, 578–583. [Google Scholar] [PubMed]
- Kim, K.H.; Lee, M.S. Autophagy-a key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Abdrakhmanov, A.; Gogvadze, V.; Zhivotovsky, B. To Eat or to Die: Deciphering Selective Forms of Autophagy. Trends Biochem. Sci. 2020, 45, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem 2017, 61, 585–596. [Google Scholar]
- Lin, S.Y.; Li, T.Y.; Liu, Q.; Zhang, C.; Li, X.; Chen, Y.; Zhang, S.M.; Lian, G.; Liu, Q.; Ruan, K.; et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 2012, 336, 477–481. [Google Scholar] [CrossRef]
- Grasso, D.; Renna, F.J.; Vaccaro, M.I. Initial Steps in Mammalian Autophagosome Biogenesis. Front. Cell Dev. Biol. 2018, 6, 146. [Google Scholar] [CrossRef]
- Chang, Y.Y.; Neufeld, T.P. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol. Biol. Cell 2009, 20, 2004–2014. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yang, P.; Huang, X.; Hu, W.; Guo, B.; Wu, F.; Lin, L.; Kovacs, A.L.; Yu, L.; Zhang, H. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev. Cell 2011, 21, 343–357. [Google Scholar] [CrossRef]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef]
- Fader, C.M.; Sanchez, D.; Furlan, M.; Colombo, M.I. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 2008, 9, 230–250. [Google Scholar] [CrossRef]
- Dzierzak, E.; Philipsen, S. Erythropoiesis: Development and differentiation. Cold Spring Harb. Perspect. Med. 2013, 3, a011601. [Google Scholar] [CrossRef]
- Chasis, J.A.; Mohandas, N. Erythroblastic islands: Niches for erythropoiesis. Blood 2008, 112, 470–478. [Google Scholar] [CrossRef]
- Baechler, B.L.; Bloemberg, D.; Quadrilatero, J. Mitophagy regulates mitochondrial network signaling, oxidative stress, and apoptosis during myoblast differentiation. Autophagy 2019, 15, 1606–1619. [Google Scholar] [CrossRef] [PubMed]
- Lampert, M.A.; Orogo, A.M.; Najor, R.H.; Hammerling, B.C.; Leon, L.J.; Wang, B.J.; Kim, T.; Sussman, M.A.; Gustafsson, A.B. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 2019, 15, 1182–1198. [Google Scholar] [CrossRef] [PubMed]
- Sin, J.; Andres, A.M.; Taylor, D.J.; Weston, T.; Hiraumi, Y.; Stotland, A.; Kim, B.J.; Huang, C.; Doran, K.S.; Gottlieb, R.A. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 2016, 12, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Kundu, M.; Lindsten, T.; Yang, C.Y.; Wu, J.; Zhao, F.; Zhang, J.; Selak, M.A.; Ney, P.A.; Thompson, C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008, 112, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lee, J.Y.; Wei, H.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, J.L. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef]
- Mortensen, M.; Simon, A.K. Nonredundant role of Atg7 in mitochondrial clearance during erythroid development. Autophagy 2010, 6, 423–425. [Google Scholar] [CrossRef]
- Stolla, M.C.; Reilly, A.; Bergantinos, R.; Stewart, S.; Thom, N.; Clough, C.A.; Wellington, R.C.; Stolitenko, R.; Abkowitz, J.L.; Doulatov, S. ATG4A regulates human erythroid maturation and mitochondrial clearance. Blood Adv. 2022, 6, 3579–3589. [Google Scholar] [CrossRef]
- Zhang, J.; Randall, M.S.; Loyd, M.R.; Dorsey, F.C.; Kundu, M.; Cleveland, J.L.; Ney, P.A. Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation. Blood 2009, 114, 157–164. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Schroder, M. Endoplasmic reticulum stress responses. Cell Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Shalev, O.; Mogilner, S.; Shinar, E.; Rachmilewitz, E.A.; Schrier, S.L. Impaired erythrocyte calcium homeostasis in beta-thalassemia. Blood 1984, 64, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2018, 70, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Xu, W.; Xu, L.; Kong, Q.; Fang, J. Mitophagy is increased during erythroid differentiation in beta-thalassemia. Int. J. Hematol. 2017, 105, 162–173. [Google Scholar] [CrossRef]
- Khandros, E.; Thom, C.S.; D’Souza, J.; Weiss, M.J. Integrated protein quality-control pathways regulate free alpha-globin in murine beta-thalassemia. Blood 2012, 119, 5265–5275. [Google Scholar] [CrossRef] [PubMed]
- Lechauve, C.; Keith, J.; Khandros, E.; Fowler, S.; Mayberry, K.; Freiwan, A.; Thom, C.S.; Delbini, P.; Romero, E.B.; Zhang, J.; et al. The autophagy-activating kinase ULK1 mediates clearance of free alpha-globin in beta-thalassemia. Sci. Transl. Med. 2019, 11, eaav4881. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Fang, J.; Menon, M.; Kapelle, W.; Bogacheva, O.; Bogachev, O.; Houde, E.; Browne, S.; Sathyanarayana, P.; Wojchowski, D.M. EPO modulation of cell-cycle regulatory genes, and cell division, in primary bone marrow erythroblasts. Blood 2007, 110, 2361–2370. [Google Scholar] [CrossRef]
- Casu, C.; Presti, V.L.; Oikonomidou, P.R.; Melchiori, L.; Abdulmalik, O.; Ramos, P.; Rivella, S. Short-term administration of JAK2 inhibitors reduces splenomegaly in mouse models of beta-thalassemia intermedia and major. Haematologica 2018, 103, e46–e49. [Google Scholar] [CrossRef]
- Taher, A.T.; Karakas, Z.; Cassinerio, E.; Siritanaratkul, N.; Kattamis, A.; Maggio, A.; Rivella, S.; Hollaender, N.; Mahuzier, B.; Gadbaw, B.; et al. Efficacy and safety of ruxolitinib in regularly transfused patients with thalassemia: Results from a phase 2a study. Blood 2018, 131, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Grace, R.F.; Rose, C.; Layton, D.M.; Galacteros, F.; Barcellini, W.; Morton, D.H.; van Beers, E.J.; Yaish, H.; Ravindranath, Y.; Kuo, K.H.M.; et al. Safety and Efficacy of Mitapivat in Pyruvate Kinase Deficiency. N. Engl. J. Med. 2019, 381, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.H.M.; Layton, D.M.; Lal, A.; Al-Samkari, H.; Bhatia, J.; Kosinski, P.A.; Tong, B.; Lynch, M.; Uhlig, K.; Vichinsky, E.P. Safety and efficacy of mitapivat, an oral pyruvate kinase activator, in adults with non-transfusion dependent alpha-thalassaemia or beta-thalassaemia: An open-label, multicentre, phase 2 study. Lancet 2022, 400, 493–501. [Google Scholar] [CrossRef]
- Huang, F.; Chen, Y.G. Regulation of TGF-beta receptor activity. Cell Biosci 2012, 2, 9. [Google Scholar] [CrossRef]
- Dussiot, M.; Maciel, T.T.; Fricot, A.; Chartier, C.; Negre, O.; Veiga, J.; Grapton, D.; Paubelle, E.; Payen, E.; Beuzard, Y.; et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in beta-thalassemia. Nat. Med. 2014, 20, 398–407. [Google Scholar] [CrossRef]
- Suragani, R.N.; Cadena, S.M.; Cawley, S.M.; Sako, D.; Mitchell, D.; Li, R.; Davies, M.V.; Alexander, M.J.; Devine, M.; Loveday, K.S.; et al. Transforming growth factor-beta superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat. Med. 2014, 20, 408–414. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Porter, J.; Origa, R.; Forni, G.L.; Voskaridou, E.; Galacteros, F.; Taher, A.T.; Arlet, J.B.; Ribeil, J.A.; Garbowski, M.; et al. Sotatercept, a novel transforming growth factor beta ligand trap, improves anemia in beta-thalassemia: A phase II, open-label, dose-finding study. Haematologica 2019, 104, 477–484. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Viprakasit, V.; Taher, A.T.; Georgiev, P.; Kuo, K.H.M.; Coates, T.; Voskaridou, E.; Liew, H.K.; Pazgal-Kobrowski, I.; Forni, G.L.; et al. A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent beta-Thalassemia. N. Engl. J. Med. 2020, 382, 1219–1231. [Google Scholar] [CrossRef]
- Taher, A.T.; Cappellini, M.D.; Kattamis, A.; Voskaridou, E.; Perrotta, S.; Piga, A.G.; Filosa, A.; Porter, J.B.; Coates, T.D.; Forni, G.L.; et al. Luspatercept for the treatment of anaemia in non-transfusion-dependent beta-thalassaemia (BEYOND): A phase 2, randomised, double-blind, multicentre, placebo-controlled trial. Lancet Haematol. 2022. ahead of print. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaichompoo, P.; Svasti, S.; Smith, D.R. The Roles of Mitophagy and Autophagy in Ineffective Erythropoiesis in β-Thalassemia. Int. J. Mol. Sci. 2022, 23, 10811. https://doi.org/10.3390/ijms231810811
Chaichompoo P, Svasti S, Smith DR. The Roles of Mitophagy and Autophagy in Ineffective Erythropoiesis in β-Thalassemia. International Journal of Molecular Sciences. 2022; 23(18):10811. https://doi.org/10.3390/ijms231810811
Chicago/Turabian StyleChaichompoo, Pornthip, Saovaros Svasti, and Duncan R. Smith. 2022. "The Roles of Mitophagy and Autophagy in Ineffective Erythropoiesis in β-Thalassemia" International Journal of Molecular Sciences 23, no. 18: 10811. https://doi.org/10.3390/ijms231810811
APA StyleChaichompoo, P., Svasti, S., & Smith, D. R. (2022). The Roles of Mitophagy and Autophagy in Ineffective Erythropoiesis in β-Thalassemia. International Journal of Molecular Sciences, 23(18), 10811. https://doi.org/10.3390/ijms231810811