Abstract
Genetic variants in gene-encoding proteins involved in cell–cell connecting structures, such as desmosomes and gap junctions, may cause a skin and/or cardiac phenotype, of which the combination is called cardiocutaneous syndrome. The cardiac phenotype is characterized by cardiomyopathy and/or arrhythmias, while the skin particularly displays phenotypes such as keratoderma, hair abnormalities and skin fragility. The reported variants associated with cardiocutaneous syndrome, in genes DSP, JUP, DSC2, KLHL24, GJA1, are classified by interpretation guidelines from the American College of Medical Genetics and Genomics. The genotype–phenotype correlation, however, remains poorly understood. By providing an overview of variants that are assessed for a functional protein pathology, we show that this number (n = 115) is low compared to the number of variants that are assessed by in silico algorithms (>5000). As expected, there is a mismatch between the prediction of variant pathogenicity and the prediction of the functional effect compared to the real functional evidence. Aiding to improve genotype–phenotype correlations, we separate variants into ‘protein reducing’ or ‘altered protein’ variants and provide general conclusions about the skin and heart phenotype involved. We conclude by stipulating that adequate prognoses can only be given, and targeted therapies can only be designed, upon full knowledge of the protein pathology through functional investigation.
1. Introduction
Pathogenic variants in genes encoding for proteins expressed in both skin and heart may cause so-called cardiocutaneous syndromes. These can be inherited in an autosomal dominant or recessive way, and can be life-threatening [1,2,3]. Cardiocutaneous syndromes usually present in childhood with the solitary or combined skin and adnexal features of skin fragility, palmoplantar keratoderma (PPK), woolly hair (WH) and alopecia [3]. When present, the skin fragility phenotype usually persists throughout life and presents as blisters, erosions and wounds with an intraepidermal level of skin separation [2,4,5,6,7]. Cardiomyopathy usually manifests later in adult life, with a high risk of death through arrhythmias or end-stage heart failure [8]. While arrhythmogenic cardiomyopathy (ACM) is the most commonly observed form, with a prevalence of 1:5000, other cardiomyopathies such as dilated (DCM; 1:250), hypertrophic (HCM; 1:500) or non-compaction (NCCM) have also been observed in patients. The underlying cause of this disease is mainly found in genes that encode for commonly shared proteins in the skin and heart, involved in the intercellular compliance network. This network consists of intracellular intermediate filaments anchored to specialized cell–cell connecting structures, called desmosomes. In 2000, the first desmosomal gene variant underlying a cardiocutaneous syndrome was identified as a homozygous DSP variant that translated to a C-terminal truncated desmoplakin protein [1]. Today, many variants have been associated with a disease phenotype, found in desmosomal genes DSP (also known as Carvajal syndrome [9,10]), JUP (also known as Naxos disease [9,11]) and DSC2 [2], the intermediate filament regulating gene KLHL24 [12], and the gap junction gene GJA1 [13] (Figure 1).
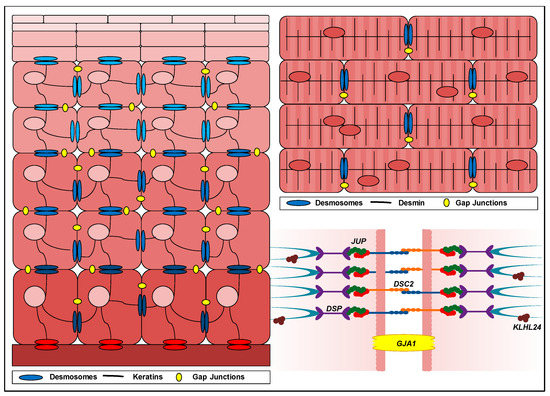
Figure 1.
Schematic overview depicting the location of proteins causative for a cardiocutaneous syndrome. The left panel depicts continuously differentiating keratinocytes in the skin, migrating from the basement membrane towards the stratum corneum, where the terminally differentiated keratinocytes are localized. Keratinocytes are interconnected by gap junctions (in yellow) and desmosomes (in blue) that anchor the keratin intermediate filament network. Hemidesmosomes (in red) connect the basal keratinocytes to the underlying dermis. The upper right panel depicts a zoomed-in fraction of the myocardial wall, where cardiomyocytes are interconnected by gap junctions and desmosomes at the intercalated disc. The lower right panel depicts a plasma membrane between two adjacent cells containing a gap junction, connexin 43 (gene GJA1, protein Cx43) and a desmosomal junction, consisting of desmoplakin (gene DSP, protein DP), plakoglobin (gene JUP, protein PG), desmocollins (subtype 2; gene DSC2, protein DC2) desmogleins (blue protein structures) and plakophilins (red protein structures). Intermediate filaments (turquoise protein structures) adhere to the desmosomal junction. Kelch-like protein 24 (gene and protein KLHL24) mostly accumulates near the plasma membrane.
While most variants in the abovementioned genes cause a dual organ phenotype, some variants are strictly confined to a skin or cardiac phenotype. This phenomenon is poorly understood. Guidelines to interpret pathogenicity of genetic variants, provided by the American College of Medical Genetics and Genomics (ACMG) [14], are currently multifaceted, meaning they take population-based frequencies, segregation analysis, prediction models and functional evidence into consideration. While population-based frequencies and segregation analysis determine disease penetrance, they do not unveil the underlying mechanism. In reality, the pathogenicity of variants is predicted by and dominated through in silico algorithms that rely on physical properties of amino acids, the conservation of residues in sequence alignments of closely related proteins or sequence and structural comparisons. However, in silico algorithms frequently contradict one another, and the actual impact on protein production and function can only be determined via functional assays. As the genotype–phenotype correlations of cardiocutaneous genes are poorly understood and therefore prognoses are difficult to establish, there is a pressing unmet need for fundamental research that determines the effect of genetic variants on the protein (function), which in turn is also paramount to the development of targeted therapies [15].
In this review, we summarized the functional evidence of 115 investigated variants in cardiocutaneous-expressed genes and compared these with their ACMG class and the prediction on protein pathology through in silico algorithms. Based on this overview and the major findings, we provide recommendations for therapeutic strategies and future directions.
2. Desmosomal Genes
Desmosomes are mirroring, transmembrane protein chains that connect the intermediate filament networks of neighbouring cells. Each chain continuously (dis)assembles due to the turnover of five desmosomal protein types: desmoplakin, plakoglobin, plakophilins, desmocollins and desmogleins (Figure 1) [16]. The expression of two genes is critical to the formation of all desmosomes: namely DSP, encoding two differently spliced desmoplakin proteins (DPI and DPII) and JUP, encoding plakoglobin (PG). Meanwhile, plakophilins, desmocollins and desmogleins are expressed in a tissue-specific manner and are therefore encoded by multiple genes. The plakophilin and desmocollin gene family each contain three subtypes (PKP1, PKP2 and PKP3, encoding proteins PP1, PP2 and PP3 and DSC1, DSC2 and DSC3, encoding proteins DC1, DC2 and DC3), while the desmoglein gene family contains four subtypes (DSG1, DSG2, DSG3 and DSG4, encoding proteins DG1, DG2, DG3 and DG4). Keratinocytes of the skin and adnexes express both DP isoforms and PG, in addition to any of the aforementioned PP1-3, DC1-3 and DG1-4 combinations. Each specific composition is in accordance with the differentiation status of keratinocytes in the epidermis [17]. Desmosomal proteins are crucial for epidermal integrity and proper epidermal proliferation and differentiation, and irregularities thereof may cause a skin phenotype [18]. Desmosomal proteins also accommodate hair growth and irregularities thereof. The hair follicle contains keratinocytes in an inner and an outer root sheath. In straight hairs, shafts are straight and homogenous, without clear delimitations, but in coiled hair, these shafts have retro-curvatures [19,20,21]. This curve is achieved via a rotation mechanism of aberrantly proliferating and differentiating cells in the inner root sheath [18,22]. Unlike the skin, the protein composition of desmosomes in cardiac tissue is fixed and consists of DPI, PG, PP2, DC2 and DG2. In the heart, desmosomal anomalies typically disrupt the mechanical continuity of cardiac muscle fibres, which is essential for proper conductance and cardiac muscle contraction. Desmosomes are crucial for the anchorage of cardiomyocytes at the intercalated disc parallel to the direction of strain, where they internally dock the desmin network [16]. Thus far, variants in desmosomal genes DSP, JUP and DSC2 have been associated with a cardiocutaneous phenotype.
2.1. Reported DSP Variants
DSP encodes for two differently spliced DP proteins: DPI (332 kDa) and a smaller DPII isoform (260 kDa) that contains a shorter rod domain [23]. The latter is created by an alternative donor splice site in exon 23. The N-terminal plakin-domain binds with PPs and PG, while domains B and C at the C-terminal side bind to intermediate filaments (Figure 2). In cardiac muscle, DSP is predominantly spliced into DPI, while the skin contains both isoforms equally. The ClinVar database reported 3290 variants in DSP. Of these, 495 were claimed (likely) pathogenic; 1026 (likely) benign; 161 show conflicting interpretations and 1608 have an unknown significance. Only 48 variants were substantiated by functional evidence, including data from transgenic mouse models (see Figure 2; full report in Table 1). This indicates that over 98% of all DSP variants were merely predicted by in silico algorithms. For the majority of variants (36/48), the predictions on protein level were correct, while only partially correct in 2/48 variants and incorrect in 5/48 variants. In 5/48 variants, the functional evidence was too elusive to draw conclusions. Moreover, the in silico prediction algorithms frequently contradicted one another, providing little help in assessing the pathogenicity of DSP variants.
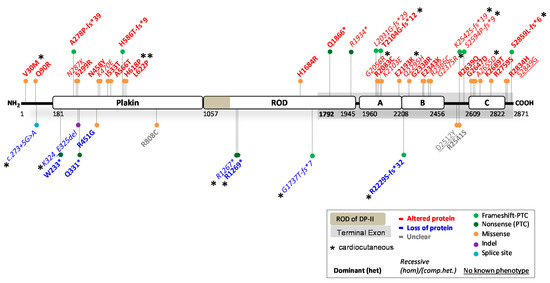
Figure 2.
Location of functionally investigated DSP variants.

Table 1.
Functionally analysed DSP variants.
2.1.1. DSP Variants Causing DP Reduction
Complete loss of DPI&II is probably incompatible with human life, as it causes early embryonic lethality in mice [55]. More importantly, it has not been functionally observed in human patients. However, several variants that cause protein reduction (<100% native DPI/DPII left) in humans and animal models have been reported [56,57,58]. In total, 9/48 variants resulted in variable degrees of DP reduction (Table 1) and inflicted disease in either a dominant (n = 5) or recessive (n = 4) mode of inheritance. This occurred due to two splice-site variants (c.273 + 5G > A and c.939 + 1G > A), four nonsense-inducing variants (c.699G > A, c.3799C > T, c.3805C > T and c.5208_5209del) located before the terminal exon, and one nonsense-inducing variant in the terminal exon (c.6687del) that was unexpectedly targeted by NMD. Moreover, one missense (c.1348C > G) and one in-frame indel variant (c.969_974del) resulted in DP protein reduction, probably due to instable protein degradation. All of the aforementioned variants affected both isoforms, except for variants c.3799C > T, c.3805C > T and c.5208_5209del. The latter are located in the ROD domain of DPI and therefore only affect DPI, but not DPII. Interpreting the phenotype of patients, DP deficiency (≤50% native DPI) seems to be associated with severe cardiomyopathy, while DP deficiency in the skin (≤50% native DPI and DPII) is mostly associated with PPK and WH. Recessive variants that caused loss of DPI but not DPII, or extreme deficient levels of both DPI and DPII (<20%), were associated with skin fragility [41].
2.1.2. DSP Variants Causing an Altered DP Protein
The majority of functionally investigated DSP variants (36/48) led to an altered DP protein (Table 1), due to 23 dominantly and 13 recessively inherited variants. Missense variants were the predominate source for altered DP proteins (n = 27), while the remaining nine variants were due to nonsense or nonsense-inducing variants. Out of the 36 variants, two variants were located near the N-terminus, ten were located in the plakin domain, three in the ROD domain of DPI, six in domain A, five in domain B, two in the linkers, five in domain C and three near the C-terminus. All but one (c.6687del) of the nonsense-inducing variants in the terminal exon of DSP skipped NMD. As expected, the phenotype of patients with an altered DP protein indicated that a recessive mode of inheritance was more severe than a dominant mode of inheritance, mostly due to absence of native protein in the former. Cardiomyopathy was observed in 31/36 of the variant carriers, while it went unobserved or unreported in the others. In the skin, 11/36 variants resulted in PPK (recessive n = 9, dominant n = 2) often with WH (recessive n = 7, dominant n = 2). However, PPK and WH were frequently not observed or not reported in the studies primarily focused on the cardiac phenotype. Furthermore, 10/36 variants caused skin fragility, mostly due to a recessive inheritance (n = 7, dominant n = 3). Variants located in the plakin domain frequently affected the binding efficiency to PG [24,25,26], PPs [59] or intermediate filament anchorage [31,60]. Variants located in domains A, B or C almost always affected the binding affinity to intermediate filaments, especially when causing severe property alterations in domains B and C or loss of these through C-terminal truncation. The functional evidence of the remaining 3/48 variants was inconclusive as to whether it resulted in protein reduction or an altered DP protein.
2.1.3. Potential Therapeutic Avenues
Given the contradicting results from in silico algorithms and several inconsistencies between the prediction and functional evidence, functional assays of the remaining DSP variants would be strongly encouraged. The nine variants causing DP reduction indicated that the disease severity tends to be dose-dependent in nature, both in the heart as well as in the skin. Hence, strategies to increase native DP protein levels, especially in the heart, would be of benefit to patients. Injections with DSP mRNA in DP-deficient zebrafish have been promising in regaining cardiac function [61]. Besides strategies like RNAi or CRISPR that eliminate protein expression from mutant alleles in patients with altered DP proteins, strategies should simultaneously aim to increase native DP protein levels. For most of the functionally investigated variants, it remains unclear whether they cause a dual organ phenotype, as it is not often assessed or specified.
2.2. Reported JUP Variants
The JUP gene encodes for the 82 kDa PG protein, also known as ɣ-catenin. PG contains an N-terminal head-domain, 12 armadillo domains and a C-terminal tail-domain (Figure 3). PG belongs to the catenin protein family and is highly homologous to β-catenin, a potent transcription factor of the canonical Wnt/β-catenin signalling pathway. PG is an important desmosomal protein, comprising the outer dense plaque of the desmosome and connecting the transmembrane DG and DC proteins to DP and PP. β-catenin and PG can be substituted for one another, as β-catenin can be incorporated into desmosomes, while PG can also act as a nuclear transcription factor [62]. The ClinVar database has reported 838 variants in JUP. Of these, 30 are claimed (likely) pathogenic; 307 (likely) benign; 70 show conflicting interpretations, and 431 have an unknown significance. Merely eight variants where substantiated by functional evidence, including data from transgenic mouse and zebrafish models (see Figure 3 and full report in Table 2). As for DSP variants, effects of over 98% of all JUP variants were merely predicted by algorithms. The predictions on protein level were correct in 4/8 variants, incorrect in 3/8 variants, while the functional data remained inconclusive in 1/8 variants.
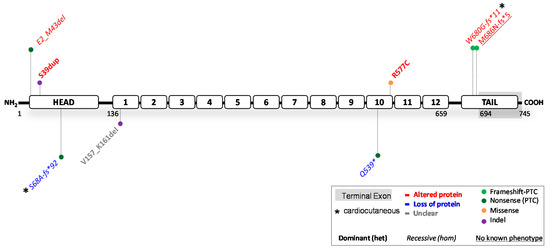
Figure 3.
Location of functionally investigated JUP variants.
Table 2.
Experimental investigation of JUP variants.
2.2.1. JUP Variants Causing PG Reduction
Complete loss of PG induces lethality within embryogenesis in mice due to severe heart defects or immediately post-natal due to severe skin fragility [74,75]. Highly suppressed PG protein levels (<10%) also lead to ACM [76], while 40% protein levels do not induce cardiac dysfunction in mice [77]. This suggests that threshold levels for cardiomyopathy in mice span between 10–40% of native protein. Two human variants (c.201del and c.1615C > T, nonsense) were reported to induce complete PG depletion in homozygous patients [66,68]. These carriers developed severe and sometimes lethal skin fragility, in combination with PPK and alopecia in homozygous c.201del carriers. Cardiomyopathy was not observed in any but one patient with old age [66]. Meanwhile, no other patients with PG reduction (≤50% protein) have been reported. The above results suggest that (near) PG depletion is strongly correlated to skin fragility. However, the dose effect of PG reduction on the development of skin features is unknown and warrants further functional studies. In contrast, while PG depletion induces cardiac lethality during embryogenesis in mice, the limited data currently suggest that the human heart may be protected, perhaps due to functional compensation of β-catenin [62,71]. To accurately assess the cardiac penetrance in patients, more variants predicted to cause reduced or depleted PG levels need to be investigated.
2.2.2. JUP Variants Causing an Altered PG Protein
Five functionally investigated variants (5/8) resulted in an altered PG protein (Table 2). These resulted from three recessively inherited nonsense-inducing variants, resulting in either a N-terminal (Glu2_Met43del) or C-terminal (Trp680Glyfs*11 and Met686Asnfs*5) truncated protein. In addition, two dominantly inherited variants, Ser39dup and Arg577Cys, caused ACM and resulted in proteins comparable to the size of native PG. Unlike the C-terminal truncations, the recessive N-terminal truncation induced skin fragility with PPK and WH, but no cardiac dysfunction. The variant causative for this N-terminal truncation c.71C > A, introduces a PTC at Ser24, but translation re-initiation took place at position Met43, which resulted in deletion of the first 42 amino acids. This suggests that any nonsense-inducing variant located between JUP:c.1_126 will likely cause translation re-initiation and a similar phenotype and effect on protein. Opposingly, the two recessive C-terminal truncations correlated with ACM, PPK and WH, but did not induce skin fragility in patients. Moreover, two homozygous PG mouse knockin Trp680Glyfs*11 models were developed, one with and one without fusion of the final five exons. In mice without fusion of exons, this variant resulted in NMD, and only very low levels of C-terminal truncated protein were expressed. These mice died on postnatal day one due to severe skin fragility, induced by depleted PG. Due to their short lifespan, the effect on cardiac function in later stages of development is unclear. In mice with fusion of exons, high levels of C-terminal truncated protein were observed, similarly as in patients. Nonetheless, even at 11 months of age, mice failed to develop cardiac dysfunction [71]. These data suggest that there may be little resemblance between the cardiac function of humans and mice with regard to JUP variants. More variants need to be functionally investigated to draw definitive conclusions.
2.2.3. Potential Therapeutic Avenues
Currently, too little functional evidence is available to adequately address potential therapeutic strategies that would benefit the cardiac function of patients with disease-causing JUP variants. Meanwhile, strategies to increase native protein in patients with skin fragility seem appropriate for all patients with PG depletion and patients with PG proteins that lack the N-terminus. Whether a skin phenotype is only observed in the case of biallelic JUP variants, as the eight functionally investigated variants now suggest, needs additional functional evidence. Furthermore, almost half of the functionally investigated variants were falsely predicted, which further pressed the need for more functional studies.
2.3. Reported DSC2 Variants
The DSC2 gene encodes for two transmembrane cadherin isoforms: DC2a (99 kDa) and DC2b (93 kDa). The DC2a isoform contains the complete intracellular segment (ICS), whereas this domain is 53 amino acids shorter in DC2b [78], due to alternative splicing of exon 16 (Figure 4). Both isoforms are first processed into a precursor protein, followed by a mature protein that can be incorporated into desmosomes. Maturely processed DC2 serves as a transmembrane desmosomal protein, important for extracellular cell–cell attachment. The ClinVar database has reported 1209 variants in DSC2. Of these, 102 were claimed (likely) pathogenic; 409 (likely) benign; 85 show conflicting interpretations and 613 have an unknown significance. Notably, merely 15 variants were substantiated by functional evidence, including data from transgenic mouse and zebrafish models (see Figure 4 and full report in Table 3). The same trend seen for DSP and JUP variants, is also observed for DSC2, indicating that over 98% of all variants have not been functionally investigated. The predictions on protein level were correct in 11/15 variants, while incorrect in 3/15 variants and unclear in 1/15 variants.
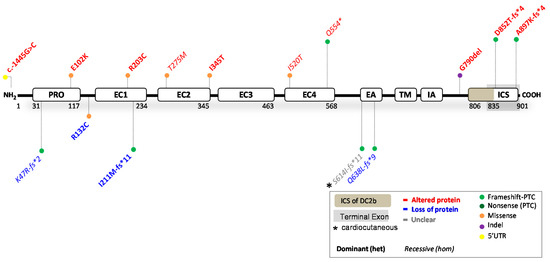
Figure 4.
Location of functionally investigated DSC2 variants.
Table 3.
Functionally analysed DSC2 variants.
2.3.1. DSC2 Variants Causing DC2 Reduction
No patients with complete absence of DC2 protein have been reported. Instead, four variants (4/15), located before the terminal exon, resulted in both DC2a and DC2b protein reduction. The recessively (c.1913_1916delAGAA; ≤10% protein left [87]) and dominantly (c.631-2A > G; 40% protein left) inherited nonsense-inducing variants both caused ACM in patients. Compound heterozygosity of out-of-frame indel variant c.140_147delAACTTGT resulted in NCCM and hypertrophy, which is the only functionally investigated DSC2 variant associated with cardiomyopathy other than ACM [80]. The missense variant c.394C > T caused 50% protein reduction via instable protein degradation and caused ACM in a dominant mode of inheritance. Altered electrical properties, a key characteristic of ACM, have been observed in patient hiPSC-CMs containing this missense variant [82]. Moreover, dominantly inherited variants c.394C > T and c.631-2A > G were also investigated in a zebrafish model. The ACM phenotype in both models was rescued by injecting human wildtype but not mutant DSC2 mRNA [82,85]. One of these studies additionally showed that gradual knockdown of DSC2 resulted in dose-dependent cardiac disease severity [85]. This seems to corroborate with the human data, suggesting that cardiomyopathy occurs in situations with ≤50% of native DC2 protein: and the higher the reduction, the more severe the phenotype. Furthermore, in mice, neither complete nor heart-specific knockout of DSC2 resulted in any altered viability or cardiac phenotype [92], which emphasizes differences in disease susceptibility among species. None of the DC2 protein-reducing variants caused a skin phenotype, indicating that near loss of the DC2 protein is well tolerated by the skin. Based on the few investigated variants and contradicting results of animal models, incisive conclusions are still difficult to draw.
2.3.2. DSC2 Variants Causing an Altered DC2 Protein
Ten variants (10/15) resulted in an altered DC2 protein (Table 3), which predominantly caused ACM via a dominant (n = 7) or recessive (n = 3) mode of inheritance. Heterozygous variant c.-1445G > C in the 5′UTR affected transcription factor binding mechanisms. Meanwhile, five variants had pronounced effects on the processing of DC2 precursor proteins. For instance, artificial transfection experiments containing missense variants Glu102Lys, Arg203Cys, Thr275Met and Ile345Thr showed punctate cytoplasmic staining, with no or partial ability to be incorporated into desmosomes [84,86,90]. Moreover, nonsense variant Gln554* escaped NMD and resulted in a C-terminal truncated protein, affecting both isoforms [86]. This variant also affected the processing of DC2 precursor proteins, and while a small proportion of maturely processed proteins was incorporated into the desmosome, a larger proportion of precursor proteins remained in the cytoplasm. It is still uncertain whether missense variant Ile520Thr will induce similar alterations that affect the processing of DC2 precursor proteins [80]. Opposingly, nonsense-inducing variants, Asp852Thrfs*4 and Ala897Lysfs*4, only affected isoform DC2a and caused a C-terminal truncated protein. Both were fully processed into a mature protein form, were incorporated into desmosomes, but lost their ability to bind to DP [84] and PG [84,90]. The latter suggests a similar perturbing binding interface for Gln554*. No protein processing information is available on variant Gly790del, other than that it is expressed and translated into a transgenic mouse model. Neither the heterozygous nor homozygous mice showed structural or functional defects in the ventricles or lethal arrhythmias, and only homozygous aged mice showed slight left ventricular dysfunction. This mouse model therefore does not represent the phenotypic severity of the heterozygous Gly790del patients with ACM. In most (7/10) variants, apart from recessive variants Gln554*, Thr275Met and Ile520Thr, ACM was observed in a dominant mode of inheritance. Only recessive inheritance of variant Ser614Ilefs*11 caused PPK and WH, but the functional data were unclear as to whether it causes protein reduction or an altered protein function in patients [87,88]. A skin phenotype was furthermore not observed or went unreported in the other variants.
2.3.3. Potential Therapeutic Avenues
Whether DSC2 variants can truly cause a cardiocutaneous phenotype remains somewhat elusive, given that only one investigated variant was associated with PPK and WH, and others that do associate with a skin phenotype were not investigated [88]. It seems that extreme deficiency in DC2 is well-compensated for by other desmocollins in the skin (i.e., DC1 and DC3). Nonetheless, more variants should be investigated to draw decisive conclusions. Meanwhile, with the limited functional evidence in mind, patients with DC2 reduction might benefit from native protein-increasing therapeutic strategies. Taken into account, over-administration may be detrimental to humans, as DSC2 overexpression caused severe cardiac dysfunction in mice [93].
3. Proteins Involved in Intermediate Filament Regulation
The intermediate filament network is part of the cell scaffold that contains multiple keratins (KRT1, KRT10, KRT5, KRT14 etc.) in keratinocytes and desmin (DES) in cardiomyocytes [94] (Figure 1). Genetic variants in genes encoding intermediate filaments may severely harm the integrity of a tissue, as it affects resistance to cellular stretch [95]. DES variants may cause striated muscle disease, and 70% of all reported DES variants have been implicated in cardiac disease [96,97]. Meanwhile, in the epidermis, different keratins are expressed solely in a differentiation-dependent manner. Basal keratinocytes mostly contain keratin 5 (KRT5) and keratin 14 (KRT14), and variants in both have been associated with EBS [4,5]. In addition, other gene variants that relate to basal keratin turnover, have been implicated in this disease [4]. Since desmin is restricted to striated muscle and keratins to epithelial cells, variants located within these genes do not cause a cardiocutaneous phenotype. However, recent findings have shown that the skin and heart have ubiquitin ligase KLHL24 as an analogous regulator of intermediate filaments in common [12].
3.1. Reported KLHL24 Variants
The gene KLHL24 encodes the 68 kDa kelch-like protein 24, referred to as KLHL24 [98]. KLHL24 is part of the kelch-like protein family, which contains 42 identified KLHLs. KLHLs play a key role in ubiquitinating many different substrates, which is mediated by the activity of E3-ligases [99]. From the N- to the C-terminal side, KLHL24 contains a BTB-domain, BACK-domain and six kelch-repeats (Figure 5). KLHL24, specifically, was implicated in the turnover of keratins via ubiquitination and subsequent proteasomal degradation [100]. The ClinVar reported 78 variants in KLHL24. Of these, 37 are claimed (likely) pathogenic; 34 (likely) benign and seven as variants of unknown significance. Eight variants with functional evidence were found (see Figure 5 and full report in Table 4). Furthermore, at least one variant has been translated into a transgenic mouse model, and a zebrafish knockout model has been developed. The predictions on protein level were probably correct in 1/8 variants and incorrect in 6/8 variants, while the evidence of one variant is somewhat inconclusive as to whether protein is produced or not.
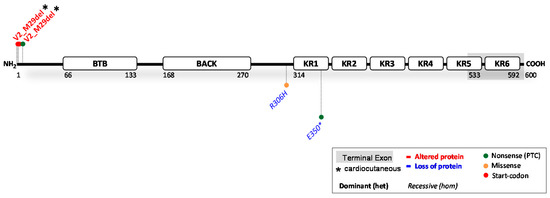
Figure 5.
Location of functionally investigated KLHL24 variants.
Table 4.
Functionally analysed KLHL24 variants.
3.1.1. KLHL24 Variants Causing KLHL24 Reduction
Two recessive variants (Table 4) that cause complete loss of KLHL24 function have been reported in patients. Both the homozygous missense (c.917G > A) and nonsense (c.1048G > T) variant resulted in HCM in patients, fitting with the higher desmin levels found in their myocardial biopsies [107]. Moreover, heterozygous parents did not display signs of cardiac disease. Furthermore, no skin phenotype was observed in heterozygous or homozygous state. Absence of KLHL24 was also linked to lethal arrhythmias in zebrafish [107]. Currently, no other investigated variants associated KLHL24 deficiency (≤50% protein) with disease, suggesting that pathological threshold levels of KLHL24 in the heart may lie somewhere between 0–50% of wildtype expression. In the skin, reduction in KLHL24 apparently does not cause a skin (fragility) phenotype [107].
3.1.2. KLHL24 Variants Causing an Altered KLHL24 Protein
Five heterozygous start-codon variants (Table 4) c.1A > T, c.1A > G, c.2T > C, c.3G > A and c.3G > T, and one predicted nonsense variant c.22A > T were associated with congenital aplasia cutis, skin fragility, PPK, alopecia and DCM [12,98,100,101,105,107,108,109]. Disregarding the inaccurate predictions, all six variants were pathogenic and resulted in translation initiation or re-initiation at Met29. Overexpression studies determined that these six variants lead to an N-terminally truncated protein Val2_Met29del, 28 amino acids shorter than the wildtype counterpart [98,100,105]. This truncated protein is less affected by auto-ubiquitination, which results in a longer half-life and disproportionate degradation of keratin 14 [98] in the skin and desmin in the heart [101,105]. In dynamically loaded engineered heart tissues derived from hiPSC from patients (c.1A > G), 10-fold lower levels of desmin were observed which caused all the clinical characteristics of DCM. This result was in line with a reduced expression and phenotype of the explanted heart of one of the patients [101]. Meanwhile, another study reported that KLHL24 is also responsible for the turnover of foetal-like keratins 7,8,17 and 18 [103], while degradation of keratin 14 may be higher in foetal-like hiPSC-derived keratinocytes compared to adult keratinocytes [102]. These data would explain why patients develop congenital aplasia cutis but only mild skin fragility later in life. Furthermore, another study investigating the occurrence of alopecia showed that KLHL24 in skin regulates hair maintenance by mediating the stability of keratin 15. The presence of mutated protein Val2_Met29del disrupted the structure of hair stem follicles, leading to alopecia in mice [104].
3.1.3. Potential Therapeutic Avenues
The abovementioned studies have indicated keratins and desmin as natural targets for KLHL24. This seems to be a tight balance as the gain-of-function KLHL24 variants cause excessive breakdown of desmin and keratins. These results furthermore indicate that patients displaying variants that result in expression of Val2_Met29del would likely benefit from KLHL24 RNAi therapies, as this has effectively regained keratin and desmin protein levels in in vitro models [98,100,101,102]. While unlikely to affect the skin, excessive RNAi can be detrimental to cardiac function, as absence of KLHL24 has been associated with HCM. Opposingly, patients lacking KLHL24 could benefit from opposing strategies that attain its expression in the heart, minding that the opposite effect (i.e.,: DCM) can be achieved by administrating too high dosages. Strategies to mitigate all patient’s symptoms thus must be precisely executed. Regarding the expression of KLHL24 in tissues: although mRNA expression of KLHL24 is high in both the skin and heart, protein levels have nearly been impossible to detect in patient-derived materials. This complicates the analysis of novel variants using Western blotting techniques to assess protein pathology. Functional interpretation must therefore shift towards artificial cell models or protein target quantification (i.e., keratins and desmin). Of the functionally assessed KLHL24 variants, it is clear that the effect of the variant on the protein is difficult, if not impossible, to predict in silico in the case of KLHL24.
4. Gap Junction Genes
Gap junctions are channels at cell–cell junctions that function for intercellular communication and exchange of molecules (Figure 1). They are formed by connexins (Cx, GJA), a family of at least 20 different transmembrane proteins that can assemble into hemichannels, also called connexomes, clusters of six proteins forming a pore between two adjacent cells [110]. Different kinds of connexins are expressed in the skin and heart [111,112,113]. Gap junctions in the heart, specifically in cardiomyocytes, are located within the intercalated disc, in close proximity to desmosomes. Moreover, connexomes have been found to be in direct communication with desmosomes and are often defective in function and/or structure in a cardiocutaneous syndrome and other cardiac pathologies [37,114,115]. Due to the versatility of the transport function of connexins, variants in these proteins can cause a broad range of distinct diseases. Connexin 43 (Cx43), encoded by GJA1, is a connexin expressed in both the skin and heart, and variants in GJA1 are known to cause a broad range of phenotypes, including PPK, alopecia and cardiac conductive disorders thus capable of causing a cardiocutaneous phenotype.
4.1. Reported GJA1 Variants
The gene GJA1 encodes a 43 kDa connexin (Cx43) protein. The hemichannel pore between adjacent cells is formed by six connexins that form a hexamere. Each individual Cx43 contains four transmembrane domains (TM1-4) linked by a cytoplasmic and two extracellular loops and their N-terminal and C-terminal fragment within the cytoplasm [116,117] (Figure 6). The ClinVar database reported 217 variants in GJA1. Of these, 58 were claimed (likely) pathogenic; 45 (likely) benign; 16 show conflicting interpretations and 98 have an unknown significance. We found 36 variants substantiated by experiments containing functional evidence (see Figure 6 and full report in Table 5). Furthermore, at least five variants have been translated into transgenic mouse models. Still, 83% of the reported GJA1 variants have not been functionally investigated. Since GJA1 has a single coding exon and can therefore not be spliced, the rate of prediction success is high, and the predictions on protein level were correct in 36/36 of the functionally investigated variants.
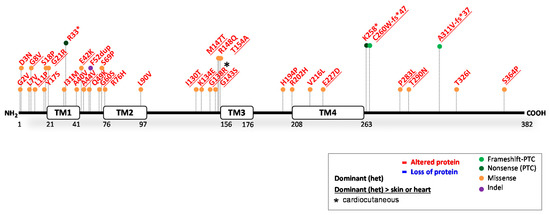
Figure 6.
Location of functionally investigated GJA1 variants.
Table 5.
Functionally analysed GJA1 variants.
4.1.1. GJA1 Variants Causing Cx43 Reduction
In humans, variants causing partial or complete Cx43 depletion have not been functionally investigated. Of note, during development, connexins, derived from other genes than GJA1, are important. Therefore, full- and heart-conditional GJA1 knockout mice die shortly after birth due to major heart defects. Due to the short lifespan of these mice, the effects of total loss of Cx43 in the skin are unclear, especially in later stages of development [162]. These data nonetheless indicate that human variants leading to complete Cx43 depletion are likely not compatible with life.
4.1.2. GJA1 Variants Causing an Altered Cx43 Protein
All 36 functionally investigated variants caused an altered, less functional gap junction, predominately due to heterozygous missense variants (31/36). In addition, four nonsense/nonsense-inducing variants and one in-frame indel variant were investigated. Over 90% of these 36 variants affected the eyes, teeth and fingers, diagnosed as the condition oculodentodigital dysplasia. Almost all of these variants were investigated solely in that context, but they can also cause keratoderma, hair abnormalities (alopecia) and cardiac problems, although they are not always specified/reported. While most in silico algorithms accurately assessed the deleterious effect of variants, contradictory predictions still occurred. Meanwhile, in at least 14 out of the 36 investigated variants, researchers clearly demonstrated the interference of mutants with normal gap junction formation, where both mutant and wildtype Cx43 integrated into hemichannels. In the skin, Cx43 plays a key role in wound healing and intercellular communication, the latter being critical for cell differentiation, proliferation, migration and ion transport, and most variants caused serious harm to these processes [13,119,141,154]. The cardiac effects of GJA1 variants in humans have not been extensively researched. However, they have been associated with arrhythmic sudden death [130,131,163,164], DCM [164] and congenital heart malformations [116,125,149,165].
4.1.3. Potential Therapeutic Avenues
Dominant-negative missense variants, causing detrimental amino acid changes in Cx43, have profound physiological consequences, regardless of the location within the protein. In almost half of the investigated variants, the mutant protein clearly disrupted the formation of normal gap junctions. It is likely that the others caused a similar protein pathology as well. Meanwhile, the pathogenicity prediction of GJA1 variants through in silico algorithms were closer to accuracy than the other cardiocutaneous genes. Cx43, namely, highly resembles 20 other human connexins, and most amino acids in Cx43 are also highly conserved among species, which makes GJA1 an ideal candidate gene for in silico predictions. Unfortunately, almost all variants with functional evidence have been studied mainly in relation to oculodentodigital dysplasia [117]. Due to the versatility of Cx43 function, further studies should widen the scope in order to establish the full role of this protein in skin and heart disease. Nonetheless, strategies to ease the overall phenotype of patients should be directed towards conserving the functional hexameric complex structure of hemichannels at all costs. Patients may therefore benefit from treatments that focus on eliminating mutant proteins (i.e., RNAi, CRISPR).
5. Discussion
A substantial number of genetic variants in the genes DSP, JUP, DSC2, KLHL24 and GJA1 have been reported to underly skin and/or cardiac disease. In this review, we show that only few of them have been functionally assessed, and when they have, the functional data show that the effect of the variant on the protein and its function are quite often not as predicted by in silico prediction programs. The conclusions that can be drawn from functional evidence are important in light of genotype–phenotype correlations (and prognosis) and of potential targeted therapies. The data of this review show that there is a need for functional studies of variants, instead of relying on in silico prediction. In addition, although data on the protein pathology of variants has begun to shape the understanding of disease, there is still a long way to go. Several state-of-the-art in vitro approaches are now available that can significantly enhance insight into the genotype–phenotype correlation and the functional effects of the variants involved in cardiocutaneous syndromes in order to understand and subsequently develop therapies for the resulting phenotypes.
The correct interpretation of variants is an important step towards understanding cardiocutaneous disease mechanisms: a factor that will provide better recommendations for variant carriers and ultimately therapeutic interventions for the patients that are at risk. However, the overview presented here points out that the functional effect of most variants still needs to be established. Moreover, predictions frequently contradicted the functional evidence, exemplified by nonsense variants that did not necessarily reflect absence of protein, NMD that was not always acting according to current assumptions, and similarly altered proteins with different deleterious effect on tissue homeostasis.
In the meantime, the interpretation of variants can be strengthened by applying a few minor additions/alterations to guidelines that assess functional studies. The ACMG guidelines, published in 2015 [14] and following recommendations from the Sequence Variant Interpretation (SVI) working group [166], classify functional studies into different categories of evidence from low to high such as: ‘supporting’, ‘moderate’ or ‘strong’. In this classification, functional assays derived from one variant carrier (patient) compared to one control individual easily fall into the category ‘supporting’. Likewise, experiments that use artificial cell models and variant transfection methods can reach a similar status. Meanwhile, in order to reach a ‘moderate’ line of evidence, studies need to include data of more than ten control individuals, in addition to data from a carrier with a known benign and known pathogenic variant. Using these criteria, the functional evidence of variants included in this overview were given a ‘supporting’ status at maximum, while they provide a wealth of information. Using current guidelines, the functional evidence of studies could not be further differentiated, even though clear distinctions can be made. The following additions/alterations to the guidelines can provide aid. For instance, isogenic controls or variant-targeting interventions that rescue the cellular phenotype are not yet regarded as a criterion in the guidelines, but they do provide significant strength to the line of evidence. These experiments remove genetic background in a more efficient way than inclusion of multiple control individuals. In addition, direct patient or patient-derived material provides stronger lines of evidence than artificial cell models, as the latter do not necessarily reflect the in vivo regulation of gene expression, mRNA splicing and protein quality-control mechanisms [25]. Finally, as the effect of genetic variants may differ per tissue, studies that include multiple cell types provide better insight into the patient as a whole and therefore also provide a stronger line of evidence.
This overview of functional evidence of desmosomal gene variants also points out that variants may exert their effect by either protein reduction or by altered protein function, and that the understanding hereof is eventually critical to the development of therapeutic interventions. For instance, KLHL24 protein activity, whether through loss or gain of function, causes a strong dose-dependent phenotype in the heart, which seems directly related to expression of its target desmin [101,107]. The relationship to keratins in the skin is less straightforward and requires more cohesive data that may now be emerging with new studies [102,103,104]. Functional studies combined also suggest that the phenotype associated with DP reduction is both dose- and organ-dependent in nature, which is important knowledge for future interventions. DC2 deficiency, however, may mostly be related to dose-dependent heart disease, yet sufficient data are lacking. Meanwhile, the phenotype associated with PG reduction is highly organ-dependent and strongly correlated with skin, but not heart disease. More supporting studies are needed to strengthen these findings. In contrast, proteins with an altered function were mostly shown to be erratic and unpredictable in nature. Variants in GJA1 showed most coherence, as expression of each altered yet stable protein will ultimately interfere with the formation of normal gap junctions. Furthermore, variants that alter the function of DP, PG and DC2, were more diverse in phenotypic results. Some general conclusions were drawn, but most require substantially more supporting evidence. A weighted summary of the functional evidence of variants to current clinical variant databases could aid in gaining imminent clarity. More importantly, supporting studies on the protein pathology of the remaining >95% of variants that have not been functionally evaluated will unravel genotype–phenotype correlations in the near future.
With emerging novel techniques and improvements in in vitro models, much more is to be gained from functional studies. Three-dimensional skin models can now be implied, which should significantly reduce the discrepancy in results observed from ex vivo skin biopsies and biopsy-derived monolayer cultures of keratinocytes. In addition, differentiation protocols of patient hiPSC-derived keratinocytes are emerging [167], which even allow for the formation of complete hair-bearing skin [168]. With these techniques, the whole epidermis can be engineered and mechanical abrasions that provoke disease can be mimicked in vitro. In addition, significant advances have been made to study heart disease. Differentiation protocols of patient hiPSC-derived cardiomyocytes are already highly advanced and broadly applied. In addition, engineered heart tissues can now be made [169] and stimulated to allow for highly aligned cardiac fibres [170]. They can also undergo pre-and afterload in a dynamic fashion, thereby increasing the fractional shortening up to 20–30%, like in the in vivo human heart [29]. In conclusion, more emphasis on functional evaluation in variant prediction guidelines and establishment of functional protein pathologies resulting from genetic variants and state-of-the art techniques to do so can significantly enhance insight into the pathophysiology of cardiocutaneous diseases, which will be paramount to the development of therapies.
6. Materials and Methods
In August 2022, the ClinVar database was used to assess the current number of reported variants [171]. From 1995 to August 2022, PubMed entries on genes DSP (DSP; DP; desmoplakin {P15924}), JUP (JUP; plakoglobin; ɣ-catenin; PG {P14923}), DSC2 (DSC2; desmocollin-2 {Q02487}), GJA1 (GJA1; Cx43; Connexin-43; gap junction protein alpha 1 {P17302}) and KLHL24 (KLHL24; Kelch-like family member 24; DRE1; KRIP6) were evaluated. PRISMA guidelines were adhered to, and the results contained the following inclusion criteria: PubMed articles containing disease variants that published quantitative immuno-blot and/or mRNA levels, obtained from patient-derived cell/tissue sources and/or transfection studies, were included. In addition, transgenic, homozygous or heterozygous animal knockout models were included, if they reflected the pathophysiology of reported human variant(s). We separated the functionally investigated variants into a ‘protein reducing’ or ‘altered protein function’ category. In the case of variants that cause protein reduction: in the heterozygous state, protein expression from one allele is lost, resulting in an expected 50% reduction of total protein. Meanwhile, in the homozygous state, this reduction is expected to be 100%. As these numbers may vary, the amount of protein reduction is summarized for each variant and potential carrier types. In the case of variants that result in a protein with an altered function: these are regarded as expressed within the cell, but they may or may not give a normal dosage. In the heterozygous state, an expected 50% of the total protein is native, while the other 50% of total protein is mutated. Homozygous carriers, however, have no native protein expression. As these numbers may vary, the size and abundancy of the expressed mutant are summarized for each variant and potential carrier type. Moreover, all the included variants were reclassified by us, according to ACMG-AMP guidelines [14] and following recommendations [166,172,173,174]. In addition, we also reported the predicted variant pathogenicity through use of in silico algorithms and the predicted variant outcome according to the following general considerations: Missense variants, which generate protein variants with a single amino acid variation, may induce drastic structural alterations or may induce structural alterations able to perturb binding interfaces. Missense variants normally do not affect protein production processes, but proteins with single amino acid variation may be degraded if severely unstable. The pathogenicity of missense variants was predicted by us using SIFT [175], PolyPhen-2 [176] and MutPred2 [177]. In-frame insertions and deletions (indel) cause an addition and/or deletion of certain amino acids in the protein. Like missense variants, these variants normally do not cause absence of protein, but proteins with in-frame indels may be degraded if severely unstable. The pathogenicity of in-frame indels was predicted by us using MutPred-Indel [178]. Nonsense variants (i.e., stop-gain variants, out-of-frame indels leading to premature downstream stop-gain codons) introduce a premature termination codon (PTC). On occasion, nonsense variants in the first exon may cause an N-terminal truncated protein, due to translation re-initiation (if another downstream start-codon is activated). Meanwhile, nonsense variants in the terminal exon normally result in a C-terminal truncated protein. Both types of truncations are expected to be expressed in the cell, but these proteins may be degraded if severely unstable. Gene transcripts with nonsense variants located between exon 1 and the terminal exon are predicted to be broken down by nonsense-mediated decay (NMD). In mammals, this system is predicted to operate when at least one intron downstream of the premature termination codon is present [179]. It is thus predicted that the truncated protein is not expressed in the cell. The pathogenicity of nonsense variants was predicted by us using MutPred-LOF software [180]. Finally, splice-site variants can affect the splicing of introns and exons, and outcomes such as (partial) intron retention or exon skipping may occur. The Human Splicing Finder and MaxEntScan software were used by us to predict the alternative splicing odds. Furthermore, there are no prediction algorithms available for either 5′UTR or 3′URT variants. Finally, the functional evidence of each variant was compared to the in silico predictions, and this was categorized as a ‘match’, if the predictions were on par with the functional evidence observed in patient-derived cells/tissues and or transfection/animal studies; ‘probably match’ if the functional predictions were on par with the functional evidence but decisive proof of the latter was incomplete; ‘mismatch’ if the functional predictions were not on par with the functional evidence; and ‘unclear’ if the functional evidence itself was inconclusive or contradictory.
Author Contributions
Conceptualization, M.C.S.C.V., M.C.B. and P.v.d.M.; Methodology, M.C.S.C.V., D.A., L.M., J.P.v.T., H.H.W.S., M.P.v.d.B., P.v.d.M. and M.C.B.; Data Curation, M.C.S.C.V., D.A. and L.M.; Formal Analysis, M.C.S.C.V., D.A. and L.M.; Validation, L.M.; Writing—Original Draft Preparation, M.C.S.C.V.; Writing—Review and Editing, M.C.S.C.V., D.A., L.M., J.P.v.T., H.H.W.S., M.P.v.d.B.; P.v.d.M. and M.C.B.; Visualization, M.C.S.C.V.; Supervision, M.C.B., P.v.d.M., J.P.v.T., M.P.v.d.B. and H.H.W.S.; Funding Acquisition, M.C.B. and P.v.d.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Vlinderkind (grant number: none, due to patient organization funding to M.C.B.], the Human Frontier Science Program (grant number RGY 0071/2014 to P.v.d.M.) and the European Research Counsel (STOP-HF (StG); grant number 715732, ERC-2016-STG to P.v.d.M.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Norgett, E.E. Recessive Mutation in Desmoplakin Disrupts Desmoplakin-Intermediate Filament Interactions and Causes Dilated Cardiomyopathy, Woolly Hair and Keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [CrossRef]
- Bolling, M.C.; Jonkman, M.F. Skin and Heart: Une Liaison Dangereuse. Exp. Dermatol. 2009, 18, 658–668. [Google Scholar] [CrossRef]
- Karmouch, J.; Zhou, Q.Q.; Miyake, C.Y.; Lombardi, R.; Kretzschmar, K.; Bannier-Hélaouët, M.; Clevers, H.; Wehrens, X.H.T.; Willerson, J.T.; Marian, A.J. Distinct Cellular Basis for Early Cardiac Arrhythmias, the Cardinal Manifestation of Arrhythmogenic Cardiomyopathy, and the Skin Phenotype of Cardiocutaneous Syndromes. Circ. Res. 2017, 121, 1346–1359. [Google Scholar] [CrossRef]
- Coulombe, P.A.; Kerns, M.L.; Fuchs, E. Epidermolysis Bullosa Simplex: A Paradigm for Disorders of Tissue Fragility. J. Clin. Investig. 2009, 119, 1784–1793. [Google Scholar] [CrossRef]
- Has, C.; Liu, L.; Bolling, M.C.; Charlesworth, A.V.; El Hachem, M.; Escámez, M.J.; Fuentes, I.; Büchel, S.; Hiremagalore, R.; Pohla-Gubo, G.; et al. Clinical Practice Guidelines for Laboratory Diagnosis of Epidermolysis Bullosa. Br. J. Dermatol. 2020, 182, 574–592. [Google Scholar] [CrossRef]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.-D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus Reclassification of Inherited Epidermolysis Bullosa and Other Disorders with Skin Fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef]
- Fine, J.-D.; Bruckner-Tuderman, L.; Eady, R.A.J.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited Epidermolysis Bullosa: Updated Recommendations on Diagnosis and Classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef]
- Asimaki, A.; Kleber, A.G.; Saffitz, J.E. Pathogenesis of Arrhythmogenic Cardiomyopathy. Can. J. Cardiol. 2015, 31, 1313–1324. [Google Scholar] [CrossRef]
- Protonotarios, N.; Tsatsopoulou, A. Naxos Disease and Carvajal Syndrome: Cardiocutaneous Disorders that Highlight the Pathogenesis and Broaden the Spectrum of Arrhythmogenic Right Ventricular Cardiomyopathy. Cardiovasc. Pathol. 2004, 13, 185–194. [Google Scholar] [CrossRef]
- Carvajal-Huerta, L. Epidermolytic Palmoplantar Keratoderma with Woolly Hair and Dilated Cardiomyopathy. J. Am. Acad. Dermatol. 1998, 39, 418–421. [Google Scholar] [CrossRef]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a Deletion in Plakoglobin in Arrhythmogenic Right Ventricular Cardiomyopathy with Palmoplantar Keratoderma and Woolly Hair (Naxos Disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Yenamandra, V.K.; van den Akker, P.C.; Lemmink, H.H.; Jan, S.Z.; Diercks, G.F.H.; Vermeer, M.; van den Berg, M.P.; van der Meer, P.; Pasmooij, A.M.G.; Sinke, R.J.; et al. Cardiomyopathy in Patients with Epidermolysis Bullosa Simplex with Mutations in KLHL24. Br. J. Dermatol. 2018, 179, 1181–1183. [Google Scholar] [CrossRef] [PubMed]
- Boyden, L.M.; Craiglow, B.G.; Zhou, J.; Hu, R.; Loring, E.C.; Morel, K.D.; Lauren, C.T.; Lifton, R.P.; Bilguvar, K.; Paller, A.S.; et al. Dominant de Novo Mutations in GJA1 Cause Erythrokeratodermia Variabilis et Progressiva, without Features of Oculodentodigital Dysplasia. J. Investig. Dermatol. 2015, 135, 1540–1547. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Franken, R.; den Hartog, A.W.; Radonic, T.; Micha, D.; Maugeri, A.; van Dijk, F.S.; Meijers-Heijboer, H.E.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; et al. Beneficial Outcome of Losartan Therapy Depends on Type of FBN1 Mutation in Marfan Syndrome. Circ. Cardiovasc. Genet. 2015, 8, 383–388. [Google Scholar] [CrossRef]
- Getsios, S.; Huen, A.C.; Green, K.J. Working out the Strength and Flexibility of Desmosomes. Nat. Rev. Mol. Cell Biol. 2004, 5, 271–281. [Google Scholar] [CrossRef]
- Nekrasova, O.; Green, K.J. Desmosome Assembly and Dynamics. Trends Cell Biol. 2013, 23, 537–546. [Google Scholar] [CrossRef]
- Wan, H.; South, A.P.; Hart, I.R. Increased Keratinocyte Proliferation Initiated through Downregulation of Desmoplakin by RNA Interference. Exp. Cell Res. 2007, 313, 2336–2344. [Google Scholar] [CrossRef]
- Bryson, W.G.; Harland, D.P.; Caldwell, J.P.; Vernon, J.A.; Walls, R.J.; Woods, J.L.; Nagase, S.; Itou, T.; Koike, K. Cortical Cell Types and Intermediate Filament Arrangements Correlate with Fiber Curvature in Japanese Human Hair. J. Struct. Biol. 2009, 166, 46–58. [Google Scholar] [CrossRef]
- Thibaut, S.; Gaillard, O.; Bouhanna, P.; Cannell, D.W.; Bernard, B.A. Human Hair Shape Is Programmed from the Bulb. Br. J. Dermatol. 2005, 152, 632–638. [Google Scholar] [CrossRef]
- Thibaut, S.; Barbarat, P.; Leroy, F.; Bernard, B.A. Human Hair Keratin Network and Curvature. Int. J. Dermatol. 2007, 46, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Gaul, R.T.; Nolan, D.R.; Ristori, T.; Bouten, C.V.C.; Loerakker, S.; Lally, C. Strain Mediated Enzymatic Degradation of Arterial Tissue: Insights into the Role of the Non-Collagenous Tissue Matrix and Collagen Crimp. Acta Biomater. 2018, 77, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Stappenbeck, T.S.; Lamb, J.A.; Corcoran, C.M.; Green, K.J. Phosphorylation of the Desmoplakin COOH Terminus Negatively Regulates Its Interaction with Keratin Intermediate Filament Networks. J. Biol. Chem. 1994, 269, 29351–29354. [Google Scholar] [CrossRef]
- Yang, Z.; Bowles, N.E.; Scherer, S.E.; Taylor, M.D.; Kearney, D.L.; Ge, S.; Nadvoretskiy, V.V.; DeFreitas, G.; Carabello, B.; Brandon, L.I.; et al. Desmosomal Dysfunction Due to Mutations in Desmoplakin Causes Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circ. Res. 2006, 99, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.; Hansen, J.; Nissen, P.; Palmfeldt, J.; Dalager, S.; Jensen, U.; Kim, W.; Heickendorff, L.; Mølgaard, H.; Jensen, H.; et al. Protein Expression Studies of Desmoplakin Mutations in Cardiomyopathy Patients Reveal Different Molecular Disease Mechanisms. Clin. Genet. 2013, 84, 20–30. [Google Scholar] [CrossRef]
- Notari, M.; Hu, Y.; Sutendra, G.; Dedeić, Z.; Lu, M.; Dupays, L.; Yavari, A.; Carr, C.A.; Zhong, S.; Opel, A.; et al. IASPP, a Previously Unidentified Regulator of Desmosomes, Prevents Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)-Induced Sudden Death. Proc. Natl. Acad. Sci. USA 2015, 112, E973–E981. [Google Scholar] [CrossRef]
- Basso, C. Ultrastructural Evidence of Intercalated Disc Remodelling in Arrhythmogenic Right Ventricular Cardiomyopathy: An Electron Microscopy Investigation on Endomyocardial Biopsies. Eur. Heart J. 2006, 27, 1847–1854. [Google Scholar] [CrossRef]
- Vermeer, M.C.S.C.; Andrei, D.; Kramer, D.; Nijenhuis, A.M.; Hoedemaekers, Y.M.; Westers, H.; Jongbloed, J.D.H.; Pas, H.H.; Berg, M.P.; Silljé, H.H.W.; et al. Functional Investigation of Two Simultaneous or Separately Segregating DSP Variants within a Single Family Supports the Theory of a Dose-dependent Disease Severity. Exp. Dermatol. 2022, 31, 970–979. [Google Scholar] [CrossRef]
- Bliley, J.M.; Vermeer, M.C.; Duffy, R.M.; Batalov, I.; Kramer, D.; Tashman, J.W.; Shiwarski, D.J.; Lee, A.; Teplenin, A.S.; Volkers, L.; et al. Dynamic Loading of Human Engineered Heart Tissue Enhances Contractile Function and Drives a Desmosome-Linked Disease Phenotype. Sci. Transl. Med. 2021, 13, eabd1817. [Google Scholar] [CrossRef]
- Lin, X.; Ma, Y.; Cai, Z.; Wang, Q.; Wang, L.; Huo, Z.; Hu, D.; Wang, J.; Xiang, M. Next-Generation Sequencing Identified Novel Desmoplakin Frame-Shift Variant in Patients with Arrhythmogenic Cardiomyopathy. BMC Cardiovasc. Disord. 2020, 20, 74. [Google Scholar] [CrossRef]
- Patel, D.M.; Dubash, A.D.; Kreitzer, G.; Green, K.J. Disease Mutations in Desmoplakin Inhibit Cx43 Membrane Targeting Mediated by Desmoplakin-EB1 Interactions. J. Cell Biol. 2014, 206, 779–797. [Google Scholar] [CrossRef] [PubMed]
- Whittock, N.V.; Ashton, G.H.S.; Dopping-Hepenstal, P.J.C.; Gratian, M.J.; Keane, F.M.; Eady, R.A.J.; McGrath, J.A. Striate Palmoplantar Keratoderma Resulting from Desmoplakin Haploinsufficiency. J. Investig. Dermatol. 1999, 113, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D. Haploinsufficiency of Desmoplakin Causes a Striate Subtype of Palmoplantar Keratoderma. Hum. Mol. Genet. 1999, 8, 143–148, Erratum in Hum. Mol. Genet. 1999, 8, 943. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Dopping-Hepenstal, P.J.C.J.C.; Gratian, M.J.J.; Stone, M.G.G.; Zhu, G.; Purkis, P.E.E.; South, A.P.P.; Keane, F.; Armstrong, D.K.B.K.B.; Buxton, R.S.S.; et al. Striate Palmoplantar Keratoderma Arising from Desmoplakin and Desmoglein 1 Mutations Is Associated with Contrasting Perturbations of Desmosomes and the Keratin Filament Network. Br. J. Dermatol. 2004, 150, 878–891. [Google Scholar] [CrossRef]
- Ng, R.; Manring, H.; Papoutsidakis, N.; Albertelli, T.; Tsai, N.; See, C.J.; Li, X.; Park, J.; Stevens, T.L.; Bobbili, P.J.; et al. Patient Mutations Linked to Arrhythmogenic Cardiomyopathy Enhance Calpain-Mediated Desmoplakin Degradation. JCI Insight 2019, 4, e128643. [Google Scholar] [CrossRef] [PubMed]
- Al-Jassar, C.; Knowles, T.; Jeeves, M.; Kami, K.; Behr, E.; Bikker, H.; Overduin, M.; Chidgey, M. The Nonlinear Structure of the Desmoplakin Plakin Domain and the Effects of Cardiomyopathy-Linked Mutations. J. Mol. Biol. 2011, 411, 1049–1061. [Google Scholar] [CrossRef]
- Boyden, L.M.; Kam, C.Y.; Hernández-Martín, A.; Zhou, J.; Craiglow, B.G.; Sidbury, R.; Mathes, E.F.; Maguiness, S.M.; Crumrine, D.A.; Williams, M.L.; et al. Dominant de Novo DSP Mutations Cause Erythrokeratodermia-Cardiomyopathy Syndrome. Hum. Mol. Genet. 2016, 25, 348–357. [Google Scholar] [CrossRef]
- Norman, M.; Simpson, M.; Mogensen, J.; Shaw, A.; Hughes, S.; Syrris, P.; Sen-Chowdhry, S.; Rowland, E.; Crosby, A.; McKenna, W.J. Novel Mutation in Desmoplakin Causes Arrhythmogenic Left Ventricular Cardiomyopathy. Circulation 2005, 112, 636–642. [Google Scholar] [CrossRef]
- Uzumcu, A.; Norgett, E.E.; Dindar, A.; Uyguner, O.; Nisli, K.; Kayserili, H.; Sahin, S.E.; Dupont, E.; Severs, N.J.; Leigh, I.M.; et al. Loss of Desmoplakin Isoform I Causes Early Onset Cardiomyopathy and Heart Failure in a Naxos-like Syndrome. J. Med. Genet. 2006, 43, e5. [Google Scholar] [CrossRef]
- Impact of the DSP-H1684R Genetic Variant on Ion Channels Activity in IPSC-Derived Cardiomyocytes. Cell. Physiol. Biochem. 2020, 54, 696–706. [CrossRef]
- Williams, T.; Machann, W.; Kühler, L.; Hamm, H.; Müller-Höcker, J.; Zimmer, M.; Ertl, G.; Ritter, O.; Beer, M.; Schönberger, J. Novel Desmoplakin Mutation: Juvenile Biventricular Cardiomyopathy with Left Ventricular Non-Compaction and Acantholytic Palmoplantar Keratoderma. Clin. Res. Cardiol. 2011, 100, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Manchón, J.; Fernández, E.; Igual, B.; Asimaki, A.; Syrris, P.; Osca, J.; Salvador, A.; Zorio, E. Miocardiopatía Arritmogénica Con Afectación Predominante del Ventrículo Izquierdo por una Mutación Nueva «sin Sentido» En Desmoplaquina. Rev. Española Cardiol. 2011, 64, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Jonkman, M.F.; Pasmooij, A.M.G.; Pasmans, S.G.M.A.; van den Berg, M.P.; Ter Horst, H.J.; Timmer, A.; Pas, H.H. Loss of Desmoplakin Tail Causes Lethal Acantholytic Epidermolysis Bullosa. Am. J. Hum. Genet. 2005, 77, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, F.; Odintsova, E.; Chidgey, M. Missense Mutations in Desmoplakin Plakin Repeat Domains Have Dramatic Effects on Domain Structure and Function. Int. J. Mol. Sci. 2022, 23, 529. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, M.G.; Sadowski, S.; Brennan, D.; Pikander, P.; Saukko, P.; Wahl, J.; Aho, H.; Heikinheimo, K.; Bruckner-Tuderman, L.; Fertala, A.; et al. Compound Heterozygous Desmoplakin Mutations Result in a Phenotype with a Combination of Myocardial, Skin, Hair, and Enamel Abnormalities. J. Investig. Dermatol. 2010, 130, 968–978. [Google Scholar] [CrossRef]
- Favre, B.; Begré, N.; Marsili, L.; van Tintelen, J.P.; Borradori, L. Desmoplakin Gene Variants and Risk for Arrhythmogenic Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e002241. [Google Scholar] [CrossRef]
- Favre, B.; Begré, N.; Borradori, L. A Recessive Mutation in the DSP Gene Linked to Cardiomyopathy, Skin Fragility and Hair Defects Impairs the Binding of Desmoplakin to Epidermal Keratins and the Muscle-Specific Intermediate Filament Desmin. Br. J. Dermatol. 2018, 179, 797–799. [Google Scholar] [CrossRef]
- Huen, A.C.; Park, J.K.; Godsel, L.M.; Chen, X.; Bannon, L.J.; Amargo, E.V.; Hudson, T.Y.; Mongiu, A.K.; Leigh, I.M.; Kelsell, D.P.; et al. Intermediate Filament–Membrane Attachments Function Synergistically with Actin-Dependent Contacts to Regulate Intercellular Adhesive Strength. J. Cell Biol. 2002, 159, 1005–1017. [Google Scholar] [CrossRef]
- Puzzi, L.; Borin, D.; Martinelli, V.; Mestroni, L.; Kelsell, D.P.; Sbaizero, O. Cellular Biomechanics Impairment in Keratinocytes Is Associated with a C-Terminal Truncated Desmoplakin: An Atomic Force Microscopy Investigation. Micron 2018, 106, 27–33. [Google Scholar] [CrossRef]
- Martherus, R.; Jain, R.; Takagi, K.; Mendsaikhan, U.; Turdi, S.; Osinska, H.; James, J.F.; Kramer, K.; Purevjav, E.; Towbin, J.A. Accelerated Cardiac Remodeling in Desmoplakin Transgenic Mice in Response to Endurance Exercise Is Associated with Perturbed Wnt/β-Catenin Signaling. Am. J. Physiol.-Heart Circ. Physiol. 2016, 38103, H174–H187. [Google Scholar] [CrossRef]
- Albrecht, L.V.; Zhang, L.; Shabanowitz, J.; Purevjav, E.; Towbin, J.A.; Hunt, D.F.; Green, K.J. GSK3- and PRMT-1-Dependent Modifications of Desmoplakin Control Desmoplakin-Cytoskeleton Dynamics. J. Cell Biol. 2015, 208, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Lapouge, K.; Fontao, L.; Champliaud, M.-F.; Jaunin, F.; Frias, M.A.; Favre, B.; Paulin, D.; Green, K.J.; Borradori, L. New Insights into the Molecular Basis of Desmoplakinand Desmin-Related Cardiomyopathies. J. Cell Sci. 2006, 119, 4974–4985. [Google Scholar] [CrossRef] [PubMed]
- Dehner, C.; Rötzer, V.; Waschke, J.; Spindler, V. A Desmoplakin Point Mutation with Enhanced Keratin Association Ameliorates Pemphigus Vulgaris Autoantibody-Mediated Loss of Cell Cohesion. Am. J. Pathol. 2014, 184, 2528–2536. [Google Scholar] [CrossRef] [PubMed]
- Camors, E.M.; Purevjav, E.; Jefferies, J.L.; Saffitz, J.E.; Gong, N.; Ryan, T.D.; Lucky, A.W.; Taylor, M.D.; Sullivan, L.M.; Mestroni, L.; et al. Early Lethality Due to a Novel Desmoplakin Variant Causing Infantile Epidermolysis Bullosa Simplex with Fragile Skin, Aplasia Cutis Congenita, and Arrhythmogenic Cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, e002800. [Google Scholar] [CrossRef]
- Ian Gallicano, G.; Kouklis, P.; Bauer, C.; Yin, M.; Vasioukhin, V.; Degenstein, L.; Fuchs, E. Desmoplakin Is Required Early in Development for Assembly of Desmosomes and Cytoskeletal Linkage. J. Cell Biol. 1998, 143, 2009–2022. [Google Scholar] [CrossRef]
- Gomes, J.; Finlay, M.; Ahmed, A.K.; Ciaccio, E.J.; Asimaki, A.; Saffitz, J.E.; Quarta, G.; Nobles, M.; Syrris, P.; Chaubey, S.; et al. Electrophysiological Abnormalities Precede Overt Structural Changes in Arrhythmogenic Right Ventricular Cardiomyopathy Due to Mutations in Desmoplakin-A Combined Murine and Human Study. Eur. Heart J. 2012, 33, 1942–1953. [Google Scholar] [CrossRef]
- Garcia-Gras, E. Suppression of Canonical Wnt/ -Catenin Signaling by Nuclear Plakoglobin Recapitulates Phenotype of Arrhythmogenic Right Ventricular Cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef]
- Cheedipudi, S.M.; Hu, J.; Fan, S.; Yuan, P.; Karmouch, J.; Czernuszewicz, G.; Robertson, M.J.; Coarfa, C.; Hong, K.; Yao, Y.; et al. Exercise Restores Dysregulated Gene Expression in a Mouse Model of Arrhythmogenic Cardiomyopathy. Cardiovasc. Res. 2019, 116, 1199–1213. [Google Scholar] [CrossRef]
- Smith, E.A.; Fuchs, E. Defining the Interactions between Intermediate Filaments and Desmosomes. J. Cell Biol. 1998, 141, 1229–1241. [Google Scholar] [CrossRef]
- Lechler, T.; Fuchs, E. Desmoplakin: An Unexpected Regulator of Microtubule Organization in the Epidermis. J. Cell Biol. 2007, 176, 147–154. [Google Scholar] [CrossRef]
- Giuliodori, A.; Beffagna, G.; Marchetto, G.; Fornetto, C.; Vanzi, F.; Toppo, S.; Facchinello, N.; Santimaria, M.; Vettori, A.; Rizzo, S.; et al. Loss of Cardiac Wnt/β-Catenin Signalling in Desmoplakin-Deficient AC8 Zebrafish Models Is Rescuable by Genetic and Pharmacological Intervention. Cardiovasc. Res. 2018, 114, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, W.; Liu, Y.; Haneline, L.S.; Shou, W. Lack of Plakoglobin in Epidermis Leads to Keratoderma. J. Biol. Chem. 2012, 287, 10435–10443. [Google Scholar] [CrossRef] [PubMed]
- Cabral, R.M.; Liu, L.; Hogan, C.; Dopping-Hepenstal, P.J.C.; Winik, B.C.; Asial, R.A.; Dobson, R.; Mein, C.A.; Baselaga, P.A.; Mellerio, J.E.; et al. Homozygous Mutations in the 5′ Region of the JUP Gene Result in Cutaneous Disease but Normal Heart Development in Children. J. Investig. Dermatol. 2010, 130, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Syrris, P.; Wichter, T.; Matthias, P.; Saffitz, J.E.; McKenna, W.J. A Novel Dominant Mutation in Plakoglobin Causes Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Asimaki, A.; Lo, D.; McKenna, W.; Saffitz, J. Disparate Effects of Different Mutations in Plakoglobin on Cell Mechanical Behavior. Cell Motil. Cytoskelet. 2008, 65, 964–978. [Google Scholar] [CrossRef]
- Vahidnezhad, H.; Youssefian, L.; Faghankhani, M.; Mozafari, N.; Saeidian, A.H.; Niaziorimi, F.; Abdollahimajd, F.; Sotoudeh, S.; Rajabi, F.; Mirsafaei, L.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy in Patients with Biallelic JUP-Associated Skin Fragility. Sci. Rep. 2020, 10, 21622. [Google Scholar] [CrossRef]
- Groeneweg, J.A.; Ummels, A.; Mulder, M.; Bikker, H.; van der Smagt, J.J.; van Mil, A.M.; Homfray, T.; Post, J.G.; Elvan, A.; van der Heijden, J.F.; et al. Functional Assessment of Potential Splice Site Variants in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Heart Rhythm 2014, 11, 2010–2017. [Google Scholar] [CrossRef]
- Pigors, M.; Kiritsi, D.; Krümpelmann, S.; Wagner, N.; He, Y.; Podda, M.; Kohlhase, J.; Hausser, I.; Bruckner-Tuderman, L.; Has, C. Lack of Plakoglobin Leads to Lethal Congenital Epidermolysis Bullosa: A Novel Clinico-Genetic Entity. Hum. Mol. Genet. 2011, 20, 1811–1819. [Google Scholar] [CrossRef]
- Liu, L.; Chen, C.; Li, Y.; Yu, R. Whole-Exome Sequencing Identified a De Novo Mutation of Junction Plakoglobin (p.R577C) in a Chinese Patient with Arrhythmogenic Right Ventricular Cardiomyopathy. Biomed. Res. Int. 2019, 2019, 9103860. [Google Scholar] [CrossRef]
- Kaplan, S.R.; Gard, J.J.; Protonotarios, N.; Tsatsopoulou, A.; Spiliopoulou, C.; Anastasakis, A.; Squarcioni, C.P.; McKenna, W.J.; Thiene, G.; Basso, C.; et al. Remodeling of Myocyte Gap Junctions in Arrhythmogenic Right Ventricular Cardiomyopathy Due to a Deletion in Plakoglobin (Naxos Disease). Heart Rhythm 2004, 1, 3–11. [Google Scholar] [CrossRef]
- Zhang, Z.; Stroud, M.J.; Zhang, J.; Fang, X.; Ouyang, K.; Kimura, K.; Mu, Y.; Dalton, N.D.; Gu, Y.; Bradford, W.H.; et al. Normalization of Naxos Plakoglobin Levels Restores Cardiac Function in Mice. J. Clin. Investig. 2015, 125, 1708–1712. [Google Scholar] [CrossRef] [PubMed]
- Protonotarios, N.; Tsatsopoulou, A.; Patsourakos, P.; Alexopoulos, D.; Gezerlis, P.; Simitsis, S.; Scampardonis, G. Cardiac Abnormalities in Familial Palmoplantar Keratosis. Heart 1986, 56, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Kapoor, S.; Plovie, E.; Karin Arndt, A.; Adams, E.; Liu, Z.; James, C.A.; Judge, D.P.; Calkins, H.; Churko, J.; et al. Identification of a New Modulator of the Intercalated Disc in a Zebrafish Model of Arrhythmogenic Cardiomyopathy. Sci. Transl. Med. 2014, 6, 240ra74. [Google Scholar] [CrossRef] [PubMed]
- Bierkamp, C.; McLaughlin, K.J.; Schwarz, H.; Huber, O.; Kemler, R. Embryonic Heart and Skin Defects in Mice Lacking Plakoglobin. Dev. Biol. 1996, 180, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, P.; Brinkmann, V.; Ledermann, B.; Behrend, M.; Grund, C.; Thalhammer, C.; Vogel, F.; Birchmeier, C.; Günthert, U.; Franke, W.W.; et al. Targeted Mutation of Plakoglobin in Mice Reveals Essential Functions of Desmosomes in the Embryonic Heart. J. Cell Biol. 1996, 135, 215–225. [Google Scholar] [CrossRef]
- Li, D.; Liu, Y.; Maruyama, M.; Zhu, W.; Chen, H.; Zhang, W.; Reuter, S.; Lin, S.-F.; Haneline, L.S.; Field, L.J.; et al. Restrictive Loss of Plakoglobin in Cardiomyocytes Leads to Arrhythmogenic Cardiomyopathy. Hum. Mol. Genet. 2011, 20, 4582–4596. [Google Scholar] [CrossRef]
- Swope, D.; Li, J.; Muller, E.J.; Radice, G.L. Analysis of a Jup Hypomorphic Allele Reveals a Critical Threshold for Postnatal Viability. Genesis 2012, 50, 717–727. [Google Scholar] [CrossRef]
- Kowalczyk, A.P.; Borgwardt, J.E.; Green, K.J. Analysis of Desmosomal Cadherin–Adhesive Function and Stoichiometry of Desmosomal Cadherin-Plakoglobin Complexes. J. Investig. Dermatol. 1996, 107, 293–300. [Google Scholar] [CrossRef][Green Version]
- Christensen, A.H.; Schmitz, B.; Andersen, C.B.; Bundgaard, H.; Brand, S.-M.; Svendsen, J.H. Functional Promoter Variant in Desmocollin-2 Contributes to Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2016, 9, 384–387. [Google Scholar] [CrossRef]
- Lin, Y.; Huang, J.; Zhu, Z.; Zhang, Z.; Xian, J.; Yang, Z.; Qin, T.; Chen, L.; Huang, J.; Huang, Y.; et al. Overlap Phenotypes of the Left Ventricular Noncompaction and Hypertrophic Cardiomyopathy with Complex Arrhythmias and Heart Failure Induced by the Novel Truncated DSC2 Mutation. Orphanet J. Rare Dis. 2021, 16, 496. [Google Scholar] [CrossRef]
- Beffagna, G.; De Bortoli, M.; Nava, A.; Salamon, M.; Lorenzon, A.; Zaccolo, M.; Mancuso, L.; Sigalotti, L.; Bauce, B.; Occhi, G.; et al. Missense Mutations in Desmocollin-2 N-Terminus, Associated with Arrhythmogenic Right Ventricular Cardiomyopathy, Affect Intracellular Localization of Desmocollin-2 in Vitro. BMC Med. Genet. 2007, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Moreau, A.; Reisqs, J.; Delanoe-Ayari, H.; Pierre, M.; Janin, A.; Deliniere, A.; Bessière, F.; Meli, A.C.; Charrabi, A.; Lafont, E.; et al. Deciphering DSC2 Arrhythmogenic Cardiomyopathy Electrical Instability: From Ion Channels to ECG and Tailored Drug Therapy. Clin. Transl. Med. 2021, 11, e319. [Google Scholar] [CrossRef] [PubMed]
- Reisqs, J.; Moreau, A.; Charrabi, A.; Sleiman, Y.; Meli, A.C.; Millat, G.; Briand, V.; Beauverger, P.; Richard, S.; Chevalier, P. The PPARγ Pathway Determines Electrophysiological Remodelling and Arrhythmia Risks in DSC2 Arrhythmogenic Cardiomyopathy. Clin. Transl. Med. 2022, 12, e748. [Google Scholar] [CrossRef] [PubMed]
- Gehmlich, K.; Syrris, P.; Peskett, E.; Evans, A.; Ehler, E.; Asimaki, A.; Anastasakis, A.; Tsatsopoulou, A.; Vouliotis, A.-I.; Stefanadis, C.; et al. Mechanistic Insights into Arrhythmogenic Right Ventricular Cardiomyopathy Caused by Desmocollin-2 Mutations. Cardiovasc. Res. 2011, 90, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Heuser, A.; Plovie, E.R.; Ellinor, P.T.; Grossmann, K.S.; Shin, J.T.; Wichter, T.; Basson, C.T.; Lerman, B.B.; Sasse-Klaassen, S.; Thierfelder, L.; et al. Mutant Desmocollin-2 Causes Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 1081–1088. [Google Scholar] [CrossRef]
- Gerull, B.; Kirchner, F.; Chong, J.X.; Tagoe, J.; Chandrasekharan, K.; Strohm, O.; Waggoner, D.; Ober, C.; Duff, H.J. Homozygous Founder Mutation in Desmocollin-2 (DSC2) Causes Arrhythmogenic Cardiomyopathy in the Hutterite Population. Circ. Cardiovasc. Genet. 2013, 6, 327–336. [Google Scholar] [CrossRef]
- Brodehl, A.; Weiss, J.; Debus, J.D.; Stanasiuk, C.; Klauke, B.; Deutsch, M.A.; Fox, H.; Bax, J.; Ebbinghaus, H.; Gärtner, A.; et al. A Homozygous DSC2 Deletion Associated with Arrhythmogenic Cardiomyopathy Is Caused by Uniparental Isodisomy. J. Mol. Cell. Cardiol. 2020, 141, 17–29. [Google Scholar] [CrossRef]
- Simpson, M.A.; Mansour, S.; Ahnood, D.; Kalidas, K.; Patton, M.A.; McKenna, W.J.; Behr, E.R.; Crosby, A.H. Homozygous Mutation of Desmocollin-2 in Arrhythmogenic Right Ventricular Cardiomyopathy with Mild Palmoplantar Keratoderma and Woolly Hair. Cardiology 2009, 113, 28–34. [Google Scholar] [CrossRef]
- Hamada, Y.; Yamamoto, T.; Nakamura, Y.; Sufu-Shimizu, Y.; Nanno, T.; Fukuda, M.; Ono, M.; Oda, T.; Okuda, S.; Ueyama, T.; et al. G790del Mutation in DSC2 Alone Is Insufficient to Develop the Pathogenesis of ARVC in a Mouse Model. Biochem. Biophys. Rep. 2020, 21, 100711. [Google Scholar] [CrossRef]
- Gehmlich, K.; Lambiase, P.D.; Asimaki, A.; Ciaccio, E.J.; Ehler, E.; Syrris, P.; Saffitz, J.E.; McKenna, W.J. A Novel Desmocollin-2 Mutation Reveals Insights into the Molecular Link between Desmosomes and Gap Junctions. Heart Rhythm 2011, 8, 711–718. [Google Scholar] [CrossRef]
- De Bortoli, M.; Beffagna, G.; Bauce, B.; Lorenzon, A.; Smaniotto, G.; Rigato, I.; Calore, M.; Li Mura, I.E.A.; Basso, C.; Thiene, G.; et al. The p.A897KfsX4 Frameshift Variation in Desmocollin-2 Is Not a Causative Mutation in Arrhythmogenic Right Ventricular Cardiomyopathy. Eur. J. Hum. Genet. 2010, 18, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Rimpler, U. Funktionelle Charakterisierung von Desmocollin 2 Während Der Embryonalentwicklung und im Adulten Herzen in Der Maus. Ph.D. Thesis, Humboldt-Universität zu Berlin, Berlin, Germany, 2014. [Google Scholar]
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.-X.; Gordon, P.M.K.; Nygren, A.; et al. Transgenic Mice Overexpressing Desmocollin-2 (DSC2) Develop Cardiomyopathy Associated with Myocardial Inflammation and Fibrotic Remodeling. PLoS ONE 2017, 12, e0174019. [Google Scholar] [CrossRef] [PubMed]
- Capetanaki, Y.; Papathanasiou, S.; Diokmetzidou, A.; Vatsellas, G.; Tsikitis, M. Desmin Related Disease: A Matter of Cell Survival Failure. Curr. Opin. Cell Biol. 2015, 32, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Burkhard, P.; Aebi, U.; Herrmann, H.; Strelkov, S.V.; Burkhard, P.; Aebi, U. Intermediate Filaments: Primary Determinants of Cell Architecture and Plasticity. J. Clin. Investig. 2009, 119, 1772–1783. [Google Scholar] [CrossRef]
- Hnia, K.; Ramspacher, C.; Vermot, J.; Laporte, J. Desmin in Muscle and Associated Diseases: Beyond the Structural Function. Cell Tissue Res. 2015, 360, 591–608. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular Insights into Cardiomyopathies Associated with Desmin (DES) Mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef]
- Lin, Z.; Li, S.; Feng, C.; Yang, S.; Wang, H.; Ma, D.; Zhang, J.; Gou, M.; Bu, D.; Zhang, T.; et al. Stabilizing Mutations of KLHL24 Ubiquitin Ligase Cause Loss of Keratin 14 and Human Skin Fragility. Nat. Genet. 2016, 48, 1504–1516. [Google Scholar] [CrossRef]
- Shi, X.; Xiang, S.; Cao, J.; Zhu, H.; Yang, B.; He, Q.; Ying, M. Kelch-like Proteins: Physiological Functions and Relationships with Diseases. Pharmacol. Res. 2019, 148, 104404. [Google Scholar] [CrossRef]
- He, Y.; Maier, K.; Leppert, J.; Hausser, I.; Schwieger-Briel, A.; Weibel, L.; Theiler, M.; Kiritsi, D.; Busch, H.; Boerries, M.; et al. Monoallelic Mutations in the Translation Initiation Codon of KLHL24 Cause Skin Fragility. Am. J. Hum. Genet. 2016, 99, 1395–1404. [Google Scholar] [CrossRef]
- Vermeer, M.C.S.C.; Bolling, M.C.; Bliley, J.M.; Gomez, K.F.A.; Pavez-Giani, M.G.; Kramer, D.; Romero-Herrera, P.H.; Westenbrink, B.D.; Diercks, G.F.H.; van den Berg, M.P.; et al. Gain-of-Function Mutation in Ubiquitin Ligase KLHL24 Causes Desmin Degradation and Dilatation in HiPSC-Derived Engineered Heart Tissues. J. Clin. Investig. 2021, 131, e140615. [Google Scholar] [CrossRef]
- Vermeer, M.C.S.C.; Silljé, H.H.W.; Pas, H.H.; Andrei, D.; van der Meer, P.; Bolling, M.C. Keratin 14 Degradation and Aging in Epidermolysis Bullosa Simplex Due to KLHL24 Gain-of-Function Variants. J. Investig. Dermatol. 2022, 142, 2271–2274.e6. [Google Scholar] [CrossRef] [PubMed]
- Logli, E.; Marzuolo, E.; D’Agostino, M.; Conti, L.A.; Lena, A.M.; Diociaiuti, A.; Dellambra, E.; Has, C.; Cianfanelli, V.; Zambruno, G.; et al. Proteasome-Mediated Degradation of Keratins 7, 8, 17 and 18 by Mutant KLHL24 in a Foetal Keratinocyte Model: Novel Insight in Congenital Skin Defects and Fragility of Epidermolysis Bullosa Simplex with Cardiomyopathy. Hum. Mol. Genet. 2021, 11, 28. [Google Scholar] [CrossRef]
- Cui, J.; Zhao, Q.; Song, Z.; Chen, Z.; Zeng, X.; Wang, C.; Lin, Z.; Wang, F.; Yang, Y. KLHL24-Mediated Hair Follicle Stem Cells Structural Disruption Causes Alopecia. J. Investig. Dermatol. 2022, 142, 2079–2087.e8. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, M.C.S.C.; Al-shinnag, M.; Silljé, H.H.W.; Gaytan, A.E.; Murrell, D.F.; McGaughran, J.; Melbourne, W.; Cowan, T.; van den Akker, P.C.; van Spaendonck-Zwarts, K.Y.; et al. A Translation Re-initiation Variant in KLHL24 Also Causes Epidermolysis Bullosa Simplex and Dilated Cardiomyopathy via Intermediate Filament Degradation. Br. J. Dermatol. 2022; Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, O.; Hübbert, L.; Nennesmo, I.; Abramsson, A.; Banote, R.; Edling, M.; Zetterberg, H.; Oldfors, A. Hypertrophic Cardiomyopathy and Abnormal Glycogen Storage in Heart and Skeletal Muscle Associated with Inactivation of KLHL24. Neuromuscul. Disord. 2016, 26, S152. [Google Scholar] [CrossRef]
- Hedberg-Oldfors, C.; Abramsson, A.; Osborn, D.P.S.; Danielsson, O.; Fazlinezhad, A.; Nilipour, Y.; Hübbert, L.; Nennesmo, I.; Visuttijai, K.; Bharj, J.; et al. Cardiomyopathy with Lethal Arrhythmias Associated with Inactivation of KLHL24. Hum. Mol. Genet. 2019, 28, 1919–1929. [Google Scholar] [CrossRef]
- Alkhalifah, A.; Chiaverini, C.; Charlesworth, A.; Has, C.; Lacour, J.-P. Burnlike Scars: A Sign Suggestive of KLHL24-Related Epidermolysis Bullosa Simplex. Pediatr. Dermatol. 2018, 35, e193–e195. [Google Scholar] [CrossRef]
- Grilletta, E.A. Cardiac Transplant for Epidermolysis Bullosa Simplex with KLHL24 Mutation–Associated Cardiomyopathy. JAAD Case Rep. 2019, 5, 912–914. [Google Scholar] [CrossRef]
- SOHL, G.; Willecke, K. Gap Junctions and the Connexin Protein Family. Cardiovasc. Res. 2004, 62, 228–232. [Google Scholar] [CrossRef]
- Zhang, X.-F.; Cui, X. Connexin 43: Key Roles in the Skin. Biomed. Rep. 2017, 6, 605–611. [Google Scholar] [CrossRef]
- Avshalumova, L.; Fabrikant, J.; Koriakos, A. Overview of Skin Diseases Linked to Connexin Gene Mutations. Int. J. Dermatol. 2014, 53, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Lilly, E.; Sellitto, C.; Milstone, L.M.; White, T.W. Connexin Channels in Congenital Skin Disorders. Semin. Cell Dev. Biol. 2016, 50, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Michela, P.; Velia, V.; Aldo, P.; Ada, P. Role of Connexin 43 in Cardiovascular Diseases. Eur. J. Pharmacol. 2015, 768, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Schinner, C.; Erber, B.M.; Yeruva, S.; Waschke, J. Regulation of Cardiac Myocyte Cohesion and Gap Junctions via Desmosomal Adhesion. Acta Physiol. 2019, 226, e13242. [Google Scholar] [CrossRef]
- Salameh, A.; Haunschild, J.; Bräuchle, P.; Peim, O.; Seidel, T.; Reitmann, M.; Kostelka, M.; Bakhtiary, F.; Dhein, S.; Dähnert, I. On the Role of the Gap Junction Protein Cx43 (GJA1) in Human Cardiac Malformations with Fallot-Pathology. A Study on Paediatric Cardiac Specimen. PLoS ONE 2014, 9, e95344. [Google Scholar] [CrossRef]
- Laird, D.W. Syndromic and Non-Syndromic Disease-Linked Cx43 Mutations. FEBS Lett. 2014, 588, 1339–1348. [Google Scholar] [CrossRef]
- Shao, Q.; Liu, Q.; Lorentz, R.; Gong, X.Q.; Bai, D.; Shaw, G.S.; Laird, D.W. Structure and Functional Studies of N-Terminal Cx43 Mutants Linked to Oculodentodigital Dysplasia. Mol. Biol. Cell 2012, 23, 3312–3321. [Google Scholar] [CrossRef]
- Churko, J.M.; Shao, Q.; Gong, X.Q.; Swoboda, K.J.; Bai, D.; Sampson, J.; Laird, D.W. Human Dermal Fibroblasts Derived from Oculodentodigital Dysplasia Patients Suggest That Patients May Have Wound-Healing Defects. Hum. Mutat. 2011, 32, 456–466. [Google Scholar] [CrossRef]
- Kelly, J.J.; Esseltine, J.L.; Shao, Q.; Jabs, E.W.; Sampson, J.; Auranen, M.; Bai, D.; Laird, D.W. Specific Functional Pathologies of Cx43 Mutations Associated with Oculodentodigital Dysplasia. Mol. Biol. Cell 2016, 27, 2172–2185. [Google Scholar] [CrossRef]
- Srinivas, M.; Jannace, T.F.; Cocozzelli, A.G.; Li, L.; Slavi, N.; Sellitto, C.; White, T.W. Connexin43 Mutations Linked to Skin Disease Have Augmented Hemichannel Activity. Sci. Rep. 2019, 9, 19. [Google Scholar] [CrossRef]
- Wang, H.; Cao, X.; Lin, Z.; Lee, M.; Jia, X.; Ren, Y.; Dai, L.; Guan, L.; Zhang, J.; Lin, X.; et al. Exome Sequencing Reveals Mutation in GJA1 as a Cause of Keratoderma-Hypotrichosis-Leukonychia Totalis Syndrome. Hum. Mol. Genet. 2015, 24, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, J.; Paznekas, W.; Seki, A.; Taffet, S.; Jabs, E.W.; Delmar, M.; Musa, H. Functional Characterization of Connexin43 Mutations Found in Patients with Oculodentodigital Dysplasia. Circ. Res. 2005, 96, e83–e91. [Google Scholar] [CrossRef] [PubMed]
- Falk, L.; Dang-Lawson, M.; Vega, J.L.; Pournia, F.; Choi, K.; Jang, C.; Naus, C.C.; Matsuuchi, L.M. Mutations of Cx43 That Affect B Cell Spreading in Response to BCR Signaling. Biol. Open 2014, 3, 185–191. [Google Scholar] [CrossRef]
- Gong, X.Q.; Shao, Q.; Langlois, S.; Bai, D.; Laird, D.W. Differential Potency of Dominant Negative Connexin43 Mutants in Oculodentodigital Dysplasia. J. Biol. Chem. 2007, 282, 19190–19202. [Google Scholar] [CrossRef]
- McLachlan, E.; Manias, J.L.; Gong, X.-Q.; Lounsbury, C.S.; Shao, Q.; Bernier, S.M.; Bai, D.; Laird, D.W. Functional Characterization of Oculodentodigital Dysplasia-Associated Cx43 Mutants. Cell Commun. Adhes. 2005, 12, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Roscoe, W.; Veitch, G.I.L.; Gong, X.Q.; Pellegrino, E.; Bai, D.; McLachlan, E.; Shao, Q.; Kidder, G.M.; Laird, D.W. Oculodentodigital Dysplasia-Causing Connexin43 Mutants Are Non-Functional and Exhibit Dominant Effects on Wild-Type Connexin43. J. Biol. Chem. 2005, 280, 11458–11466. [Google Scholar] [CrossRef]
- Dobrowolski, R.; Sommershof, A.; Willecke, K. Some Oculodentodigital Dysplasia-Associated Cx43 Mutations Cause Increased Hemichannel Activity in Addition to Deficient Gap Junction Channels. J. Membr. Biol. 2007, 219, 9–17. [Google Scholar] [CrossRef]
- Huang, T.; Shao, Q.; MacDonald, A.; Xin, L.; Lorentz, R.; Bai, D.; Laird, D.W. Autosomal Recessive GJA1 (Cx43) Gene Mutations Cause Oculodentodigital Dysplasia by Distinct Mechanisms. J. Cell Sci. 2013, 126, 2857–2866. [Google Scholar] [CrossRef]
- Van Norstrand, D.W.; Asimaki, A.; Rubinos, C.; Dolmatova, E.; Srinivas, M.; Tester, D.J.; Saffitz, J.E.; Duffy, H.S.; Ackerman, M.J. Connexin43 Mutation Causes Heterogeneous Gap Junction Loss and Sudden Infant Death. Circulation 2012, 125, 474–481. [Google Scholar] [CrossRef]
- Lübkemeier, I.; Bosen, F.; Kim, J.S.; Sasse, P.; Malan, D.; Fleischmann, B.K.; Willecke, K. Human Connexin43E42K Mutation from a Sudden Infant Death Victim Leads to Impaired Ventricular Activation and Neonatal Death in Mice. Circ. Cardiovasc. Genet. 2015, 8, 21–29. [Google Scholar] [CrossRef]
- Tong, D.; Lu, X.; Wang, H.-X.; Plante, I.; Lui, E.; Laird, D.W.; Bai, D.; Kidder, G.M. A Dominant Loss-of-Function GJA1 (Cx43) Mutant Impairs Parturition in the Mouse1. Biol. Reprod. 2009, 80, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Manias, J.L.; Plante, I.; Gong, X.-Q.; Shao, Q.; Churko, J.; Bai, D.; Laird, D.W. Fate of Connexin43 in Cardiac Tissue Harbouring a Disease-Linked Connexin43 Mutant. Cardiovasc. Res. 2008, 80, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Shao, Q.; Barr, K.; Simek, J.; Fishman, G.I.; Laird, D.W. Myogenic Bladder Defects in Mouse Models of Human Oculodentodigital Dysplasia. Biochem. J. 2014, 457, 441–449. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, E.; Plante, I.; Shao, Q.; Tong, D.; Kidder, G.M.; Bernier, S.M.; Laird, D.W. ODDD-Linked Cx43 Mutants Reduce Endogenous Cx43 Expression and Function in Osteoblasts and Inhibit Late Stage Differentiation. J. Bone Miner. Res. 2008, 23, 928–938. [Google Scholar] [CrossRef]
- Kozoriz, M.G.; Lai, S.; Vega, J.L.; Sáez, J.C.; Sin, W.C.; Bechberger, J.F.; Naus, C.C. Cerebral Ischemic Injury Is Enhanced in a Model of Oculodentodigital Dysplasia. Neuropharmacology 2013, 75, 549–556. [Google Scholar] [CrossRef]
- Gregory, M.; Kahiri, C.N.; Barr, K.J.; Smith, C.E.; Hermo, L.; Cyr, D.G.; Kidder, G.M. Male Reproductive System Defects and Subfertility in a Mutant Mouse Model of Oculodentodigital Dysplasia. Int. J. Androl. 2011, 34, e630–e641. [Google Scholar] [CrossRef]
- Lorentz, R.; Shao, Q.; Huang, T.; Fishman, G.I.; Laird, D.W. Characterization of Gap Junction Proteins in the Bladder of Cx43 Mutant Mouse Models of Oculodentodigital Dysplasia. J. Membr. Biol. 2012, 245, 345–355. [Google Scholar] [CrossRef][Green Version]
- Tuomi, J.M.; Tyml, K.; Jones, D.L. Atrial Tachycardia/Fibrillation in the Connexin 43 G60S Mutant (Oculodentodigital Dysplasia) Mouse. Am. J. Physiol. Circ. Physiol. 2011, 300, H1402–H1411. [Google Scholar] [CrossRef][Green Version]
- Toth, K.; Shao, Q.; Lorentz, R.; Laird, D.W. Decreased Levels of Cx43 Gap Junctions Result in Ameloblast Dysregulation and Enamel Hypoplasia in Gja1Jrt/+ Mice. J. Cell. Physiol. 2010, 223, 601–609. [Google Scholar] [CrossRef]
- Churko, J.M.; Kelly, J.J.; Macdonald, A.; Lee, J.; Sampson, J.; Bai, D.; Laird, D.W. The G60S Cx43 Mutant Enhances Keratinocyte Proliferation and Differentiation. Exp. Dermatol. 2012, 21, 612–618. [Google Scholar] [CrossRef]
- Churko, J.M.; Chan, J.; Shao, Q.; Laird, D.W. The G60S Connexin43 Mutant Regulates Hair Growth and Hair Fiber Morphology in a Mouse Model of Human Oculodentodigital Dysplasia. J. Investig. Dermatol. 2011, 131, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Winterhager, E.; Gellhaus, A.; Blois, S.M.; Hill, L.A.; Barr, K.J.; Kidder, G.M. Decidual Angiogenesis and Placental Orientation Are Altered in Mice Heterozygous for a Dominant Loss-of-Function Gja1 (Connexin43) Mutation1. Biol. Reprod. 2013, 89, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Abitbol, J.M.; Kelly, J.J.; Barr, K.J.; Allman, B.L.; Laird, D.W. Mice Harbouring an Oculodentodigital Dysplasia-Linked Cx43 G60S Mutation Have Severe Hearing Loss. J. Cell Sci. 2018, 131, jcs214635. [Google Scholar] [CrossRef] [PubMed]
- Flenniken, A.M.; Osborne, L.R.; Anderson, N.; Ciliberti, N.; Fleming, C.; Gittens, J.E.I.; Gong, X.Q.; Kelsey, L.B.; Lounsbury, C.; Moreno, L.; et al. A Gja1 Missense Mutation in a Mouse Model of Oculodentodigital Dysplasia. Development 2005, 132, 4375–4386. [Google Scholar] [CrossRef]
- Tong, D.; Colley, D.; Thoo, R.; Li, T.Y.; Plante, I.; Laird, D.W.; Bai, D.; Kidder, G.M. Oogenesis Defects in a Mutant Mouse Model of Oculodentodigital Dysplasia. DMM Dis. Model. Mech. 2009, 2, 157–167. [Google Scholar] [CrossRef][Green Version]
- Hong, H.M.; Yang, J.J.; Shieh, J.C.; Li, M.L.; Li, S.Y. Novel Mutations in the Connexin43 (GJA1) and GJA1 Pseudogene May Contribute to Nonsyndromic Hearing Loss. Hum. Genet. 2010, 127, 545–551. [Google Scholar] [CrossRef]
- Kalcheva, N.; Qu, J.; Sandeep, N.; Garcia, L.; Zhang, J.; Wang, Z.; Lampe, P.D.; Suadicani, S.O.; Spray, D.C.; Fishman, G.I. Gap Junction Remodeling and Cardiac Arrhythmogenesis in a Murine Model of Oculodentodigital Dysplasia. Proc. Natl. Acad. Sci. USA 2007, 104, 20512–20516. [Google Scholar] [CrossRef]
- Paznekas, W.A.; Boyadjiev, S.A.; Shapiro, R.E.; Daniels, O.; Wollnik, B.; Keegan, C.E.; Innis, J.W.; Dinulos, M.B.; Christian, C.; Hannibal, M.C.; et al. Connexin 43 (GJA1) Mutations Cause the Pleiotropic Phenotype of Oculodentodigital Dysplasia. Am. J. Hum. Genet. 2003, 72, 408–418. [Google Scholar] [CrossRef]
- Dobrowolski, R.; Sasse, P.; Schrickel, J.W.; Watkins, M.; Kim, J.-S.; Rackauskas, M.; Troatz, C.; Ghanem, A.; Tiemann, K.; Degen, J.; et al. The Conditional Connexin43G138R Mouse Mutant Represents a New Model of Hereditary Oculodentodigital Dysplasia in Humans. Hum. Mol. Genet. 2008, 17, 539–554. [Google Scholar] [CrossRef]
- Dobrowolski, R.; Hertig, G.; Lechner, H.; Wörsdörfer, P.; Wulf, V.; Dicke, N.; Eckert, D.; Bauer, R.; Schorle, H.; Willecke, K. Loss of Connexin43-Mediated Gap Junctional Coupling in the Mesenchyme of Limb Buds Leads to Altered Expression of Morphogens in Mice. Hum. Mol. Genet. 2009, 18, 2899–2911. [Google Scholar] [CrossRef]
- Zheng, L.; Chenavas, S.; Kieken, F.; Trease, A.; Brownell, S.; Anbanandam, A.; Sorgen, P.L.; Spagnol, G. Calmodulin Directly Interacts with the Cx43 Carboxyl-Terminus and Cytoplasmic Loop Containing Three ODDD-Linked Mutants (M147T, R148Q, and T154A) That Retain α-Helical Structure, but Exhibit Loss-of-Function and Cellular Trafficking Defects. Biomolecules 2020, 10, 1452. [Google Scholar] [CrossRef] [PubMed]
- Esseltine, J.L.; Shao, Q.; Brooks, C.; Sampson, J.; Betts, D.H.; Séguin, C.A.; Laird, D.W. Connexin43 Mutant Patient-Derived Induced Pluripotent Stem Cells Exhibit Altered Differentiation Potential. J. Bone Miner. Res. 2017, 32, 1368–1385. [Google Scholar] [CrossRef] [PubMed]
- Kretz, M.; Maass, K.; Willecke, K. Expression and Function of Connexins in the Epidermis, Analyzed with Transgenic Mouse Mutants. Eur. J. Cell Biol. 2004, 83, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Moorer, M.C.; Hebert, C.; Tomlinson, R.E.; Iyer, S.R.; Chason, M.; Stains, J.P. Defective Signaling, Osteoblastogenesis and Bone Remodeling in a Mouse Model of Connexin 43 C-Terminal Truncation. J. Cell Sci. 2017, 130, 531–540. [Google Scholar] [CrossRef]
- Gong, X.Q.; Shao, Q.; Lounsbury, C.S.; Bai, D.; Laird, D.W. Functional Characterization of a GJA1 Frameshift Mutation Causing Oculodentodigital Dysplasia and Palmoplantar Keratoderma. J. Biol. Chem. 2006, 281, 31801–31811. [Google Scholar] [CrossRef]
- Li, C.; Liang, J.; Chen, P.; Zeng, K.; Xue, R.; Tian, X.; Liang, L.; Wang, Q.; Shi, M.; Zhang, X. Two de Novo GJA1 Mutation in Two Sporadic Patients with Erythrokeratodermia Variabilis et Progressiva. Mol. Genet. Genomic Med. 2019, 7, e670. [Google Scholar] [CrossRef]
- Lucaciu, S.A.; Shao, Q.; Figliuzzi, R.; Barr, K.; Bai, D.; Laird, D.W. Interrogation of Carboxy-Terminus Localized GJA1 Variants Associated with Erythrokeratodermia Variabilis et Progressiva. Int. J. Mol. Sci. 2022, 23, 486. [Google Scholar] [CrossRef]
- Thibodeau, I.L.; Xu, J.; Li, Q.; Liu, G.; Lam, K.; Veinot, J.P.; Birnie, D.H.; Jones, D.L.; Krahn, A.D.; Lemery, R.; et al. Paradigm of Genetic Mosaicism and Lone Atrial Fibrillation: Physiological Characterization of a Connexin 43-Deletion Mutant Identified from Atrial Tissue. Circulation 2010, 122, 236–244. [Google Scholar] [CrossRef]
- Britz-Cunningham, S.H.; Shah, M.M.; Zuppan, C.W.; Fletcher, W.H. Mutations of the Connexin43 Gap-Junction Gene in Patients with Heart Malformations and Defects of Laterality. N. Engl. J. Med. 1995, 332, 1323–1330. [Google Scholar] [CrossRef]
- Ek-Vitorín, J.F.; Calero, G.; Morley, G.E.; Coombs, W.; Taffet, S.M.; Delmar, M. PH Regulation of Connexin43: Molecular Analysis of the Gating Particle. Biophys. J. 1996, 71, 1273–1284. [Google Scholar] [CrossRef]
- Langlois, S.; Maher, A.C.; Manias, J.L.; Shao, Q.; Kidder, G.M.; Laird, D.W. Connexin Levels Regulate Keratinocyte Differentiation in the Epidermis. J. Biol. Chem. 2007, 282, 30171–30180. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wu, Y.; Zhang, L.; Zheng, J.; Tang, S.; Cheng, J. GJA1 Gene Variations in Sudden Unexplained Nocturnal Death Syndrome in the Chinese Han Population. Forensic Sci. Int. 2017, 270, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Wittlieb-Weber, C.A.; Haude, K.M.; Fong, C.-T.; Vinocur, J.M. A Novel GJA1 Mutation Causing Familial Oculodentodigital Dysplasia with Dilated Cardiomyopathy and Arrhythmia. Heart Case Rep. 2016, 2, 32–35. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Izumi, K.; Lippa, A.M.; Wilkens, A.; Feret, H.A.; McDonald-McGinn, D.M.; Zackai, E.H. Congenital Heart Defects in Oculodentodigital Dysplasia: Report of Two Cases. Am. J. Med. Genet. Part A 2013, 161, 3150–3154. [Google Scholar] [CrossRef]
- Brnich, S.E.; Abou Tayoun, A.N.; Couch, F.J.; Cutting, G.R.; Greenblatt, M.S.; Heinen, C.D.; Kanavy, D.M.; Luo, X.; McNulty, S.M.; Starita, L.M.; et al. Recommendations for Application of the Functional Evidence PS3/BS3 Criterion Using the ACMG/AMP Sequence Variant Interpretation Framework. Genome Med. 2020, 12, 3. [Google Scholar] [CrossRef]
- Kidwai, F.K.; Liu, H.; Toh, W.S.; Fu, X.; Jokhun, D.S.; Movahednia, M.M.; Li, M.; Zou, Y.; Squier, C.A.; Phan, T.T.; et al. Differentiation of Human Embryonic Stem Cells into Clinically Amenable Keratinocytes in an Autogenic Environment. J. Investig. Dermatol. 2013, 133, 618–628. [Google Scholar] [CrossRef]
- Lee, J.; Rabbani, C.C.; Gao, H.; Steinhart, M.R.; Woodruff, B.M.; Pflum, Z.E.; Kim, A.; Heller, S.; Liu, Y.; Shipchandler, T.Z.; et al. Hair-Bearing Human Skin Generated Entirely from Pluripotent Stem Cells. Nature 2020, 582, 399–404. [Google Scholar] [CrossRef]
- Weinberger, F.; Mannhardt, I.; Eschenhagen, T. Engineering Cardiac Muscle Tissue: A Maturating Field of Research. Circ. Res. 2017, 120, 1487–1500. [Google Scholar] [CrossRef]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.J.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced Maturation of Human Cardiac Tissue Grown from Pluripotent Stem Cells. Nature 2018, 556, 239–243. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public Archive of Relationships among Sequence Variation and Human Phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M. Recommendations for Interpreting the Loss of Function PVS1 ACMG/AMP Variant Criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G.; Harrison, S.M. The ACMG/AMP Reputable Source Criteria for the Interpretation of Sequence Variants. Genet. Med. 2018, 20, 1687–1688. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.M.; Biesecker, L.G.; Rehm, H.L. Overview of Specifications to the ACMG/AMP Variant Interpretation Guidelines. Curr. Protoc. Hum. Genet. 2019, 103, e93. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the Effects of Coding Non-Synonymous Variants on Protein Function Using the SIFT Algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. MutPred2: Inferring the Molecular and Phenotypic Impact of Amino Acid Variants. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef] [PubMed]
- Pagel, K.A.; Antaki, D.; Lian, A.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; Mooney, S.D.; Radivojac, P. Pathogenicity and Functional Impact of Non-Frameshifting Insertion/Deletion Variation in the Human Genome. PLoS Comput. Biol. 2019, 15, e1007112. [Google Scholar] [CrossRef]
- Frischmeyer, P.A. Nonsense-Mediated MRNA Decayin Health and Disease. Hum. Mol. Genet. 1999, 8, 1893–1900. [Google Scholar] [CrossRef]
- Pagel, K.A.; Pejaver, V.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; Mooney, S.D.; Radivojac, P. When Loss-of-Function Is Loss of Function: Assessing Mutational Signatures and Impact of Loss-of-Function Genetic Variants. Bioinformatics 2017, 33, i389–i398. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).