
Rodent Models of Post-Stroke Dementia


Abstract
1. Introduction
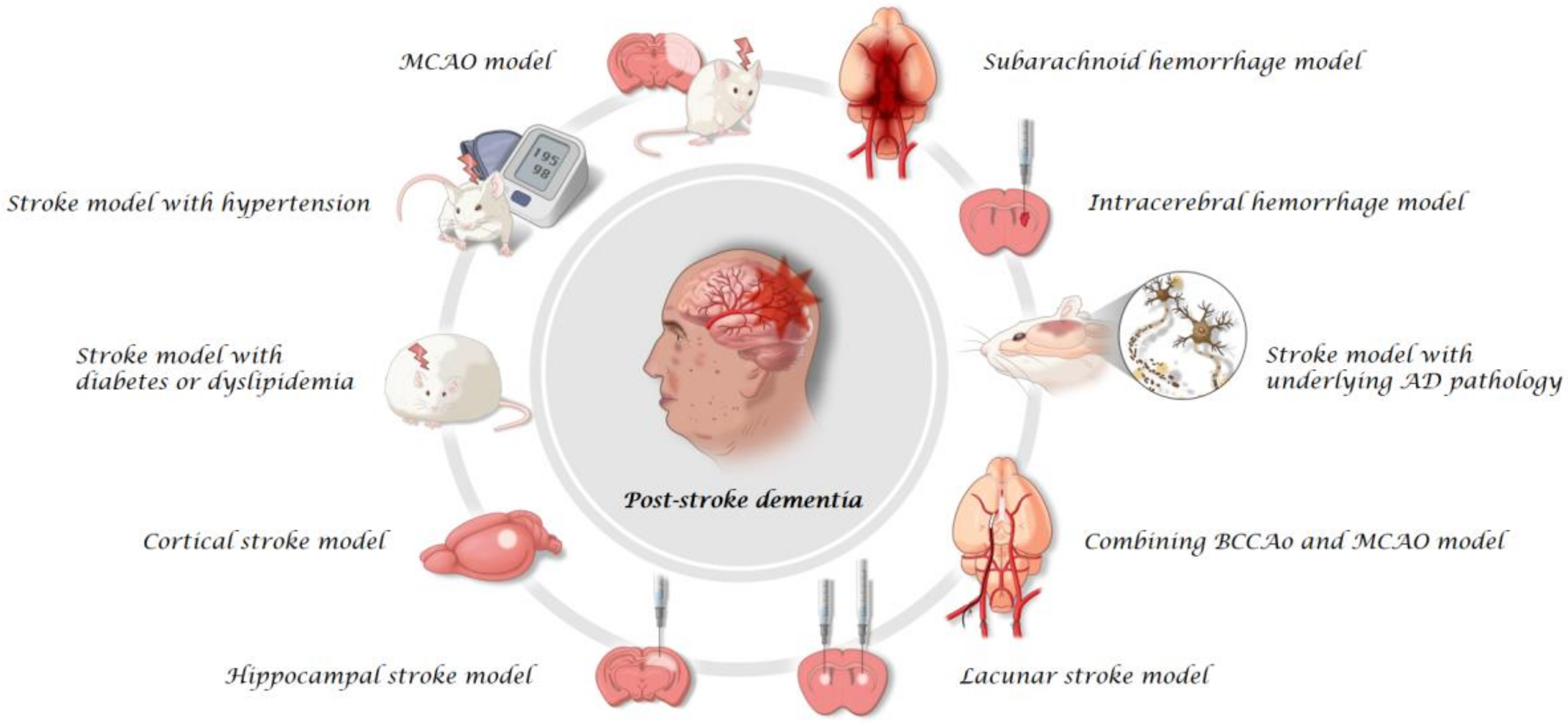
2. Rodent Models
2.1. MCAO Model
2.2. Stroke Model with Comorbid Conditions
2.3. Cortical Stroke Model
2.4. Hippocampal Stroke Model
2.5. Lacunar Stroke Model
2.6. Combining BCCAo and MCAO Model
2.7. Stroke Model with Underlying AD Pathology
2.8. ICH Model
2.9. SAH Model
3. Challenges: Investigating the Pathophysiology of PSD
4. Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Murray, C.J.; Lopez, A.D. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997, 349, 1436–1442. [Google Scholar] [CrossRef]
- Ivan, C.S.; Seshadri, S.; Beiser, A.; Au, R.; Kase, C.S.; Kelly-Hayes, M.; Wolf, P.A. Dementia after stroke: The Framingham Study. Stroke 2004, 35, 1264–1268. [Google Scholar] [CrossRef]
- Pantoni, L. Have Stroke Neurologists Entered the Arena of Stroke-Related Cognitive Dysfunctions? Not Yet, but They Should! Stroke 2017, 48, 1441–1442. [Google Scholar] [CrossRef] [PubMed]
- Pendlebury, S.T.; Rothwell, P.M. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: A systematic review and meta-analysis. Lancet Neurol. 2009, 8, 1006–1018. [Google Scholar] [CrossRef]
- Weaver, N.A.; Kuijf, H.J.; Aben, H.P.; Abrigo, J.; Bae, H.-J.; Barbay, M.; Best, J.G.; Bordet, R.; Chappell, F.M.; Chen, C.P.L.H.; et al. Strategic infarct locations for post-stroke cognitive impairment: A pooled analysis of individual patient data from 12 acute ischaemic stroke cohorts. Lancet Neurol. 2021, 20, 448–459. [Google Scholar] [CrossRef]
- Kim, K.Y.; Shin, K.Y.; Chang, K.-A. Potential Biomarkers for Post-Stroke Cognitive Impairment: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2022, 23, 602. [Google Scholar] [CrossRef]
- Censori, B.; Manara, O.; Agostinis, C.; Camerlingo, M.; Casto, L.; Galavotti, B.; Partziguian, T.; Servalli, M.C.; Cesana, B.; Belloni, G.; et al. Dementia After First Stroke. Stroke 1996, 27, 1205–1210. [Google Scholar] [CrossRef]
- Mok, V.; Lam, B.Y.K.; Wong, A.; Ko, H.; Markus, H.S.; Wong, L.K.S. Early-onset and delayed-onset poststroke dementia—revisiting the mechanisms. Nat. Rev. Neurol. 2017, 13, 148–159. [Google Scholar] [CrossRef]
- Chow, W.Z.; Ong, L.K.; Kluge, M.G.; Gyawali, P.; Walker, F.R.; Nilsson, M. Similar cognitive deficits in mice and humans in the chronic phase post-stroke identified using the touchscreen-based paired-associate learning task. Sci. Rep. 2020, 10, 19545. [Google Scholar] [CrossRef]
- Hennerici, M.G. What are the mechanisms for post-stroke dementia? Lancet Neurol. 2009, 8, 973–975. [Google Scholar] [CrossRef]
- Leys, D.; Henon, H.; Mackowiak-Cordoliani, M.A.; Pasquier, F. Poststroke dementia. Lancet Neurol. 2005, 4, 752–759. [Google Scholar] [CrossRef]
- Pasquier, F.; Leys, D. Why are stroke patients prone to develop dementia? J. Neurol. 1997, 244, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.-R.; Kwon, K.J.; Park, S.H.; Jeon, W.K.; Han, S.-H.; Kim, H.Y.; Han, J.-S. Alternations of Septal-hippocampal System in the Adult Wistar Rat with Spatial Memory Impairments Induced by Chronic Cerebral Hypoperfusion. Exp. Neurobiol. 2011, 20, 92–99. [Google Scholar] [CrossRef]
- Chin, Y.; Kishi, M.; Sekino, M.; Nakajo, F.; Abe, Y.; Terazono, Y.; Hiroyuki, O.; Kato, F.; Koizumi, S.; Gachet, C.; et al. Involvement of glial P2Y1 receptors in cognitive deficit after focal cerebral stroke in a rodent model. J. Neuroinflamm. 2013, 10, 860. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Huang, R.; Shetty, R.A.; Thangthaeng, N.; Liu, R.; Chen, Z.; Sumien, N.; Rutledge, M.; Dillon, G.H.; Yuan, F.; et al. Transient focal cerebral ischemia induces long-term cognitive function deficit in an experimental ischemic stroke model. Neurobiol. Dis. 2013, 59, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, G.; Liu, H.; Chang, H.; Wilson, J.X. Folic acid enhances Notch signaling, hippocampal neurogenesis, and cognitive function in a rat model of cerebral ischemia. Nutr. Neurosci. 2012, 15, 55–61. [Google Scholar] [CrossRef]
- Zhang, T.; Pan, B.-S.; Sun, G.-C.; Sun, X.; Sun, F.-Y. Diabetes synergistically exacerbates poststroke dementia and tau abnormality in brain. Neurochem. Int. 2010, 56, 955–961. [Google Scholar] [CrossRef]
- Ward, R.; Valenzuela, J.P.; Li, W.; Dong, G.; Fagan, S.C.; Ergul, A. Poststroke cognitive impairment and hippocampal neurovascular remodeling: The impact of diabetes and sex. Am. J. Physiol. Circ. Physiol. 2018, 315, H1402–H1413. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, M.; Yang, S.; Chen, X.; Wu, J.; Wen, M.; Yan, K.; Bi, X. Enriched environment improves post-stroke cognitive impairment and inhibits neuroinflammation and oxidative stress by activating Nrf2-ARE pathway. Int. J. Neurosci. 2021, 131, 641–649. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, X.; Dong, J.; Liu, W.-C.; Song, M.; Sun, Y.; Shu, H.; Towse, C.-L.; Liu, W.; Liu, C.-F.; et al. Inhibition of Reactive Astrocytes with Fluorocitrate Ameliorates Learning and Memory Impairment Through Upregulating CRTC1 and Synaptophysin in Ischemic Stroke Rats. Cell. Mol. Neurobiol. 2019, 39, 1151–1163. [Google Scholar] [CrossRef]
- Ward, R.; Li, W.; Abdul, Y.; Jackson, L.; Dong, G.; Jamil, S.; Filosa, J.; Fagan, S.C.; Ergul, A. NLRP3 inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharmacol. Res. 2019, 142, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Yonemori, F.; Yamada, H.; Yamaguchi, T.; Uemura, A.; Tamura, A. Spatial Memory Disturbance after Focal Cerebral Ischemia in Rats. J. Cereb. Blood Flow Metab. 1996, 16, 973–980. [Google Scholar] [CrossRef]
- Andersen, M.B.; Zimmer, J.; Sams-Dodd, F. Specific Behavioral Effects Related to Age and Cerebral Ischemia in Rats. Pharmacol. Biochem. Behav. 1999, 62, 673–682. [Google Scholar] [CrossRef]
- Ward, N.M.; Sharkey, J.; Marston, H.M.; Brown, V. Simple and choice reaction-time performance following occlusion of the anterior cerebral arteries in the rat. Exp. Brain Res. 1998, 123, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Diederich, K.; Strecker, J.-K.; Geng, B.; Hoppen, M.; Duning, T.; Schäbitz, W.-R.; Minnerup, J. Progressive Cognitive Deficits in a Mouse Model of Recurrent Photothrombotic Stroke. Stroke 2015, 46, 1127–1131. [Google Scholar] [CrossRef]
- Endepols, H.; Mertgens, H.; Backes, H.; Himmelreich, U.; Neumaier, B.; Graf, R.; Mies, G. Longitudinal assessment of infarct progression, brain metabolism and behavior following anterior cerebral artery occlusion in rats. J. Neurosci. Methods 2015, 253, 279–291. [Google Scholar] [CrossRef][Green Version]
- Fan, W.; Zhang, Y.; Li, X.; Xu, C. S-oxiracetam Facilitates Cognitive Restoration after Ischemic Stroke by Activating α7nAChR and the PI3K-Mediated Pathway. Neurochem. Res. 2021, 46, 888–904. [Google Scholar] [CrossRef]
- Sayed, M.A.; Eldahshan, W.; Abdelbary, M.; Pillai, B.; Althomali, W.; Johnson, M.H.; Arbab, A.S.; Ergul, A.; Fagan, S.C. Stroke promotes the development of brain atrophy and delayed cell death in hypertensive rats. Sci. Rep. 2020, 10, 20233. [Google Scholar] [CrossRef]
- Niedowicz, D.M.; Reeves, V.L.; Platt, T.L.; Kohler, K.; Beckett, T.L.; Powell, D.K.; Lee, T.L.; Sexton, T.R.; Song, E.S.; Brewer, L.D.; et al. Obesity and diabetes cause cognitive dysfunction in the absence of accelerated beta-amyloid deposition in a novel murine model of mixed or vascular dementia. Acta Neuropathol. Commun. 2014, 2, 64. [Google Scholar] [CrossRef]
- Zhang, T.; Pan, B.S.; Zhao, B.; Zhang, L.M.; Huang, Y.L.; Sun, F.Y. Exacerbation of poststroke dementia by type 2 diabetes is associated with synergistic increases of beta-secretase activation and beta-amyloid generation in rat brains. Neuroscience 2009, 161, 1045–1056. [Google Scholar] [CrossRef]
- Houlton, J.; Barwick, D.; Clarkson, A.N. Frontal cortex stroke-induced impairment in spatial working memory on the trial-unique nonmatching-to-location task in mice. Neurobiol. Learn. Mem. 2020, 177, 107355. [Google Scholar] [CrossRef] [PubMed]
- Houlton, J.; Zhou, L.Y.Y.; Barwick, D.; Gowing, E.K.; Clarkson, A.N. Stroke Induces a BDNF-Dependent Improvement in Cognitive Flexibility in Aged Mice. Neural Plast. 2019, 2019, 1460890. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Bezanilla, S.; Hood, R.J.; Collins-Praino, L.E.; Turner, R.J.; Walker, F.R.; Nilsson, M.; Ong, L.K. More than motor impairment: A spatiotemporal analysis of cognitive impairment and associated neuropathological changes following cortical photothrombotic stroke. J. Cereb. Blood Flow Metab. 2021, 41, 2439–2455. [Google Scholar] [CrossRef] [PubMed]
- Ermine, C.M.; Nithianantharajah, J.; O’Brien, K.; Kauhausen, J.A.; Frausin, S.; Oman, A.; Parsons, M.W.; Brait, V.H.; Brodtmann, A.; Thompson, L.H. Hemispheric cortical atrophy and chronic microglial activation following mild focal ischemic stroke in adult male rats. J. Neurosci. Res. 2021, 99, 3222–3237. [Google Scholar] [CrossRef]
- Sadigh-Eteghad, S.; Geranmayeh, M.H.; Majdi, A.; Salehpour, F.; Mahmoudi, J.; Farhoudi, M. Intranasal cerebrolysin improves cognitive function and structural synaptic plasticity in photothrombotic mouse model of medial prefrontal cortex ischemia. Neuropeptides 2018, 71, 61–69. [Google Scholar] [CrossRef]
- Ishii, D.; Osaki, H.; Yozu, A.; Ishibashi, K.; Kawamura, K.; Yamamoto, S.; Miyata, M.; Kohno, Y. Ipsilesional spatial bias after a focal cerebral infarction in the medial agranular cortex: A mouse model of unilateral spatial neglect. Behav. Brain Res. 2020, 401, 113097. [Google Scholar] [CrossRef]
- Déziel, R.A.; Ryan, C.L.; Tasker, R.A. Ischemic lesions localized to the medial prefrontal cortex produce selective deficits in measures of executive function in rats. Behav. Brain Res. 2015, 293, 54–61. [Google Scholar] [CrossRef]
- Song, M.-K.; Kim, E.-J.; Kim, J.-K.; Lee, S.-G. Effects of exercise timing and intensity on neuroplasticity in a rat model of cerebral infarction. Brain Res. Bull. 2020, 160, 50–55. [Google Scholar] [CrossRef]
- Song, M.-K.; Kim, E.-J.; Kim, J.-K.; Park, H.-K.; Lee, S.-G. Effect of regular swimming exercise to duration-intensity on neurocognitive function in cerebral infarction rat model. Neurol. Res. 2018, 41, 37–44. [Google Scholar] [CrossRef]
- Wang, X.; Chen, A.; Wu, H.; Ye, M.; Cheng, H.; Jiang, X.; Wang, X.; Zhang, X.; Wu, D.; Gu, X.; et al. Enriched environment improves post-stroke cognitive impairment in mice by potential regulation of acetylation homeostasis in cholinergic circuits. Brain Res. 2016, 1650, 232–242. [Google Scholar] [CrossRef]
- Rogers, D.C.; Hunter, A. Photothrombotic Lesions of the Rat Cortex Impair Acquisition of the Water Maze. Pharmacol. Biochem. Behav. 1997, 56, 747–754. [Google Scholar] [CrossRef]
- Seyedaghamiri, F.; Hosseini, L.; Kazmi, S.; Mahmoudi, J.; Shanehbandi, D.; Ebrahimi-Kalan, A.; Rahbarghazi, R.; Sadigh-Eteghad, S.; Farhoudi, M. Varenicline improves cognitive impairment in a mouse model of mPFC ischemia: The possible roles of inflammation, apoptosis, and synaptic factors. Brain Res. Bull. 2022, 181, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Ong, L.K.; Zhao, Z.; Kluge, M.; Walker, F.R.; Nilsson, M. Chronic stress exposure following photothrombotic stroke is associated with increased levels of Amyloid beta accumulation and altered oligomerisation at sites of thalamic secondary neurodegeneration in mice. J. Cereb. Blood Flow Metab. 2016, 37, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Vahid-Ansari, F.; Albert, P.R. Chronic Fluoxetine Induces Activity Changes in Recovery From Poststroke Anxiety, Depression, and Cognitive Impairment. Neurotherapeutics 2017, 15, 200–215. [Google Scholar] [CrossRef]
- Hosseini, S.M.; Pourbadie, H.G.; Sayyah, M.; Zibaii, M.I.; Naderi, N. Neuroprotective effect of monophosphoryl lipid A, a detoxified lipid A derivative, in photothrombotic model of unilateral selective hippocampal ischemia in rat. Behav. Brain Res. 2018, 347, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Faraji, J.; Ejaredar, M.; Metz, G.A.; Sutherland, R.J. Chronic stress prior to hippocampal stroke enhances post-stroke spatial deficits in the ziggurat task. Neurobiol. Learn. Mem. 2011, 95, 335–345. [Google Scholar] [CrossRef]
- El Amki, M.; Clavier, T.; Perzo, N.; Bernard, R.; Guichet, P.O.; Castel, H. Hypothalamic, thalamic and hippocampal lesions in the mouse MCAO model: Potential involvement of deep cerebral arteries? J. Neurosci. Methods 2015, 254, 80–85. [Google Scholar] [CrossRef]
- Finney, C.A.; Morris, M.J.; Westbrook, R.F.; Jones, N.M. Hippocampal silent infarct leads to subtle cognitive decline that is associated with inflammation and gliosis at twenty-four hours after injury in a rat model. Behav. Brain Res. 2020, 401, 113089. [Google Scholar] [CrossRef]
- Blasi, F.; Hérisson, F.; Wang, S.; Mao, J.; Ayata, C. Late-Onset Thermal Hypersensitivity after Focal Ischemic Thalamic Infarcts as a Model for Central Post-Stroke Pain in Rats. J. Cereb. Blood Flow Metab. 2015, 35, 1100–1103. [Google Scholar] [CrossRef]
- Cordova, C.A.; Jackson, D.; Langdon, K.D.; Hewlett, K.A.; Corbett, D. Impaired executive function following ischemic stroke in the rat medial prefrontal cortex. Behav. Brain Res. 2014, 258, 106–111. [Google Scholar] [CrossRef]
- Abdul, Y.; Jamil, S.; He, L.; Li, W.; Ergul, A. Endothelin-1 (ET-1) promotes a proinflammatory microglia phenotype in diabetic conditions. Can. J. Physiol. Pharmacol. 2020, 98, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Ueki, A.; Rosen, L.; Andbjer, B.; Agnati, L.F.; Hallstrom, A.; Goiny, M.; Tanganelli, S.; Ungerstedt, U.; Fuxe, K. Evidence for a preventive action of the vigilance-promoting drug modafinil against striatal ischemic injury induced by endothelin-1 in the rat. Exp. Brain Res. 1993, 96, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Kurosawa, N.; Cintra, A.; Hallström, Å.; Goiny, M.; Rosén, L.; Agnati, L.F.; Ungerstedt, U. Involvement of local ischemia in endothelin-1 induced lesions of the neostriatum of the anaesthetized rat. Exp. Brain Res. 1992, 88, 131–139. [Google Scholar] [CrossRef]
- Muñeton-Gomez, V.C.; Doncel-Pérez, E.; Fernandez, A.P.; Serrano, J.; Pozo-Rodrigálvarez, A.; Vellosillo-Huerta, L.; Taylor, J.S.; Cardona-Gomez, G.P.; Nieto-Sampedro, M.; Martínez-Murillo, R. Neural differentiation of transplanted neural stem cells in a rat model of striatal lacunar infarction: Light and electron microscopic observations. Front. Cell. Neurosci. 2012, 6, 30. [Google Scholar] [CrossRef]
- Martínez-Murillo, R.; Fernández, A.P.; Serrano, J.; Rodrigo, J.; Salas, E.; Mourelle, M.; Martínez, A. The nitric oxide donor LA 419 decreases brain damage in a focal ischemia model. Neurosci. Lett. 2007, 415, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Frost, S.B.; Barbay, S.; Mumert, M.L.; Stowe, A.; Nudo, R.J. An animal model of capsular infarct: Endothelin-1 injections in the rat. Behav. Brain Res. 2006, 169, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F.; Wei, Y.; Balkaya, M.; Tikka, S.; Mandeville, J.B.; Waeber, C.; Ayata, C.; Moskowitz, M.A. Recognition Memory Impairments After Subcortical White Matter Stroke in Mice. Stroke 2014, 45, 1468–1473. [Google Scholar] [CrossRef]
- Hainsworth, A.H.; Brittain, J.F.; Khatun, H. Pre-clinical models of human cerebral small vessel disease: Utility for clinical application. J. Neurol. Sci. 2012, 322, 237–240. [Google Scholar] [CrossRef]
- Amtul, Z.; Hill, D.J.; Arany, E.J.; Cechetto, D.F. Altered Insulin/Insulin-Like Growth Factor Signaling in a Comorbid Rat model of Ischemia and beta-Amyloid Toxicity. Sci. Rep. 2018, 8, 5136. [Google Scholar] [CrossRef]
- Amtul, Z.; Whitehead, S.N.; Keeley, R.J.; Bechberger, J.; Fisher, A.L.; McDonald, R.J.; Naus, C.C.; Munoz, D.G.; Cechetto, D.F. Comorbid rat model of ischemia and beta-amyloid toxicity: Striatal and cortical degeneration. Brain Pathol. 2015, 25, 24–32. [Google Scholar] [CrossRef]
- Whitehead, S.N.; Cheng, G.; Hachinski, V.C.; Cechetto, D.F. Progressive Increase in Infarct Size, Neuroinflammation, and Cognitive Deficits in the Presence of High Levels of Amyloid. Stroke 2007, 38, 3245–3250. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, S.; Cheng, G.L.; Hachinski, V.; Cechetto, D.F. Interaction between a rat model of cerebral ischemia and beta-amyloid toxicity-II. Effects of triflusal. Stroke 2005, 36, 1782–1789. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, S.N.; Hachinski, V.C.; Cechetto, D.F. Interaction between a rat model of cerebral ischemia and beta-amyloid toxicity-Inflammatory responses. Stroke 2005, 36, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Washida, K.; Hattori, Y.; Ihara, M. Animal Models of Chronic Cerebral Hypoperfusion: From Mouse to Primate. Int. J. Mol. Sci. 2019, 20, 6176. [Google Scholar] [CrossRef] [PubMed]
- Du, S.Q.; Wang, X.R.; Xiao, L.Y.; Tu, J.F.; Zhu, W.; He, T.; Liu, C.Z. Molecular Mechanisms of Vascular Dementia: What Can Be Learned from Animal Models of Chronic Cerebral Hypoperfusion? Mol. Neurobiol. 2017, 54, 3670–3682. [Google Scholar] [CrossRef]
- Bin Back, D.; Kwon, K.J.; Choi, D.-H.; Shin, C.Y.; Lee, J.; Han, S.-H.; Kim, H.Y. Chronic cerebral hypoperfusion induces post-stroke dementia following acute ischemic stroke in rats. J. Neuroinflamm. 2017, 14, 216. [Google Scholar] [CrossRef]
- Back, D.; Choi, B.-R.; Han, J.-S.; Kwon, K.; Choi, D.-H.; Shin, C.; Lee, J.; Kim, H. Characterization of Tauopathy in a Rat Model of Post-Stroke Dementia Combining Acute Infarct and Chronic Cerebral Hypoperfusion. Int. J. Mol. Sci. 2020, 21, 6929. [Google Scholar] [CrossRef]
- Liu, M.; Beckett, T.L.; Thomason, L.A.; Dorr, A.; Stefanovic, B.; McLaurin, J. Covert strokes prior to Alzheimer’s disease onset accelerate peri-lesional pathology but not cognitive deficits in an inducible APP mouse model. Brain Res. 2021, 1754, 147233. [Google Scholar] [CrossRef]
- James, M.L.; Warner, D.S.; Laskowitz, D.T. Preclinical Models of Intracerebral Hemorrhage: A Translational Perspective. Neurocrit. Care 2007, 9, 139–152. [Google Scholar] [CrossRef]
- Andaluz, N.; Zuccarello, M.; Wagner, K.R. Experimental animal models of intracerebral hemorrhage. Neurosurg. Clin. North Am. 2002, 13, 385–393. [Google Scholar] [CrossRef]
- Chen, Y.; Chang, J.; Wei, J.; Feng, M.; Wang, R. Assessing the Evolution of Intracranial Hematomas by using Animal Models: A Review of the Progress and the Challenges. Metab. Brain Dis. 2021, 36, 2205–2214. [Google Scholar] [CrossRef] [PubMed]
- Shi, E.; Shi, K.; Qiu, S.; Sheth, K.N.; Lawton, M.T.; Ducruet, A.F. Chronic inflammation, cognitive impairment, and distal brain region alteration following intracerebral hemorrhage. FASEB J. 2019, 33, 9616–9626. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Dong, S.; Zheng, Q.; Zhang, L.; Tan, X.; Zou, J.; Yan, B.; Chen, Y. FTY720 attenuates iron deposition and glial responses in improving delayed lesion and long-term outcomes of collagenase-induced intracerebral hemorrhage. Brain Res. 2019, 1718, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Hartman, R.; Lekic, T.; Rojas, H.; Tang, J.; Zhang, J.H. Assessing functional outcomes following intracerebral hemorrhage in rats. Brain Res. 2009, 1280, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Titova, E.; Ostrowski, R.P.; Zhang, J.H.; Tang, J. Experimental models of subarachnoid hemorrhage for studies of cerebral vasospasm. Neurol. Res. 2009, 31, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Turan, N.; Miller, B.A.; Heider, R.A.; Nadeem, M.; Sayeed, I.; Stein, D.G.; Pradilla, G. Neurobehavioral testing in subarachnoid hemorrhage: A review of methods and current findings in rodents. J. Cereb. Blood Flow Metab. 2016, 37, 3461–3474. [Google Scholar] [CrossRef]
- Tso, M.K.; Macdonald, R.L. Subarachnoid Hemorrhage: A Review of Experimental Studies on the Microcirculation and the Neurovascular Unit. Transl. Stroke Res. 2014, 5, 174–189. [Google Scholar] [CrossRef]
- Choi, B.-R.; Kim, D.-H.; Bin Back, D.; Kang, C.H.; Moon, W.-J.; Han, J.-S.; Choi, D.-H.; Kwon, K.J.; Shin, C.Y.; Kim, B.-R.; et al. Characterization of White Matter Injury in a Rat Model of Chronic Cerebral Hypoperfusion. Stroke 2016, 47, 542–547. [Google Scholar] [CrossRef]
- Choi, B.R.; Lee, S.R.; Han, J.S.; Woo, S.K.; Kim, K.M.; Choi, D.H.; Kwon, K.J.; Han, S.H.; Shin, C.Y.; Lee, J.; et al. Synergistic memory impairment through the interaction of chronic cerebral hypoperfusion and amlyloid toxicity in a rat model. Stroke 2011, 42, 2595–2604. [Google Scholar] [CrossRef]
- Norrving, B. Evolving Concept of Small Vessel Disease through Advanced Brain Imaging. J. Stroke 2015, 17, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wong, A.; Au, L.; Yang, J.; Wang, Z.; Leung, E.Y.; Chen, S.; Ho, C.L.; Mok, V.C. Influence of Amyloid-beta on Cognitive Decline After Stroke/Transient Ischemic Attack: Three-Year Longitudinal Study. Stroke 2015, 46, 3074–3080. [Google Scholar] [CrossRef] [PubMed]
- Nihashi, T.; Inao, S.; Kawai, T.; Sugimoto, T.; Niwa, M.; Hata, N.; Hayashi, S.; Yoshida, J.; Kajita, Y.; Kabeya, R. Expression and Distribution of Beta Amyloid Precursor Protein and Beta Amyloid Peptide in Reactive Astrocytes After Transient Middle Cerebral Artery Occlusion. Acta Neurochir. 2001, 143, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, M.; Makinen, P.; Peraniemi, S.; Sivenius, J.; van Groen, T.; Soininen, H.; Jolkkonen, J. Focal cerebral ischemia in rats alters APP processing and expression of Abeta peptide degrading enzymes in the thalamus. Neurobiol. Dis. 2009, 35, 103–113. [Google Scholar] [CrossRef]
- van Groen, T.; Puurunen, K.; Maki, H.M.; Sivenius, J.; Jolkkonen, J. Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke 2005, 36, 1551–1556. [Google Scholar] [CrossRef]
- Aam, S.; Einstad, M.S.; Munthe-Kaas, R.; Lydersen, S.; Ihle-Hansen, H.; Knapskog, A.-B.; Ellekjær, H.; Seljeseth, Y.; Saltvedt, I. Post-stroke Cognitive Impairment—Impact of Follow-Up Time and Stroke Subtype on Severity and Cognitive Profile: The Nor-COAST Study. Front. Neurol. 2020, 11, 699. [Google Scholar] [CrossRef]
- Potter, T.; Lioutas, V.-A.; Tano, M.; Pan, A.; Meeks, J.; Woo, D.; Seshadri, S.; Selim, M.; Vahidy, F. Cognitive Impairment After Intracerebral Hemorrhage: A Systematic Review of Current Evidence and Knowledge Gaps. Front. Neurol. 2021, 12, 71663. [Google Scholar] [CrossRef]
- Donnellan, C.; Werring, D. Cognitive impairment before and after intracerebral haemorrhage: A systematic review. Neurol. Sci. 2019, 41, 509–527. [Google Scholar] [CrossRef]
- Biffi, A.; Bailey, D.; Anderson, C.D.; Ayres, A.M.; Gurol, E.M.; Greenberg, S.M.; Rosand, J.; Viswanathan, A. Risk Factors Associated With Early vs Delayed Dementia After Intracerebral Hemorrhage. JAMA Neurol. 2016, 73, 969–976. [Google Scholar] [CrossRef]
- Pasi, M.; Sugita, L.; Xiong, L.; Charidimou, A.; Boulouis, G.; Pongpitakmetha, T.; Singh, S.; Kourkoulis, C.; Schwab, K.; Greenberg, S.M.; et al. Association of Cerebral Small Vessel Disease and Cognitive Decline After Intracerebral Hemorrhage. Neurology 2020, 96, e182–e192. [Google Scholar] [CrossRef]
- Gong, L.; Gu, Y.; Yu, Q.; Wang, H.; Zhu, X.; Dong, Q.; Xu, R.; Zhao, Y.; Liu, X. Prognostic Factors for Cognitive Recovery Beyond Early Poststroke Cognitive Impairment (PSCI): A Prospective Cohort Study of Spontaneous Intracerebral Hemorrhage. Front. Neurol. 2020, 11, 278. [Google Scholar] [CrossRef] [PubMed]
- Bahader, G.A.; Nash, K.M.; Almarghalani, D.A.; Alhadidi, Q.; McInerney, M.F.; Shah, Z.A. Type-I diabetes aggravates post-hemorrhagic stroke cognitive impairment by augmenting oxidative stress and neuroinflammation in mice. Neurochem. Int. 2021, 149, 105151. [Google Scholar] [CrossRef] [PubMed]
- Bahadar, G.A.; Shah, Z.A. Intracerebral Hemorrhage and Diabetes Mellitus: Blood-Brain Barrier Disruption, Pathophysiology and Cognitive Impairments. CNS Neurol. Disord. Drug Targets 2021, 20, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Charidimou, A.; Pasi, M.; Boulouis, G.; Pongpitakmetha, T.; Schirmer, M.D.; Singh, S.; Benson, E.; Gurol, E.M.; Rosand, J.; et al. Predictors for Late Post-Intracerebral Hemorrhage Dementia in Patients with Probable Cerebral Amyloid Angiopathy. J. Alzheimer’s Dis. 2019, 71, 435–442. [Google Scholar] [CrossRef]
- Langa, K.M.; Foster, N.L.; Larson, E.B. Mixed dementia: Emerging concepts and therapeutic implications. JAMA 2004, 292, 2901–2908. [Google Scholar] [CrossRef]
- O’Brien, J.T.; Thomas, A. Vascular dementia. Lancet 2015, 386, 1698–1706. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Schroder, E.; Walker, L.C.; Kessler, C. beta-Amyloid precursor protein and ss-amyloid peptide immunoreactivity in the rat brain after middle cerebral artery occlusion: Effect of age. Stroke 1998, 29, 2196–2202. [Google Scholar] [CrossRef]
- Ji, C.; Yu, X.; Xu, W.; Lenahan, C.; Tu, S.; Shao, A. The role of glymphatic system in the cerebral edema formation after ischemic stroke. Exp. Neurol. 2021, 340, 113685. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Xing, S.; Liang, Z.; Zeng, J. Secondary neurodegeneration in remote regions after focal cerebral infarction: A new target for stroke management? Stroke 2012, 43, 1700–1705. [Google Scholar] [CrossRef]
- Zhang, Y.; Xing, S.; Zhang, J.; Li, J.; Li, C.; Pei, Z. Reduction of beta-amyloid deposits by gamma-secretase inhibitor is associated with the attenuation of secondary damage in the ipsilateral thalamus and sensory functional improvement after focal cortical infarction in hypertensive rats. J. Cereb. Blood Flow Metab. 2011, 31, 572–579. [Google Scholar] [CrossRef]
- Loos, M.; Dihné, M.; Block, F. Tumor necrosis factor-α expression in areas of remote degeneration following middle cerebral artery occlusion of the rat. Neuroscience 2003, 122, 373–380. [Google Scholar] [CrossRef]
- Ermine, C.M.; Somaa, F.; Wang, T.-Y.; Kagan, B.J.; Parish, C.L.; Thompson, L.H. Long-Term Motor Deficit and Diffuse Cortical Atrophy Following Focal Cortical Ischemia in Athymic Rats. Front. Cell. Neurosci. 2019, 13, 552. [Google Scholar] [CrossRef] [PubMed]
- Doyle, K.P.; Buckwalter, M.S. Immunological mechanisms in poststroke dementia. Curr. Opin. Neurol. 2020, 33, 30–36. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Stroke Types | Rodent Models * | Experimental Cognitive Outcome Measure | Clinical PSD Subtypes * | Associated Clinical Conditions | References ** |
|---|---|---|---|---|---|
| Ischemic stroke | MCAO model | MWM, YMT, NOR, NSR, CCT [14] | Large hemispheric infarct dementia Multiple territorial infarct dementia | Intra- or extracranial atherosclerosis, or embolic source | [14,15,16,17,18,19,20,21,22,23,24,25,26,27] |
| MCAO model with comorbid conditions | MWM, YMT, NOR, NSR | PSD with comorbid conditions | Aging, hypertension, diabetes, or dyslipidemia | [11,17,18,21,23,28,29,30] | |
| Cortical stroke model | MWM, NOR, TUNL [31], VDT [32,33,34], WWW [35], 8ARM [36], SST [37] | Small cortical infarct dementia, cortical vascular dementia, strategic infarct dementia | Single embolism, or small cortical branch occlusion | [24,25,31,32,33,34,35,36,37,38,39,40,41,42,43,44] | |
| Hippocampal stroke model | MWM, YMT, NOR, PAT [45], RAWMT [45], ZT [46] | Strategic infarct dementia | Cognitive pathway involvements such as thalamus, hippocampus, corpus callosum, Papez circuit, or other sophisticated areas | [14,15,16,18,19,20,24,27,31,32,35,36,42,45,46,47,48,49] | |
| Lacunar infarct model | NOR, SMT, PWT [49], ASST [50] | PSD with single or multiple lacunar infarcts or with underlying small vessel disease | Arteriolosclerosis, hypertensive angiopathy, single or multiple lacunar status, or vascular dementia | [49,50,51,52,53,54,55,56,57,58,59,60,61,62,63] | |
| Combining BCCAo and MCAO model | MWM, NOR | PSD in patients with chronic cerebral hypoperfusion | Severe carotid artery stenosis, chronic heart failure, white matter hyperintensities, subcortical ischemic vascular dementia, or impaired glymphatic clearance | [64,65,66,67] | |
| Stroke model with underlying AD pathology | YMT, NBT, MBT [68] | PSD in patients with underlying AD pathology | Impaired amyloid metabolism, mild cognitive impairment, AD, medial temporal lobe atrophy, brain atrophy, or mixed dementia | [59,60,61,62,63,68] | |
| Hemorrhagic model | Intracerebral hemorrhage model | MWM, SMFT | PSD in patients with intracerebral hemorrhage | Hypertensive angiopathy, cerebral amyloid angiopathy, or cerebral microbleeds | [69,70,71,72,73,74] |
| Subarachnoid hemorrhage model | PSD in patients with subarachnoid hemorrhage | Aneurysmal rupture, arteriovenous malformation, or superficial cortical hemosiderosis | [75,76,77] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.Y.; Back, D.B.; Choi, B.-R.; Choi, D.-H.; Kwon, K.J. Rodent Models of Post-Stroke Dementia. Int. J. Mol. Sci. 2022, 23, 10750. https://doi.org/10.3390/ijms231810750
Kim HY, Back DB, Choi B-R, Choi D-H, Kwon KJ. Rodent Models of Post-Stroke Dementia. International Journal of Molecular Sciences. 2022; 23(18):10750. https://doi.org/10.3390/ijms231810750
Chicago/Turabian StyleKim, Hahn Young, Dong Bin Back, Bo-Ryoung Choi, Dong-Hee Choi, and Kyoung Ja Kwon. 2022. "Rodent Models of Post-Stroke Dementia" International Journal of Molecular Sciences 23, no. 18: 10750. https://doi.org/10.3390/ijms231810750
APA StyleKim, H. Y., Back, D. B., Choi, B.-R., Choi, D.-H., & Kwon, K. J. (2022). Rodent Models of Post-Stroke Dementia. International Journal of Molecular Sciences, 23(18), 10750. https://doi.org/10.3390/ijms231810750