

Regulation of the Soluble Amyloid Precursor Protein α (sAPPα) Levels by Acetylcholinesterase and Brain-Derived Neurotrophic Factor in Lung Cancer Cell Media

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
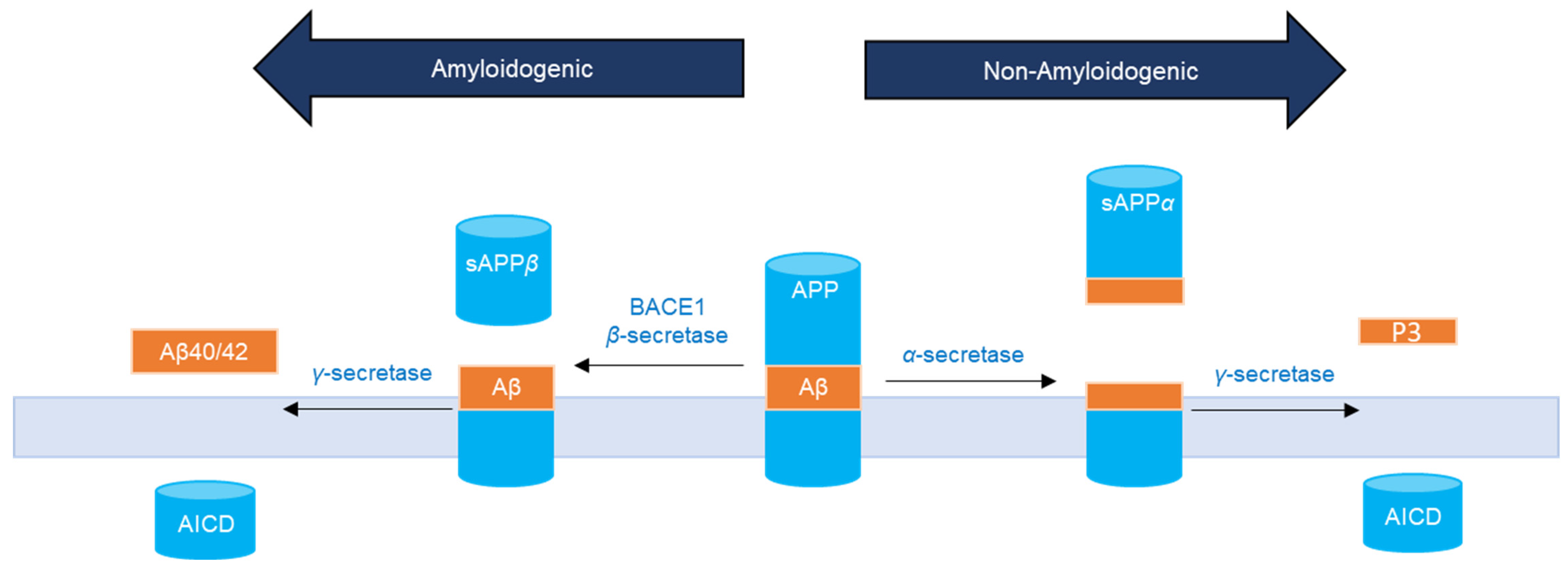
:1. Introduction
2. Results
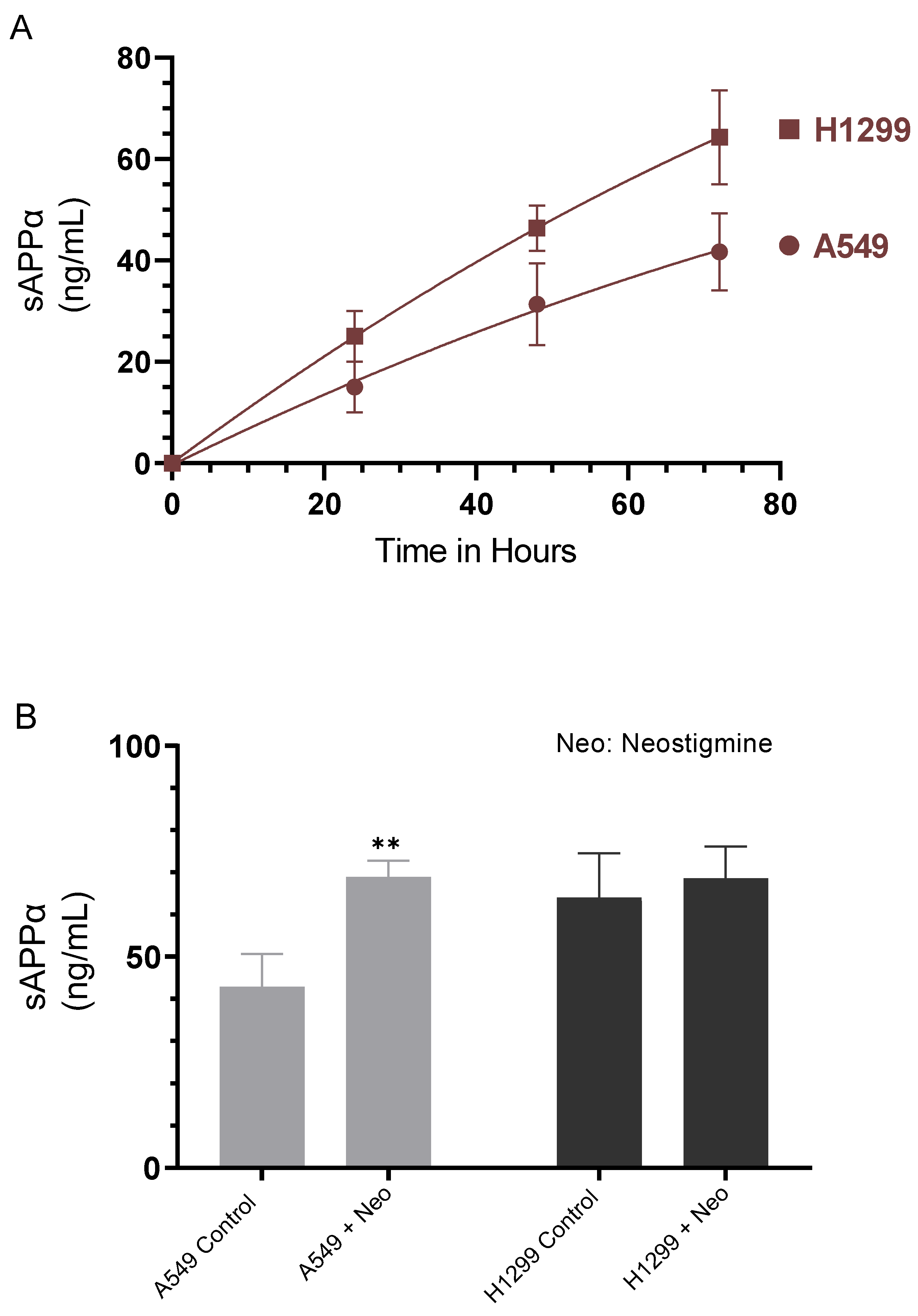
2.1. Cell Treatment with the AChE Inhibitor, Neostigmine, Led to Increased sAPPα Levels in A549 Cell Media That Were Comparable to Those Detected in the Media of H1299 cells
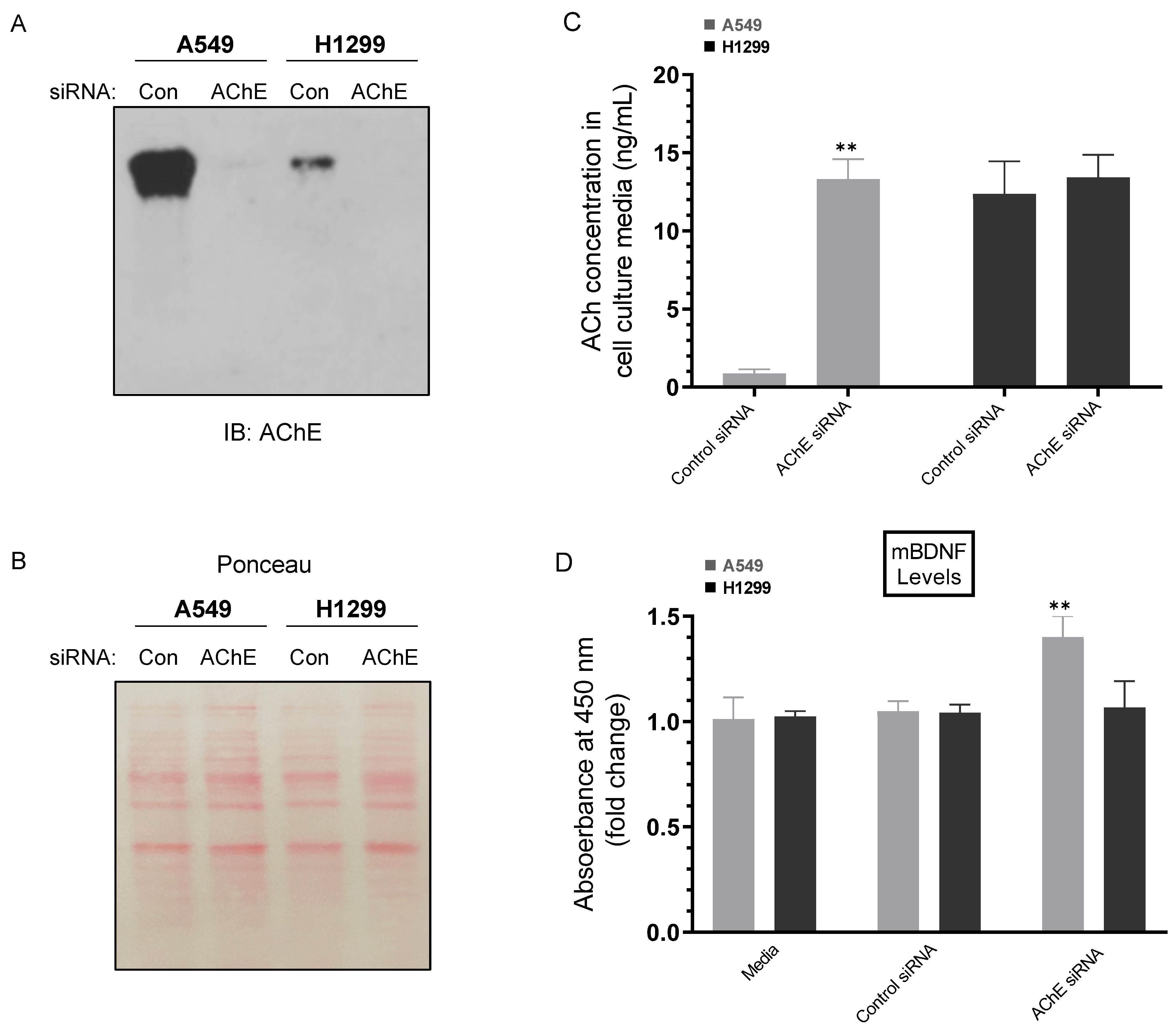
2.2. Knockdown of AChE Led to Increased ACh and mBDNF Levels in A549 Cell Media
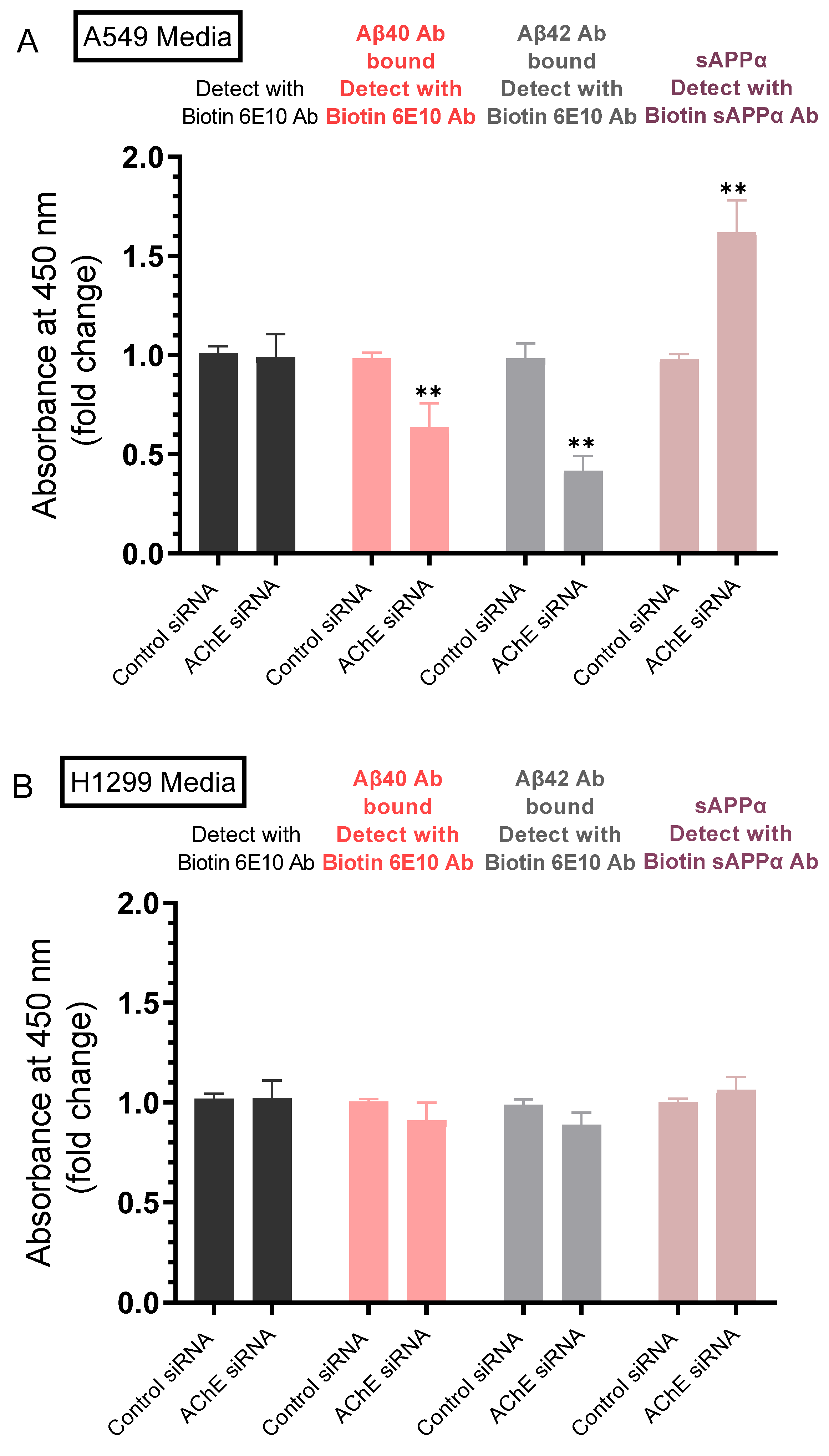
2.3. Lower Levels of Aβ40 and Aβ42 Were Observed in A549 Cell Media upon Transfection with AChE siRNA and Correlated with Higher sAPPα Levels
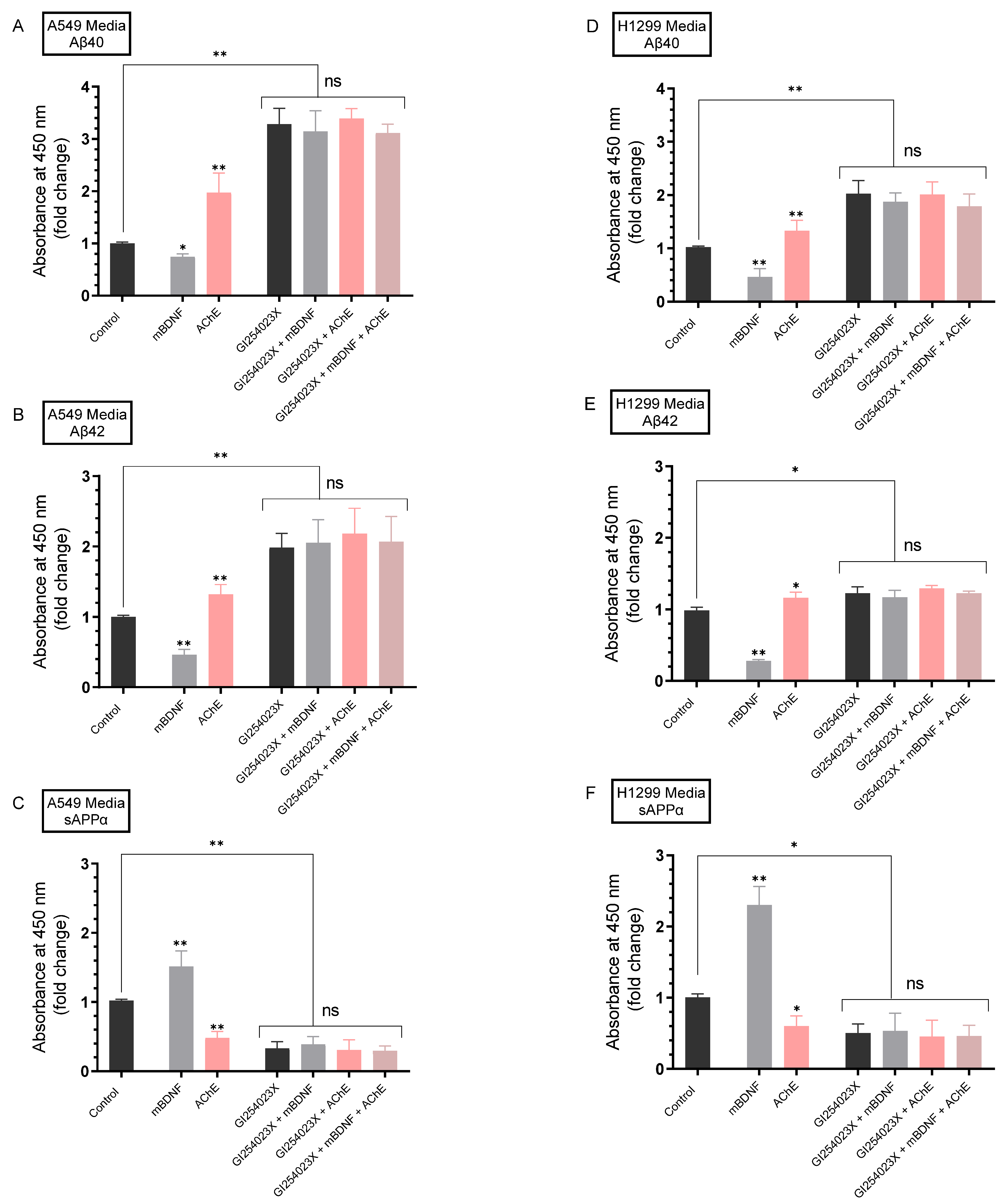
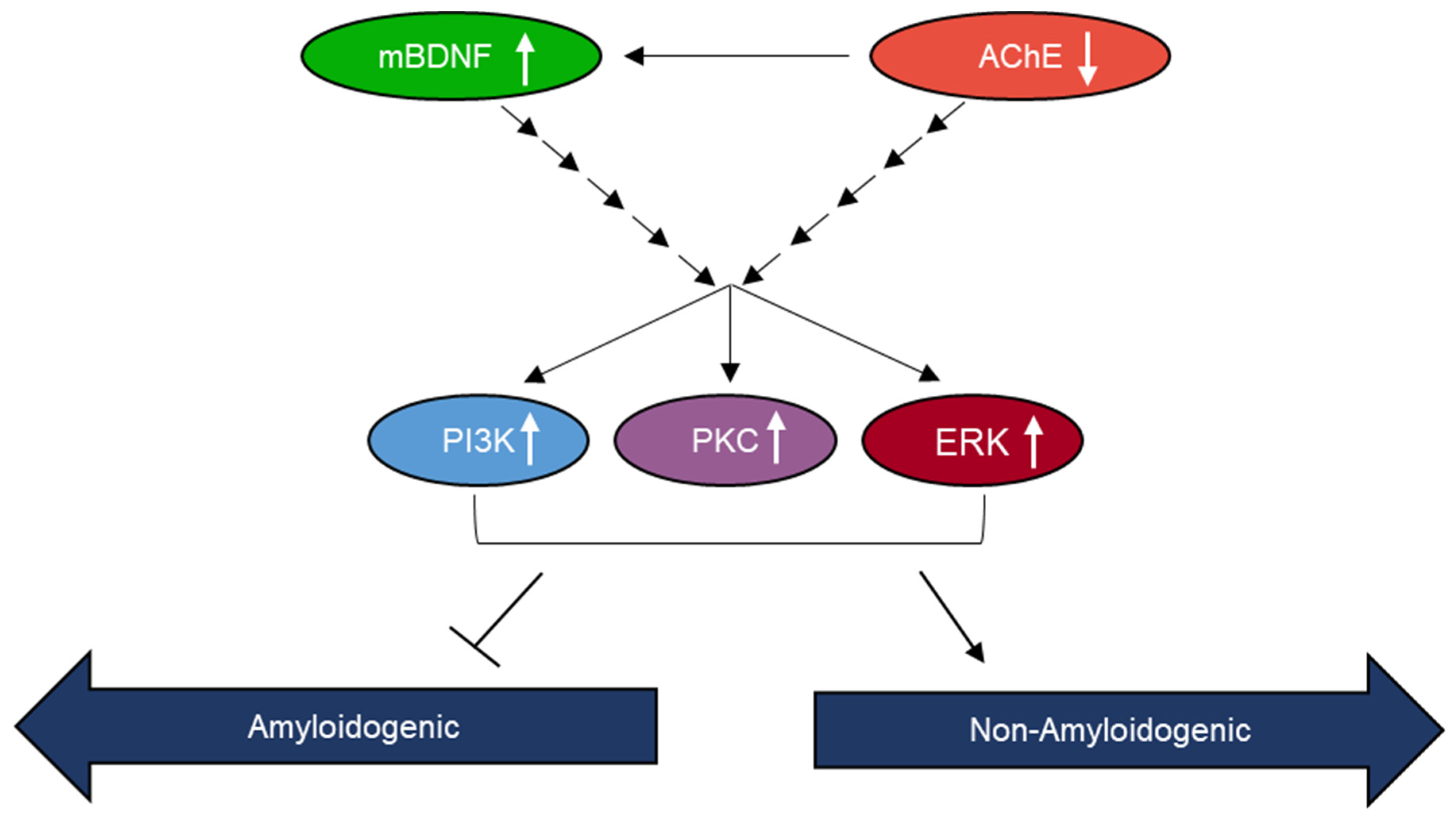
2.4. AChE and mBDNF Have Opposite Effects on the Levels of Aβ and sAPPα and Operate through a Mechanism Involving α-Secretase Activity
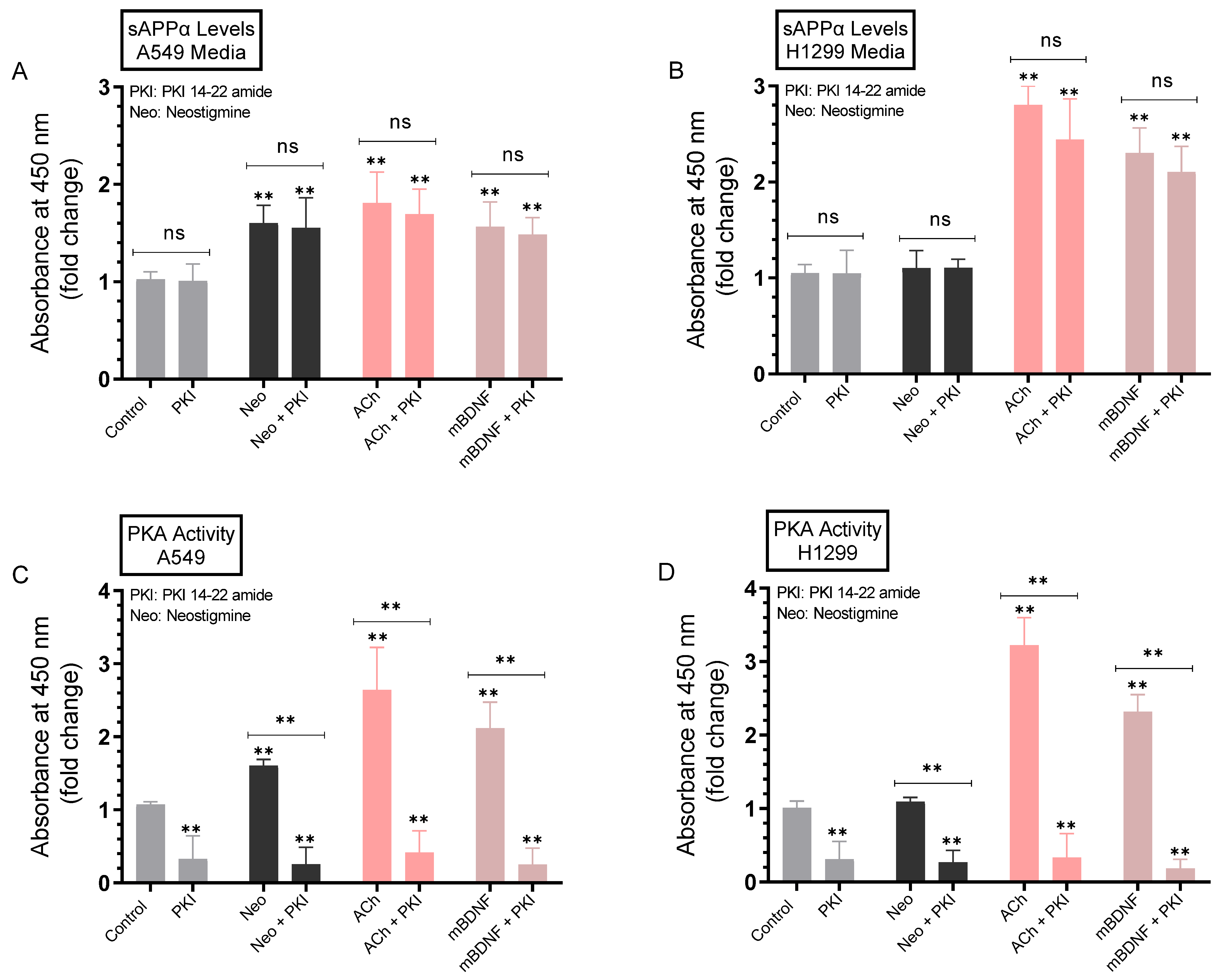
2.5. Minimal Effects Were Observed on the Levels of sAPPα in A549 and H1299 Cell Media upon Blocking Protein Kinase A (PKA) Activity
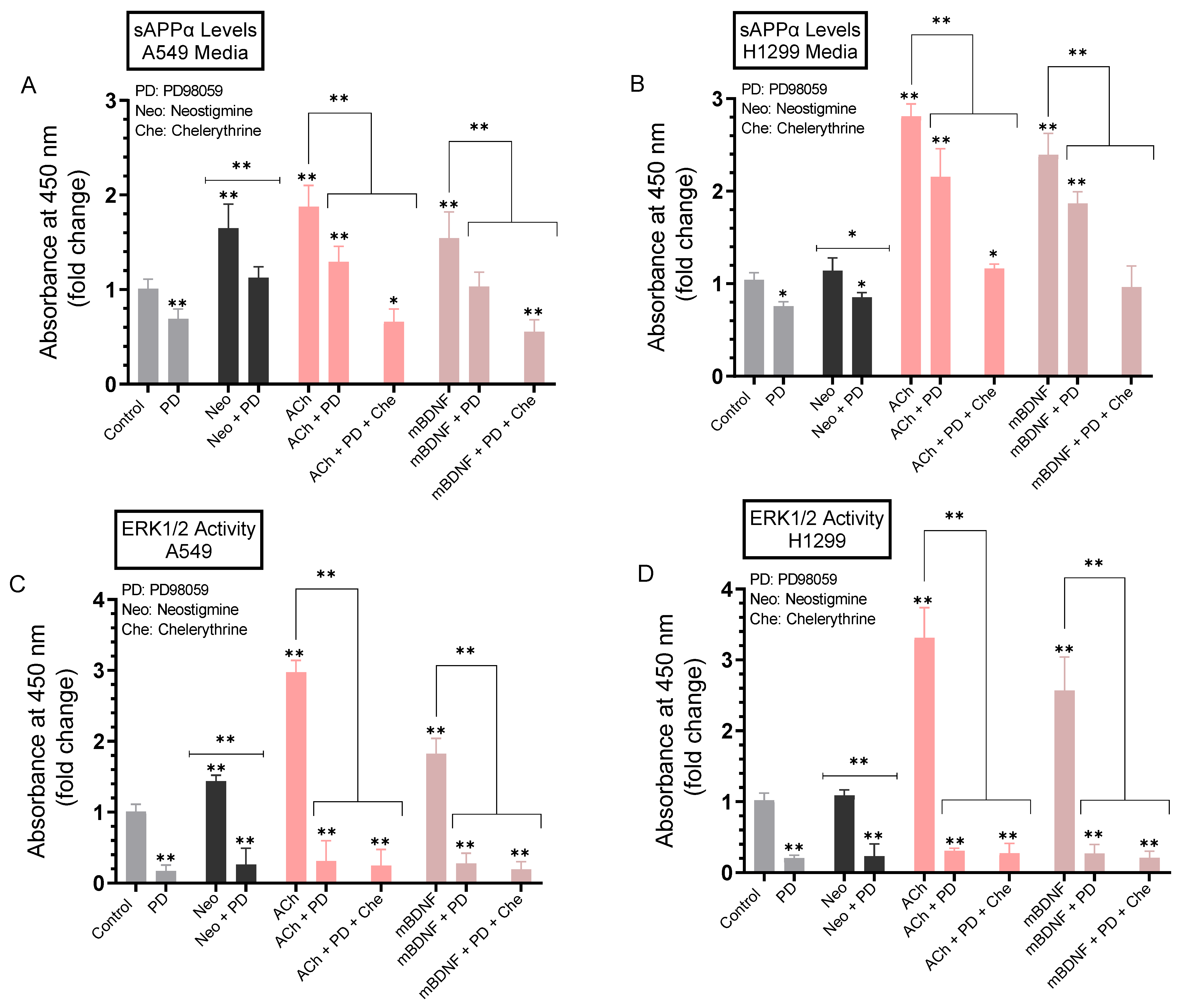
2.6. The Levels of sAPPα Decreased in the Media of A549 and H1299 Cells upon Treatment with the PKC Inhibitor, Chelerythrine
2.7. A more Pronounced Decrease in the Levels of sAPPα Was Observed in A549 and H1299 Cell Media When the Activities of ERK1/2 and PKC Were Blocked Using a Combination of PD98059 and Chelerythrine
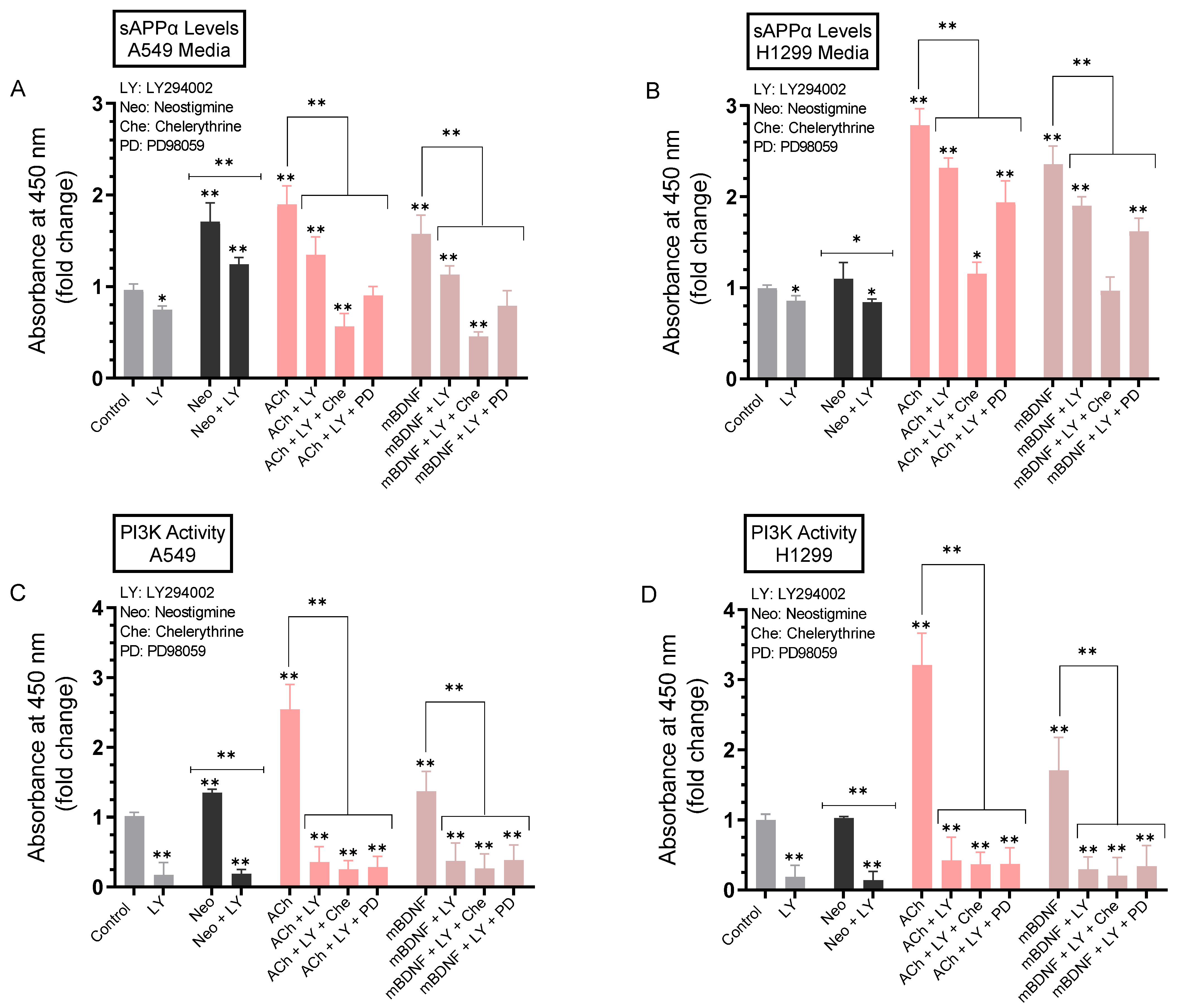
2.8. The Levels of sAPPα Were Decreased to a Greater Extent in the Media upon Co-Treatment of A549 and H1299 Cells with ACh or mBDNF, the PI3K Inhibitor, LY294002, and Either the ERK1/2 Inhibitor or PKC Inhibitor Compared to Cell Co-Treatment with LY294002 Alone
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. ELISA
4.4. Quantitation of ACh Concentrations
4.5. Quantitative Determination of sAPPα
4.6. Quantitation of Aβ
4.7. PKC Assay
4.8. PKA Assay
4.9. ERK Assay
4.10. PI3K Assay
4.11. Quantitation of mBDNF
4.12. Western Blotting
4.13. SiRNA Transfection
4.14. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Majd, S.; Power, J.; Majd, Z. Alzheimer’s Disease and Cancer: When Two Monsters Cannot Be Together. Front. Neurosci. 2019, 13, 155. [Google Scholar] [CrossRef]
- Ganguli, M. Cancer and Dementia: It’s Complicated. Alzheimer Dis. Assoc. Disord. 2015, 29, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Okereke, O.I.; Meadows, M.-E. More Evidence of an Inverse Association between Cancer and Alzheimer Disease. JAMA Netw. Open 2019, 2, e196167. [Google Scholar] [CrossRef]
- Lee, H.N.; Jeong, M.S.; Jang, S.B. Molecular Characteristics of Amyloid Precursor Protein (APP) and Its Effects in Cancer. Int. J. Mol. Sci. 2021, 22, 4999. [Google Scholar] [CrossRef] [PubMed]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef]
- Dawkins, E.; Small, D.H. Insights into the physiological function of the β-amyloid precursor protein: Beyond Alzheimer’s disease. J. Neurochem. 2014, 129, 756–769. [Google Scholar] [CrossRef]
- Pandey, P.; Sliker, B.; Peters, H.L.; Tuli, A.; Herskovitz, J.; Smits, K.; Purohit, A.; Singh, R.K.; Dong, J.; Batra, S.K.; et al. Amyloid precursor protein and amyloid precursor-like protein 2 in cancer. Oncotarget 2016, 7, 19430–19444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-Q.; Qu, D.-H.; Wang, K. Therapeutic approaches to Alzheimer’s disease through stimulating of non-amyloidogenic processing of amyloid precursor protein. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2389–2403. [Google Scholar] [PubMed]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. Neuromol. Med. 2010, 12, 1–12. [Google Scholar] [CrossRef]
- Tsang, J.Y.S.; Lee, M.A.; Chan, T.-H.; Li, J.; Ni, Y.-B.; Shao, Y.; Chan, S.-K.; Cheungc, S.-Y.; Lau, K.-F.; Tse, G.M.K. Proteolytic cleavage of amyloid precursor protein by ADAM10 mediates proliferation and migration in breast cancer. EBioMedicine 2018, 38, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Racchi, M.; Govoni, S. Rationalizing a pharmacological intervention on the amyloid precursor protein metabolism. Trends Pharmacol. Sci. 1999, 20, 418–423. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F. Alpha-secretase cleavage of the amyloid precursor protein: Proteolysis regulated by signaling pathways and protein trafficking. Curr. Alzheimer Res. 2012, 9, 165–177. [Google Scholar] [CrossRef]
- Leroy, B.; Girard, L.; Hollestelle, A.; Minna, J.D.; Gazdar, A.F.; Soussi, T. Analysis of TP53 Mutation Status in Human Cancer Cell Lines: A Reassessment. Hum. Mutat. 2014, 35, 756–765. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Moon, H.; Chun, K.-H.; Chang, Y.-S.; Hassan, K.; Ji, L.; Lotan, R.; Khuri, F.R.; Hong, W.K. Effects of Insulin-like Growth Factor Binding Protein-3 and Farnesyltransferase Inhibitor SCH66336 on Akt Expression and Apoptosis in Non–Small-Cell Lung Cancer Cells. J. Natl. Cancer Inst. 2004, 96, 1536–1548. [Google Scholar] [CrossRef]
- Dorandish, S.; Williams, A.; Atali, S.; Sendo, S.; Price, D.; Thompson, C.; Guthrie, J.; Heyl, D.; Evans, H.G. Regulation of amyloid-β levels by matrix metalloproteinase-2/9 (MMP2/9) in the media of lung cancer cells. Sci. Rep. 2021, 11, 9708. [Google Scholar] [CrossRef]
- Price, D.; Muterspaugh, R.; Clegg, B.; Williams, A.; Stephens, A.; Guthrie, J.; Heyl, D.; Guy Evans, H. IGFBP-3 Blocks Hyaluronan-CD44 Signaling, Leading to Increased Acetylcholinesterase Levels in A549 Cell Media and Apoptosis in a p53-Dependent Manner. Sci. Rep. 2020, 10, 5083–5099. [Google Scholar] [CrossRef]
- Small, D.H.; Michaelson, S.; Sberna, G. Non-classical actions of cholinesterases: Role in cellular differentiation, tumorigenesis and Alzheimer’s disease. Neurochem. Int. 1996, 28, 453–483. [Google Scholar] [CrossRef]
- Xi, H.-J.; Wu, R.-P.; Liu, J.-J.; Zhang, L.-J.; Li, Z.-S. Role of acetylcholinesterase in lung cancer. Thorac. Cancer 2015, 6, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic-Pasti, T.; Leskovac, A.; Momic, T.; Petrovic, S.; Vasic, V. Modulators of Acetylcholinesterase Activity: From Alzheimer’s Disease to Anti-Cancer Drugs. Curr. Med. Chem. 2017, 24, 3283–3309. [Google Scholar] [CrossRef]
- Zhang, X.-J.; Greenberg, D.S. Acetylcholinesterase Involvement in Apoptosis. Front. Mol. Neurosci. 2012, 5, 40. [Google Scholar] [CrossRef]
- Dorandish, S.; Atali, S.; Ray, R.; Al Khashali, H.; Coleman, K.-L.; Guthrie, J.; Heyl, D.; Evans, H.G. Differences in the Relative Abundance of ProBDNF and Mature BDNF in A549 and H1299 Human Lung Cancer Cell Media. Int. J. Mol. Sci. 2021, 22, 7059. [Google Scholar] [CrossRef]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef]
- Frisch, S.M.; Schaller, M.; Cieply, B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J. Cell. Sci. 2013, 126, 21–29. [Google Scholar] [CrossRef]
- De la Cruz-Morcillo, M.A.; Berger, J.; Sánchez-Prieto, R.; Saada, S.; Naves, T.; Guillaudeau, A.; Perraud, A.; Sindou, P.; Lacroix, A.; Descazeaud, A.; et al. p75 neurotrophin receptor and pro-BDNF promote cell survival and migration in clear cell renal cell carcinoma. Oncotarget 2016, 7, 34480–34497. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Hui, L.-P.; Li, C.-Y.; Gao, J.; Cui, Z.-S.; Qiu, X.-S. More expression of BDNF associates with lung squamous cell carcinoma and is critical to the proliferation and invasion of lung cancer cells. BMC Cancer 2016, 16, 171. [Google Scholar] [CrossRef]
- Meng, L.; Liu, B.; Ji, R.; Jiang, X.; Yan, X.; Xin, Y. Targeting the BDNF/TrkB pathway for the treatment of tumors. Oncol. Lett. 2019, 17, 2031–2039. [Google Scholar] [CrossRef] [Green Version]
- Hansel, D.E.; Rahman, A.; Wehner, S.; Herzog, V.; Yeo, C.J.; Maitra, A. Increased expression and processing of the Alzheimer amyloid precursor protein in pancreatic cancer may influence cellular proliferation. Cancer Res. 2003, 63, 7032–7037. [Google Scholar]
- Meng, J.Y.; Kataoka, H.; Itoh, H.; Koono, M. Amyloid beta protein precursor is involved in the growth of human colon carcinoma cell in vitro and in vivo. Int. J. Cancer 2001, 92, 31–39. [Google Scholar] [CrossRef]
- Saitoh, T.; Sundsmo, M.; Roch, J.M.; Kimura, N.; Cole, G.; Schubert, D.; Oltersdorf, T.; Schenk, D.B. Secreted form of amyloid beta protein precursor is involved in the growth regulation of fibroblasts. Cell 1989, 58, 615–622. [Google Scholar] [CrossRef]
- Fisher, A. Cholinergic modulation of amyloid precursor protein processing with emphasis on M1 muscarinic receptor: Perspectives and challenges in treatment of Alzheimer’s disease. J. Neurochem. 2012, 120 (Suppl. S1), 22–33. [Google Scholar] [CrossRef] [PubMed]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.-S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef]
- Mousavi, M.; Hellström-Lindahl, E. Nicotinic receptor agonists and antagonists increase sAPPalpha secretion and decrease Abeta levels in vitro. Neurochem. Int. 2009, 54, 237–244. [Google Scholar] [CrossRef]
- Mori, F.; Lai, C.C.; Fusi, F.; Giacobini, E. Cholinesterase inhibitors increase secretion of APPs in rat brain cortex. Neuroreport 1995, 6, 633–636. [Google Scholar] [CrossRef]
- Neely, G.A.; Sabir, S.; Kohli, A. Neostigmine. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2021. [Google Scholar]
- Song, P.; Sekhon, H.S.; Fu, X.W.; Maier, M.; Jia, Y.; Duan, J.; Proskosil, B.J.; Gravett, C.; Lindstrom, J.; Mark, G.P.; et al. Activated cholinergic signaling provides a target in squamous cell lung carcinoma. Cancer Res. 2008, 68, 4693–4700. [Google Scholar] [CrossRef]
- Lau, J.K.; Brown, K.C.; Dasgupta, P. Measurement of Acetylcholine from Cell Lines. Bio-Protocol 2013, 3, e1007. [Google Scholar] [CrossRef]
- Friedman, J.R.; Richbart, S.D.; Merritt, J.C.; Brown, K.C.; Nolan, N.A.; Akers, A.T.; Lau, J.K.; Robateau, Z.R.; Miles, S.L.; Dasgupta, P. Acetylcholine signaling system in progression of lung cancers. Pharmacol. Ther. 2019, 194, 222–254. [Google Scholar] [CrossRef]
- Zanini, D.; Schmatz, R.; Pelinson, L.P.; Pimentel, V.C.; da Costa, P.; Cardoso, A.M.; Martins, C.C.; Schetinger, C.C.; Baldissareli, J.; do Carmo Araújo, M.; et al. Ectoenzymes and cholinesterase activity and biomarkers of oxidative stress in patients with lung cancer. Mol. Cell. Biochem. 2013, 374, 137–148. [Google Scholar] [CrossRef]
- Lu, L.; Zhang, X.; Zhang, B.; Wu, J.; Zhang, X. Synaptic acetylcholinesterase targeted by microRNA-212 functions as a tumor suppressor in non-small cell lung cancer. Int. J. Biochem. Cell Biol. 2013, 45, 2530–2540. [Google Scholar] [CrossRef] [PubMed]
- Martínez-López de Castro, A.; Nieto-Cerón, S.; Aurelio, P.-C.; Galbis-Martínez, L.; Latour-Pérez, J.; Torres-Lanzas, J.; Tovar-Zapata, I.; Martínez-Hernández, P.; Rodríguez-López, J.N.; Cabezas-Herrera, J. Cancer-associated differences in acetylcholinesterase activity in bronchial aspirates from patients with lung cancer. Clin. Sci. 2008, 115, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Yang, L.; Zhao, Q.; Caen, J.P.; He, H.Y.; Jin, Q.H.; Guo, L.H.; Alemany, M.; Zhang, L.Y.; Shi, Y.F. Induction of acetylcholinesterase expression during apoptosis in various cell types. Cell Death Differ. 2002, 9, 790–800. [Google Scholar] [CrossRef]
- Price, D.; Dorandish, S.; Williams, A.; Iwaniec, B.; Stephens, A.; Marshall, K.; Guthrie, J.; Heyl, D.; Evans, H.G. Humanin Blocks the Aggregation of Amyloid-β Induced by Acetylcholinesterase, an Effect Abolished in the Presence of IGFBP-3. Biochemistry 2020, 59, 1981–2002. [Google Scholar] [CrossRef]
- Atali, S.; Dorandish, S.; Devos, J.; Williams, A.; Price, D.; Taylor, J.; Guthrie, J.; Heyl, D.; Evans, H.G. Interaction of amyloid beta with humanin and acetylcholinesterase is modulated by ATP. FEBS Open Bio 2020, 10, 2805–2823. [Google Scholar] [CrossRef]
- Leyhe, T.; Stransky, E.; Eschweiler, G.W.; Buchkremer, G.; Laske, C. Increase of BDNF serum concentration during donepezil treatment of patients with early Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 2008, 258, 124–128. [Google Scholar] [CrossRef]
- Li, C.; Wang, T.; Jiang, N.; Yu, P.; Du, Y.; Ren, R.; Fu, F. Steady and fluctuant methods of inhibition of acetylcholinesterase differentially regulate neurotrophic factors in the hippocampus of juvenile mice. Exp. Ther. Med. 2012, 3, 269–272. [Google Scholar] [CrossRef]
- Lukiw, W.J. Amyloid beta (Aβ) peptide modulators and other current treatment strategies for Alzheimer’s disease (AD). Expert Opin. Emerg. Drugs 2012, 17, 43–60. [Google Scholar] [CrossRef]
- Hunter, S.; Brayne, C. Do anti-amyloid beta protein antibody cross reactivities confound Alzheimer disease research? J. Negat. Results Biomed. 2017, 16, 1. [Google Scholar] [CrossRef]
- Baghallab, I.; Reyes-Ruiz, J.M.; Abulnaja, K.; Huwait, E.; Glabe, C. Epitomic Characterization of the Specificity of the Anti-Amyloid Aβ Monoclonal Antibodies 6E10 and 4G8. J. Alzheimers Dis. 2018, 66, 1235–1244. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, M.; Kandimalla, K.K.; Wengenack, T.M.; Howell, K.G.; Poduslo, J.F. Surface plasmon resonance binding kinetics of Alzheimer’s disease amyloid beta peptide-capturing and plaque-binding monoclonal antibodies. Biochemistry 2009, 48, 10405–10415. [Google Scholar] [CrossRef]
- Miller, M.A.; Sullivan, R.J.; Lauffenburger, D.A. Molecular Pathways: Receptor Ectodomain Shedding in Treatment, Resistance, Monitoring of Cancer. Clin. Cancer Res. 2017, 23, 623–629. [Google Scholar] [CrossRef]
- Asai, M.; Hattori, C.; Szabó, B.; Sasagawa, N.; Maruyama, K.; Tanuma, S.; Ishiura, S. Putative function of ADAM9, ADAM10, ADAM17 as APP alpha-secretase. Biochem. Biophys. Res. Commun. 2003, 301, 231–235. [Google Scholar] [CrossRef]
- Pruessmeyer, J.; Ludwig, A. The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin. Cell Dev. Biol. 2009, 20, 164–174. [Google Scholar] [CrossRef]
- Wetzel, S.; Seipold, L.; Saftig, P. The metalloproteinase ADAM10: A useful therapeutic target? Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2071–2081. [Google Scholar] [CrossRef]
- Kuhn, P.-H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Rossner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef]
- Yoneyama, T.; Gorry, M.; Sobo-Vujanovic, A.; Lin, Y.; Vujanovic, L.; Gaither-Davis, A.; Moss, M.L.; Miller, M.A.; Griffith, L.G.; Lauffenburger, D.A.; et al. ADAM10 Sheddase Activity is a Potential Lung-Cancer Biomarker. J. Cancer 2018, 9, 2559–2570. [Google Scholar] [CrossRef]
- Costantini, C.; Weindruch, R.; Della Valle, G.; Puglielli, L. A TrkA-to-p75NTR molecular switch activates amyloid beta-peptide generation during aging. Biochem. J. 2005, 391, 59–67. [Google Scholar] [CrossRef]
- Triaca, V.; Calissano, P. Impairment of the nerve growth factor pathway driving amyloid accumulation in cholinergic neurons: The incipit of the Alzheimer’s disease story? Neural Regen. Res. 2016, 11, 1553–1556. [Google Scholar] [CrossRef]
- Nigam, S.M.; Xu, S.; Kritikou, J.S.; Marosi, K.; Brodin, L.; Mattson, M.P. Exercise and BDNF reduce Aβ production by enhancing α-secretase processing of APP. J. Neurochem. 2017, 142, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Kolisnyk, B.; Al-Onaizi, M.; Soreq, L.; Barbash, S.; Bekenstein, U.; Haberman, N.; Hanin, G.; Kish, M.T.; Souza da Silva, J.; Fahnestock, M.; et al. Cholinergic Surveillance over Hippocampal RNA Metabolism and Alzheimer’s-Like Pathology. Cereb. Cortex 2017, 27, 3553–3567. [Google Scholar] [CrossRef]
- Kojro, E.; Postina, R.; Buro, C.; Meiringer, C.; Gehrig-Burger, K.; Fahrenholz, F. The neuropeptide PACAP promotes the alpha-secretase pathway for processing the Alzheimer amyloid precursor protein. FASEB J. 2006, 20, 512–514. [Google Scholar] [CrossRef]
- Sobol, A.; Galluzzo, P.; Weber, M.J.; Alani, S.; Bocchetta, M. Depletion of Amyloid Precursor Protein (APP) causes G0 arrest in non-small cell lung cancer (NSCLC) cells. J. Cell Physiol. 2015, 230, 1332–1341. [Google Scholar] [CrossRef]
- Lim, S.; Yoo, B.K.; Kim, H.-S.; Gilmore, H.L.; Lee, Y.; Lee, H.; Kim, S.-J.; Letterio, J.; Lee, H. Amyloid-β precursor protein promotes cell proliferation and motility of advanced breast cancer. BMC Cancer 2014, 14, 928. [Google Scholar] [CrossRef]
- Grossberg, G.T. Cholinesterase Inhibitors for the Treatment of Alzheimer’s Disease. Curr. Ther. Res. Clin. Exp. 2003, 64, 216–235. [Google Scholar] [CrossRef]
- Racchi, M.; Sironi, M.; Caprera, A.; König, G.; Govoni, S. Short- and long-term effect of acetylcholinesterase inhibition on the expression and metabolism of the amyloid precursor protein. Mol. Psychiatry 2001, 6, 520–528. [Google Scholar] [CrossRef]
- Soreq, H.; Seidman, S. Acetylcholinesterase—New roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar] [CrossRef]
- Spindel, E.R. Cholinergic Targets in Lung Cancer. Curr. Pharm. Des. 2016, 22, 2152–2159. [Google Scholar] [CrossRef]
- Song, P.; Sekhon, H.S.; Proskocil, B.; Blusztajn, J.K.; Mark, G.P.; Spindel, E.R. Synthesis of acetylcholine by lung cancer. Life Sci. 2003, 72, 2159–2168. [Google Scholar] [CrossRef]
- Cheng, K.; Samimi, R.; Xie, G.; Shant, J.; Drachenberg, C.; Wade, M.; Davis, R.J.; Nomikos, G.; Raufman, J.-P. Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G591–G597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Xia, H.; Tang, Q.; Xu, H.; Wei, G.; Chen, Y.; Dai, X.; Gong, Q.; Bi, F. Acetylcholine acts through M3 muscarinic receptor to activate the EGFR signaling and promotes gastric cancer cell proliferation. Sci. Rep. 2017, 7, 40802. [Google Scholar] [CrossRef] [PubMed]
- Egleton, R.D.; Brown, K.C.; Dasgupta, P. Nicotinic acetylcholine receptors in cancer: Multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol. Sci. 2008, 29, 151–158. [Google Scholar] [CrossRef]
- Russo, P.; Cardinale, A.; Margaritora, S.; Cesario, A. Nicotinic receptor and tobacco-related cancer. Life Sci. 2012, 91, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.A.; Kawashima, K.; Kirkpatrick, C.J.; Meurs, H.; Wessler, I. The non-neuronal cholinergic system: Basic science, therapeutic implications and new perspectives. Life Sci. 2012, 91, 969–972. [Google Scholar] [CrossRef]
- Trombino, S.; Bisio, A.; Catassi, A.; Cesario, A.; Falugi, C.; Russo, P. Role of the non-neuronal human cholinergic system in lung cancer and mesothelioma: Possibility of new therapeutic strategies. Curr. Med. Chem. Anticancer Agents 2004, 4, 535–542. [Google Scholar] [CrossRef]
- Kotani, S.; Yamauchi, T.; Teramoto, T.; Ogura, H. Pharmacological evidence of cholinergic involvement in adult hippocampal neurogenesis in rats. Neuroscience 2006, 142, 505–514. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, L.; Yan, H.; Wang, Y.; Tang, X. Effects of huperzine A on memory deficits and neurotrophic factors production after transient cerebral ischemia and reperfusion in mice. Pharmacol. Biochem. Behav. 2006, 83, 603–611. [Google Scholar] [CrossRef]
- Rossner, S.; Ueberham, U.; Schliebs, R.; Perez-Polo, J.R.; Bigl, V. The regulation of amyloid precursor protein metabolism by cholinergic mechanisms and neurotrophin receptor signaling. Prog. Neurobiol. 1998, 56, 541–569. [Google Scholar] [CrossRef]
- Habib, A.; Sawmiller, D.; Tan, J. Restoring sAPPα functions as a potential treatment for Alzheimer’s disease. J. Neurosci. Res. 2017, 95, 973–991. [Google Scholar] [CrossRef]
- Hung, A.Y.; Haass, C.; Nitsch, R.M.; Qiu, W.Q.; Citron, M.; Wurtman, R.J.; Growdon, J.H.; Selkoe, D.J. Activation of protein kinase C inhibits cellular production of the amyloid beta-protein. J. Biol. Chem. 1993, 268, 22959–22962. [Google Scholar] [CrossRef]
- Takagi, K.; Ito, S.; Miyazaki, T.; Miki, Y.; Shibahara, Y.; Ishida, T.; Watanabe, M.; Inoue, S.; Sasano, H.; Suzuki, T. Amyloid precursor protein in human breast cancer: An androgen-induced gene associated with cell proliferation. Cancer Sci. 2013, 104, 1532–1538. [Google Scholar] [CrossRef]
- Matrone, C.; Ciotti, M.T.; Mercanti, D.; Marolda, R.; Calissano, P. NGF and BDNF signaling control amyloidogenic route and Abeta production in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 13139–13144. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.S.; West, K.A.; Blumberg, P.M.; Dennis, P.A. Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKCdelta promotes cellular survival and chemotherapeutic resistance. Cancer Res. 2003, 63, 780–786. [Google Scholar] [PubMed]
- Cisse, M.; Braun, U.; Leitges, M.; Fisher, A.; Pages, G.; Checler, F.; Vincent, B. ERK1-independent α-secretase cut of β-amyloid precursor protein via M1 muscarinic receptors and PKCα/ε. Mol. Cell Neurosci. 2011, 47, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Gardoni, F.; Di Luca, M. Molecular rationale for the pharmacological treatment of Alzheimer’s disease. Drugs Aging 2005, 22 (Suppl. S1), 27–37. [Google Scholar] [CrossRef]
- Muterspaugh, R.; Price, D.; Esckilsen, D.; McEachern, S.; Guthrie, J.; Heyl, D.; Evans, H.G. Interaction of Insulin-Like Growth Factor-Binding Protein 3 with Hyaluronan and Its Regulation by Humanin and CD44. Biochemistry 2018, 57, 5726–5737. [Google Scholar] [CrossRef]
- Dorandish, S.; Devos, J.; Clegg, B.; Price, D.; Muterspaugh, R.; Guthrie, J.; Heyl, D.L.; Evans, H.G. Biochemical determinants of the IGFBP-3-hyaluronan interaction. FEBS Open Bio 2020, 10, 1668–1684. [Google Scholar] [CrossRef]
- Heyl, D.L.; Iwaniec, B.; Esckilsen, D.; Price, D.; Guttikonda, P.; Cooper, J.; Lombardi, J.; Milletti, M.; Evans, H.G. Using Small Peptide Segments of Amyloid-β and Humanin to Examine their Physical Interactions. Protein Pept. Lett. 2019, 26, 502–511. [Google Scholar] [CrossRef]
- LeVine, H. Alzheimer’s beta-peptide oligomer formation at physiologic concentrations. Anal. Biochem. 2004, 335, 81–90. [Google Scholar] [CrossRef]
- Izzo, N.J.; Staniszewski, A.; To, L.; Fa, M.; Teich, A.F.; Saeed, F.; Wostein, H.; Walko, T.; Vaswani, A.; Wardius, M.; et al. Alzheimer’s Therapeutics Targeting Amyloid Beta 1–42 Oligomers I: Abeta 42 Oligomer Binding to Specific Neuronal Receptors Is Displaced by Drug Candidates that Improve Cognitive Deficits. PLoS ONE 2014, 9, e111898. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Lindholm, K.; Yang, L.-B.; Yue, X.; Citron, M.; Yan, R.; Beach, T.; Sue, L.; Sabbagh, M.; Cai, H.; et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2004, 101, 3632–3637. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zhang, C.; Chen, Y.; Zhou, T. Upregulation of Acetylcholinesterase Mediated by p53 Contributes to Cisplatin-Induced Apoptosis in Human Breast Cancer Cell. J. Cancer 2015, 6, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Khashali, H.; Ray, R.; Coleman, K.-L.; Atali, S.; Haddad, B.; Wareham, J.; Guthrie, J.; Heyl, D.; Evans, H.G. Regulation of the Soluble Amyloid Precursor Protein α (sAPPα) Levels by Acetylcholinesterase and Brain-Derived Neurotrophic Factor in Lung Cancer Cell Media. Int. J. Mol. Sci. 2022, 23, 10746. https://doi.org/10.3390/ijms231810746
Al Khashali H, Ray R, Coleman K-L, Atali S, Haddad B, Wareham J, Guthrie J, Heyl D, Evans HG. Regulation of the Soluble Amyloid Precursor Protein α (sAPPα) Levels by Acetylcholinesterase and Brain-Derived Neurotrophic Factor in Lung Cancer Cell Media. International Journal of Molecular Sciences. 2022; 23(18):10746. https://doi.org/10.3390/ijms231810746
Chicago/Turabian StyleAl Khashali, Hind, Ravel Ray, Kai-Ling Coleman, Sarah Atali, Ben Haddad, Jadziah Wareham, Jeffrey Guthrie, Deborah Heyl, and Hedeel Guy Evans. 2022. "Regulation of the Soluble Amyloid Precursor Protein α (sAPPα) Levels by Acetylcholinesterase and Brain-Derived Neurotrophic Factor in Lung Cancer Cell Media" International Journal of Molecular Sciences 23, no. 18: 10746. https://doi.org/10.3390/ijms231810746
APA StyleAl Khashali, H., Ray, R., Coleman, K.-L., Atali, S., Haddad, B., Wareham, J., Guthrie, J., Heyl, D., & Evans, H. G. (2022). Regulation of the Soluble Amyloid Precursor Protein α (sAPPα) Levels by Acetylcholinesterase and Brain-Derived Neurotrophic Factor in Lung Cancer Cell Media. International Journal of Molecular Sciences, 23(18), 10746. https://doi.org/10.3390/ijms231810746