
High-Content Drug Discovery Targeting Molecular Bladder Cancer Subtypes

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
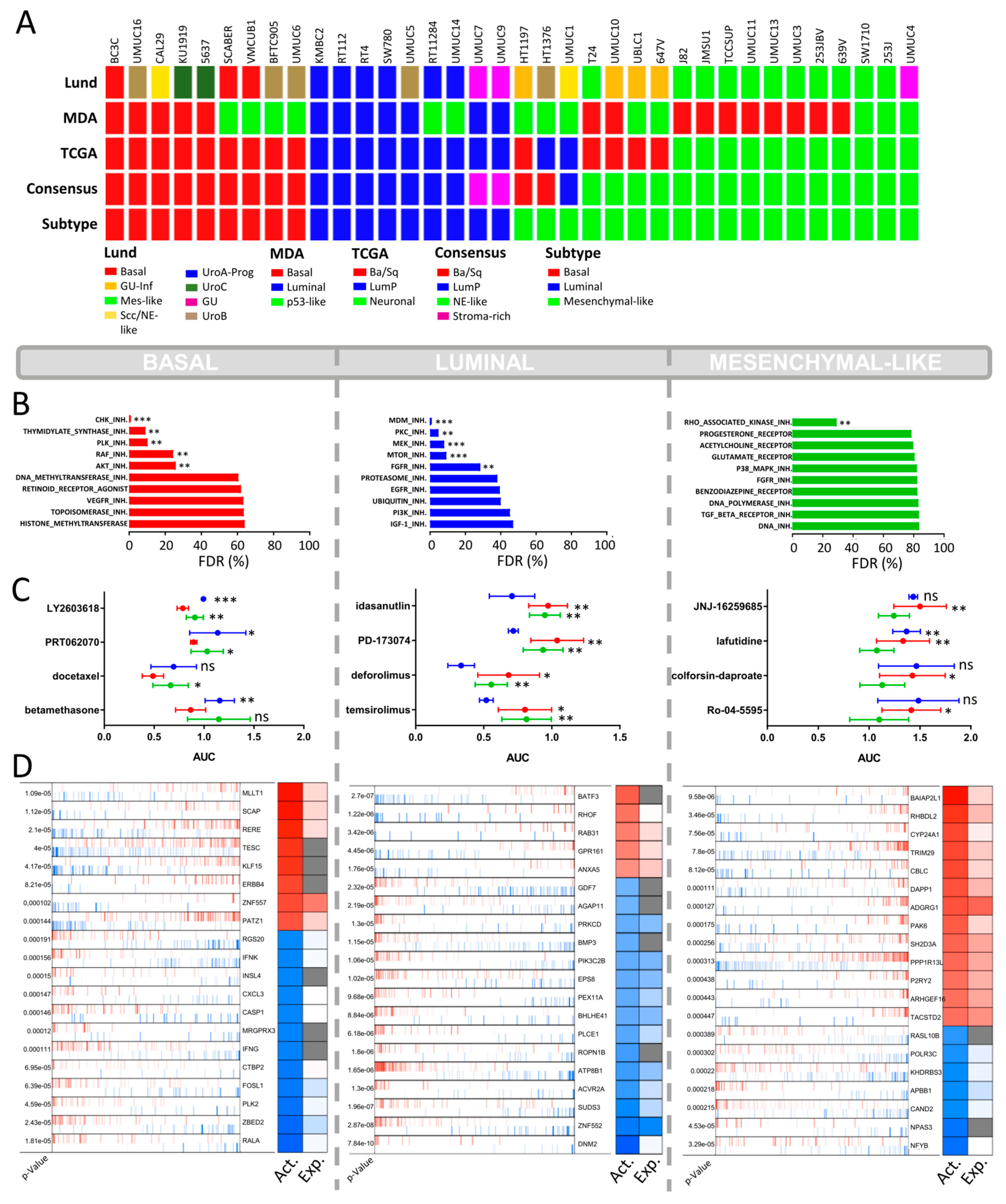
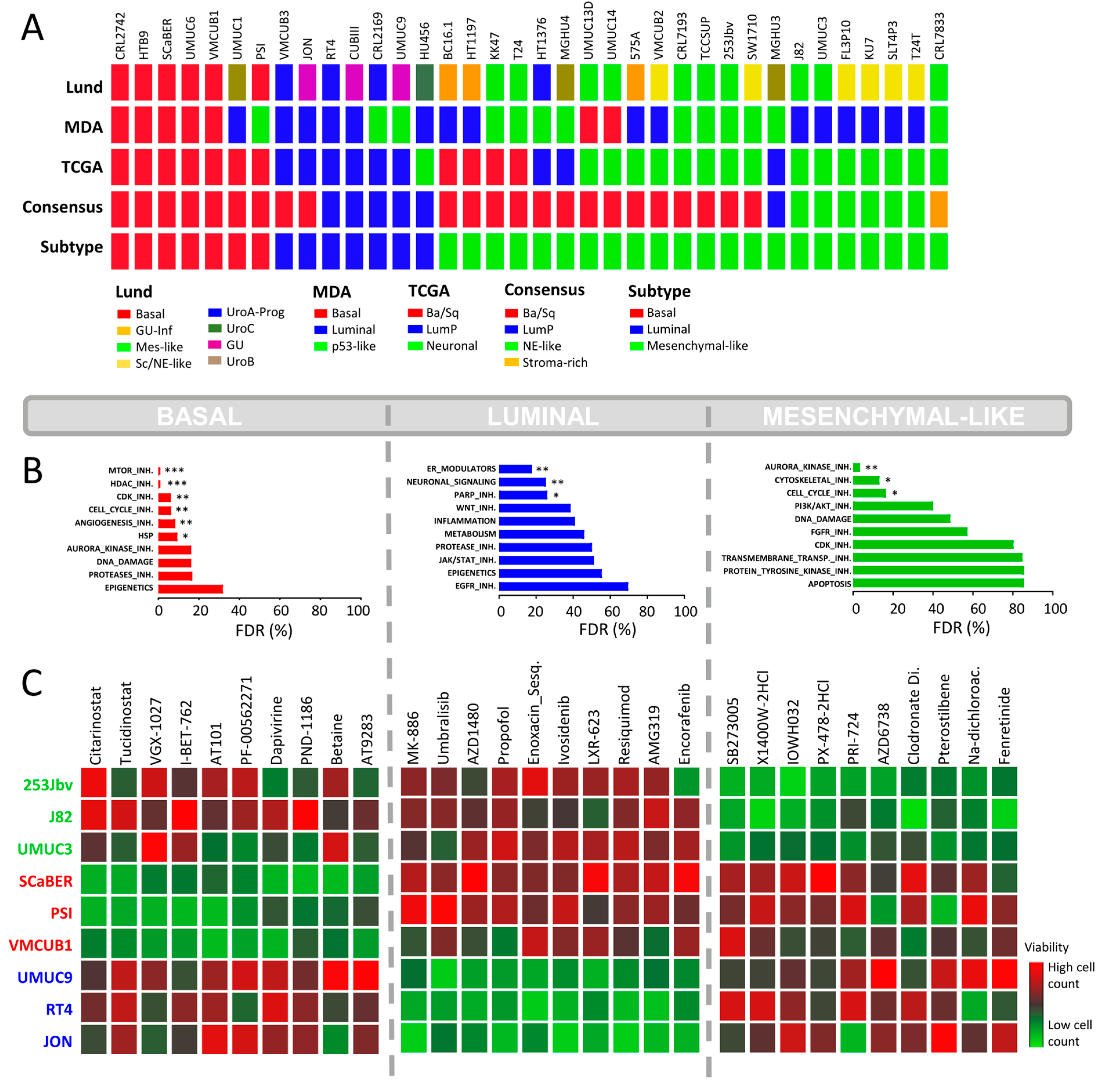
2.1. Molecular Subtyping of Bladder Cancer Cell Lines
2.2. CCLE BLCA Cell Lines: Subtype-Specific Drug Sensitivities
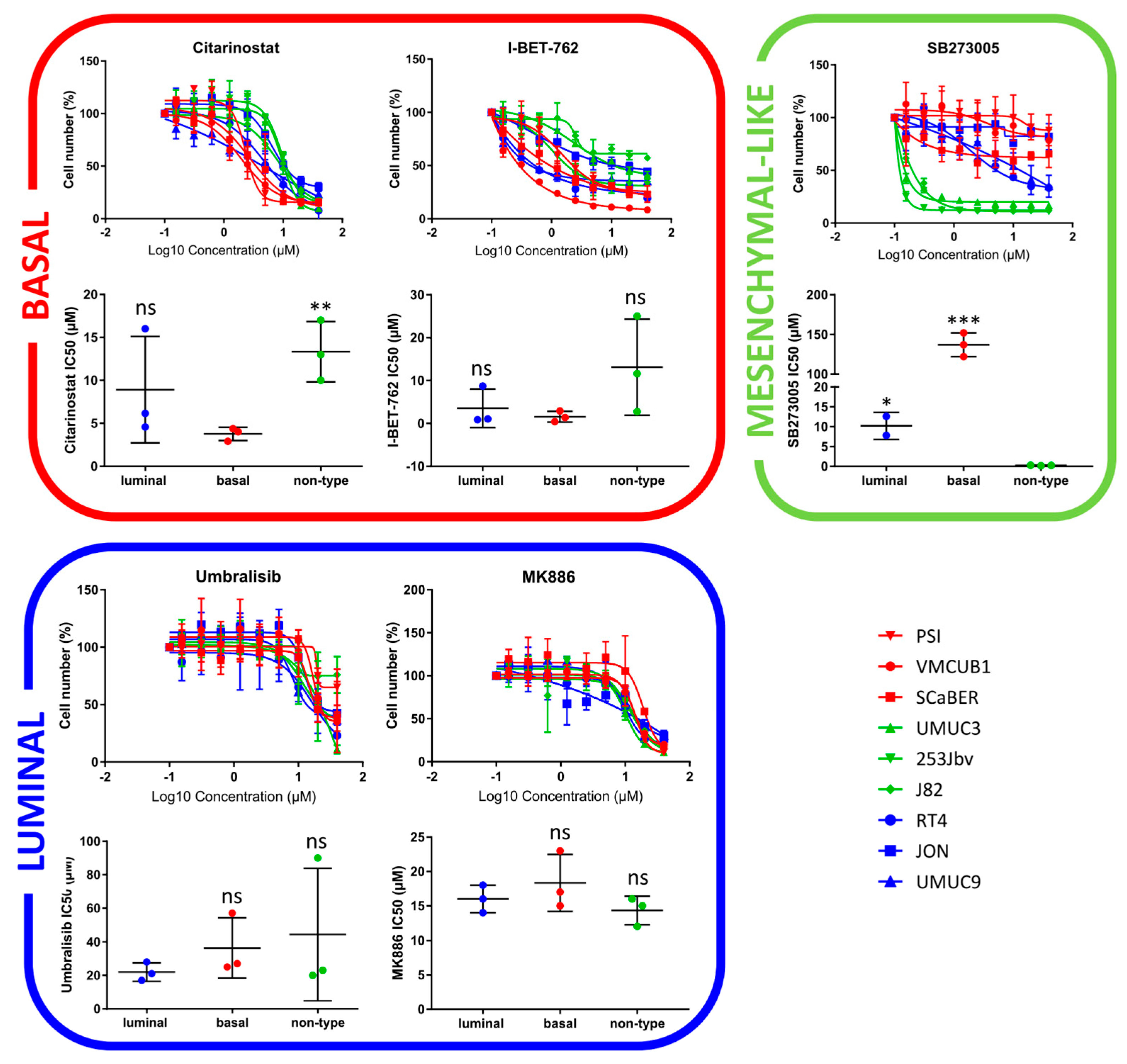
2.3. Focused In-House HCS against BLCA Subtypes
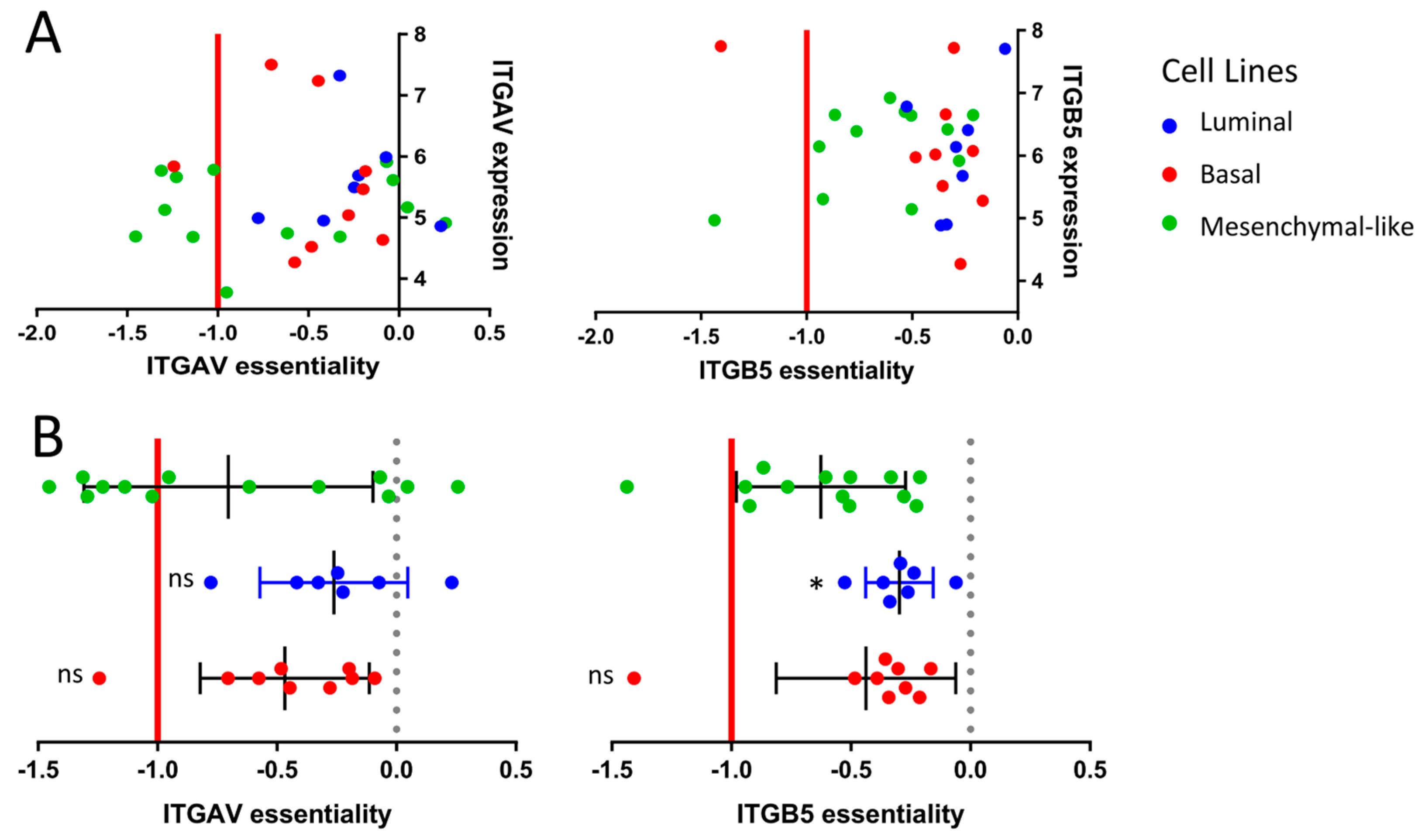
2.4. Drug Target Validation by Regulon Analysis and Functional Genomics
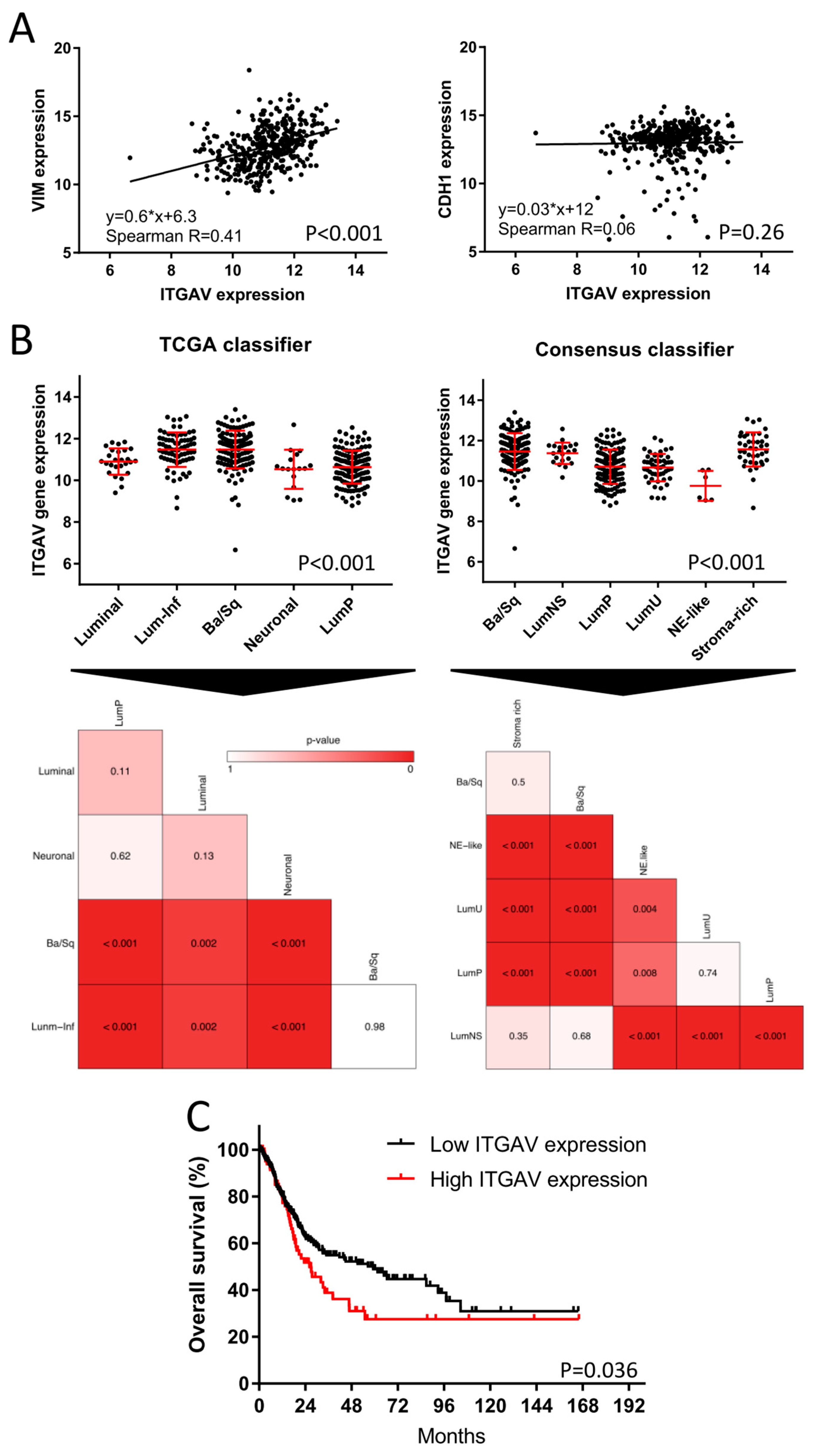
2.5. TCGA Bladder Cancer Cohort: Clinical Impact of ITGAV
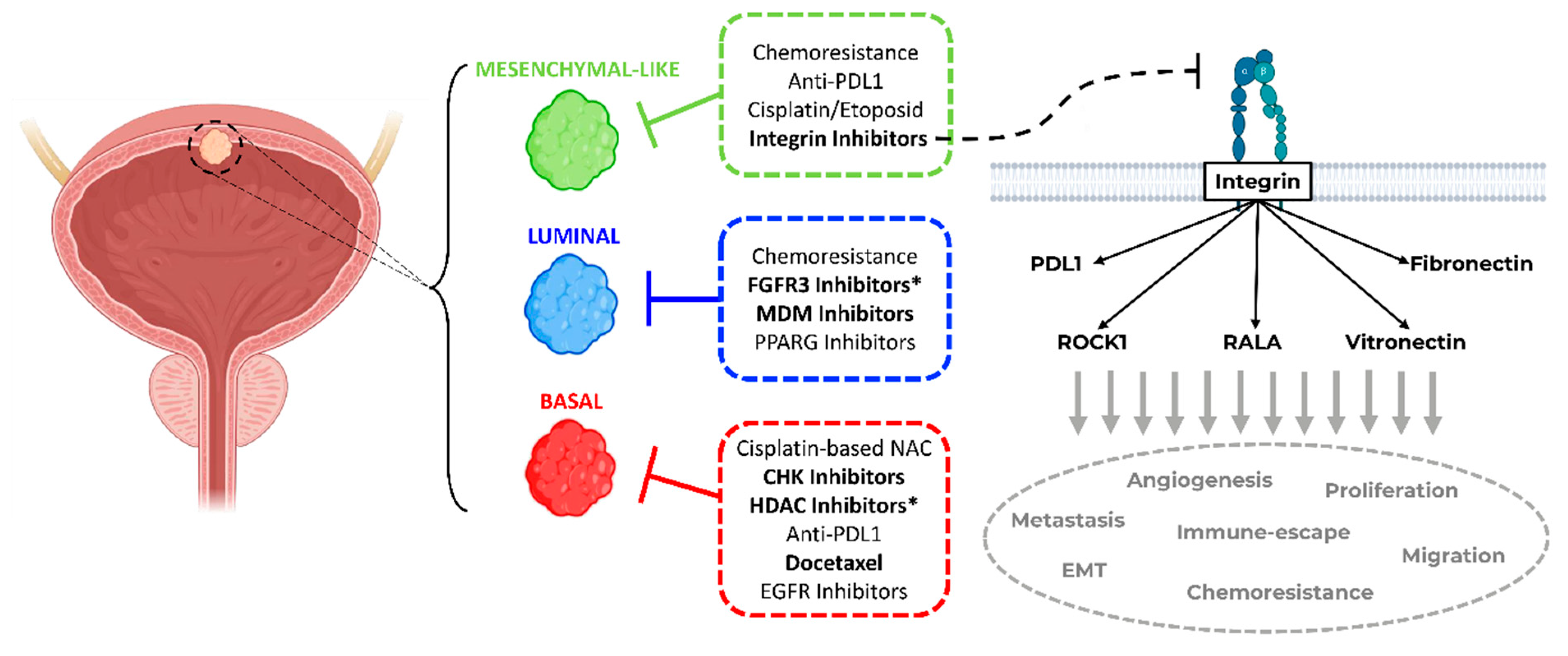
3. Discussion
4. Methods and Materials
4.1. Transcriptome Expression Analyses and Molecular Subtyping
4.2. Drug Set and Gene Set Enrichment Analyses
4.3. Regulon Analysis
4.4. Functional Genomics
4.5. HCS Drug Discovery
4.6. Clinical Data and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bladder Cancer Survival Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/bladder-cancer/survival (accessed on 1 March 2022).
- Siefker-Radtke, A.O.; Necchi, A.; Park, S.H.; García-Donas, J.; Huddart, R.A.; Burgess, E.F.; Fleming, M.T.; Rezazadeh Kalebasty, A.; Mellado, B.; Varlamov, S.; et al. Efficacy and Safety of Erdafitinib in Patients with Locally Advanced or Metastatic Urothelial Carcinoma: Long-Term Follow-up of a Phase 2 Study. Lancet Oncol. 2022, 23, 248–258. [Google Scholar] [CrossRef]
- Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Vuky, J.; Powles, T.; Plimack, E.R.; Hahn, N.M.; de Wit, R.; Pang, L.; et al. First-Line Pembrolizumab in Cisplatin-Ineligible Patients with Locally Advanced and Unresectable or Metastatic Urothelial Cancer (KEYNOTE-052): A Multicentre, Single-Arm, Phase 2 Study. Lancet Oncol. 2017, 18, 1483–1492. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef]
- Kamoun, A.; de Reyniès, A.; Allory, Y.; Sjödahl, G.; Robertson, A.G.; Seiler, R.; Hoadley, K.A.; Groeneveld, C.S.; Al-Ahmadie, H.; Choi, W.; et al. A Consensus Molecular Classification of Muscle-Invasive Bladder Cancer. Eur. Urol. 2020, 77, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Necchi, A.; Raggi, D.; Gallina, A.; Ross, J.S.; Farè, E.; Giannatempo, P.; Marandino, L.; Colecchia, M.; Lucianò, R.; Bianchi, M.; et al. Impact of Molecular Subtyping and Immune Infiltration on Pathological Response and Outcome Following Neoadjuvant Pembrolizumab in Muscle-Invasive Bladder Cancer. Eur. Urol. 2020, 77, 701–710. [Google Scholar] [CrossRef]
- McConkey, D.J.; Choi, W.; Shen, Y.; Lee, I.-L.; Porten, S.; Matin, S.F.; Kamat, A.M.; Corn, P.; Millikan, R.E.; Dinney, C.; et al. A Prognostic Gene Expression Signature in the Molecular Classification of Chemotherapy-Naïve Urothelial Cancer Is Predictive of Clinical Outcomes from Neoadjuvant Chemotherapy: A Phase 2 Trial of Dose-Dense Methotrexate, Vinblastine, Doxorubicin, and Cisplatin with Bevacizumab in Urothelial Cancer. Eur. Urol. 2016, 69, 855–862. [Google Scholar] [CrossRef]
- Seiler, R.; Gibb, E.A.; Wang, N.Q.; Oo, H.Z.; Lam, H.-M.; van Kessel, K.E.; Voskuilen, C.S.; Winters, B.; Erho, N.; Takhar, M.M.; et al. Divergent Biological Response to Neoadjuvant Chemotherapy in Muscle-Invasive Bladder Cancer. Clin. Cancer Res. 2019, 25, 5082–5093. [Google Scholar] [CrossRef]
- Zuiverloon, T.C.M.; de Jong, F.C.; Costello, J.C.; Theodorescu, D. Systematic Review: Characteristics and Preclinical Uses of Bladder Cancer Cell Lines. Bladder. Cancer 2018, 4, 169–183. [Google Scholar] [CrossRef]
- Lee, J.K.; Havaleshko, D.M.; Cho, H.; Weinstein, J.N.; Kaldjian, E.P.; Karpovich, J.; Grimshaw, A.; Theodorescu, D. A Strategy for Predicting the Chemosensitivity of Human Cancers and Its Application to Drug Discovery. Proc. Natl. Acad. Sci. USA 2007, 104, 13086–13091. [Google Scholar] [CrossRef]
- Havaleshko, D.M.; Cho, H.; Conaway, M.; Owens, C.R.; Hampton, G.; Lee, J.K.; Theodorescu, D. Prediction of Drug Combination Chemosensitivity in Human Bladder Cancer. Mol. Cancer Ther. 2007, 6, 578–586. [Google Scholar] [CrossRef][Green Version]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-Generation Characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, D.; Tao, D.; Xiang, W.; Xiao, X.; Wang, M.; Wang, L.; Luo, G.; Li, Y.; Zeng, F.; et al. BRD4 Regulates EZH2 Transcription through Upregulation of C-MYC and Represents a Novel Therapeutic Target in Bladder Cancer. Mol. Cancer Ther. 2016, 15, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Ler, L.D.; Ghosh, S.; Chai, X.; Thike, A.A.; Heng, H.L.; Siew, E.Y.; Dey, S.; Koh, L.K.; Lim, J.Q.; Lim, W.K.; et al. Loss of Tumor Suppressor KDM6A Amplifies PRC2-Regulated Transcriptional Repression in Bladder Cancer and Can Be Targeted through Inhibition of EZH2. Sci. Transl. Med. 2017, 9, eaai8312. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.E.; Hong, A.; Jana, B. Elucidation of Novel Molecular Targets for Therapeutic Strategies in Urothelial Carcinoma: A Literature Review. Front. Oncol. 2021, 11, 705294. [Google Scholar] [CrossRef]
- Grivas, P.; Mortazavi, A.; Picus, J.; Hahn, N.M.; Milowsky, M.I.; Hart, L.L.; Alva, A.; Bellmunt, J.; Pal, S.K.; Bambury, R.M.; et al. Mocetinostat for Patients with Previously Treated, Locally Advanced/Metastatic Urothelial Carcinoma and Inactivating Alterations of Acetyltransferase Genes. Cancer 2019, 125, 533–540. [Google Scholar] [CrossRef]
- Grivas, P.; Loriot, Y.; Morales-Barrera, R.; Teo, M.Y.; Zakharia, Y.; Feyerabend, S.; Vogelzang, N.J.; Grande, E.; Adra, N.; Alva, A.; et al. Efficacy and Safety of Rucaparib in Previously Treated, Locally Advanced or Metastatic Urothelial Carcinoma from a Phase 2, Open-Label Trial (ATLAS). BMC Cancer 2021, 21, 593. [Google Scholar] [CrossRef]
- Choi, W.; Porten, S.; Kim, S.; Willis, D.; Plimack, E.R.; Hoffman-Censits, J.; Roth, B.; Cheng, T.; Tran, M.; Lee, I.-L.; et al. Identification of Distinct Basal and Luminal Subtypes of Muscle-Invasive Bladder Cancer with Different Sensitivities to Frontline Chemotherapy. Cancer Cell 2014, 25, 152–165. [Google Scholar] [CrossRef]
- Efstathiou, J.A.; Mouw, K.W.; Gibb, E.A.; Liu, Y.; Wu, C.-L.; Drumm, M.R.; da Costa, J.B.; du Plessis, M.; Wang, N.Q.; Davicioni, E.; et al. Impact of Immune and Stromal Infiltration on Outcomes Following Bladder-Sparing Trimodality Therapy for Muscle-Invasive Bladder Cancer. Eur. Urol. 2019, 76, 59–68. [Google Scholar] [CrossRef]
- Marzouka, N.-A.-D.; Eriksson, P.; Rovira, C.; Liedberg, F.; Sjödahl, G.; Höglund, M. A Validation and Extended Description of the Lund Taxonomy for Urothelial Carcinoma Using the TCGA Cohort. Sci. Rep. 2018, 8, 3737. [Google Scholar] [CrossRef]
- Tate, T.; Xiang, T.; Wobker, S.E.; Zhou, M.; Chen, X.; Kim, H.; Batourina, E.; Lin, C.-S.; Kim, W.Y.; Lu, C.; et al. Pparg Signaling Controls Bladder Cancer Subtype and Immune Exclusion. Nat. Commun. 2021, 12, 6160. [Google Scholar] [CrossRef]
- Petrylak, D.P.; de Wit, R.; Chi, K.N.; Drakaki, A.; Sternberg, C.N.; Nishiyama, H.; Castellano, D.; Hussain, S.; Fléchon, A.; Bamias, A.; et al. Ramucirumab plus Docetaxel versus Placebo plus Docetaxel in Patients with Locally Advanced or Metastatic Urothelial Carcinoma after Platinum-Based Therapy (RANGE): A Randomised, Double-Blind, Phase 3 Trial. Lancet 2017, 390, 2266–2277. [Google Scholar] [CrossRef]
- Chène, P. Inhibiting the P53–MDM2 Interaction: An Important Target for Cancer Therapy. Nat. Rev. Cancer 2003, 3, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Rinaldetti, S.; Rempel, E.; Worst, T.S.; Eckstein, M.; Steidler, A.; Weiss, C.A.; Bolenz, C.; Hartmann, A.; Erben, P. Subclassification, Survival Prediction and Drug Target Analyses of Chemotherapy-Naïve Muscle-Invasive Bladder Cancer with a Molecular Screening. Oncotarget 2018, 9, 25935–25945. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. EMT, the Cytoskeleton, and Cancer Cell Invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef]
- SB273005 Datasheet. Available online: https://www.selleckchem.com/datasheet/sb273005-S754002-DataSheet.html (accessed on 18 June 2022).
- Wang, L.; Rajan, H.; Pitman, J.L.; McKeown, M.; Tsai, C.-C. Histone Deacetylase-Associating Atrophin Proteins Are Nuclear Receptor Corepressors. Genes Dev. 2006, 20, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Charroux, B.; Kerridge, S.; Tsai, C.-C. Atrophin Recruits HDAC1/2 and G9a to Modify Histone H3K9 and to Determine Cell Fates. EMBO Rep. 2008, 9, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, N.; Meier, J.A.; Scott, D.W.; Norambuena, A.; White, M.A.; Schwartz, M.A. RalA-Exocyst Complex Regulates Integrin-Dependent Membrane Raft Exocytosis and Growth Signaling. Curr. Biol. 2010, 20, 75–79. [Google Scholar] [CrossRef]
- Pawar, A.; Meier, J.A.; Dasgupta, A.; Diwanji, N.; Deshpande, N.; Saxena, K.; Buwa, N.; Inchanalkar, S.; Schwartz, M.A.; Balasubramanian, N. Ral-Arf6 Crosstalk Regulates Ral Dependent Exocyst Trafficking and Anchorage Independent Growth Signalling. Cell. Signal. 2016, 28, 1225–1236. [Google Scholar] [CrossRef]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. A B3 Integrin-KRAS-RalB Complex Drives Tumor Stemness and Resistance to EGFR Inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef]
- Wong, P.-P.; Muñoz-Félix, J.M.; Hijazi, M.; Kim, H.; Robinson, S.D.; De Luxán-Delgado, B.; Rodríguez-Hernández, I.; Maiques, O.; Meng, Y.-M.; Meng, Q.; et al. Cancer Burden Is Controlled by Mural Cell-Β3-Integrin Regulated Crosstalk with Tumor Cells. Cell 2020, 181, 1346–1363.e21. [Google Scholar] [CrossRef]
- Kudo, M.; Melton, A.C.; Chen, C.; Engler, M.B.; Huang, K.E.; Ren, X.; Wang, Y.; Bernstein, X.; Li, J.T.; Atabai, K.; et al. IL-17A Produced by Aβ T Cells Drives Airway Hyper-Responsiveness in Mice and Enhances Mouse and Human Airway Smooth Muscle Contraction. Nat. Med. 2012, 18, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Afrin, S.; Islam, M.S.; Patzkowsky, K.; Malik, M.; Catherino, W.H.; Segars, J.H.; Borahay, M.A. Simvastatin Ameliorates Altered Mechanotransduction in Uterine Leiomyoma Cells. Am. J. Obstet. Gynecol. 2020, 223, 733.e1–733.e14. [Google Scholar] [CrossRef] [PubMed]
- Esquer, H.; Zhou, Q.; Nemkov, T.; Abraham, A.D.; Rinaldetti, S.; Chen, Y.-C.; Zhang, X.; Orman, M.V.; D’Alessandro, A.; Ferrer, M.; et al. Isolating and Targeting the Real-Time Plasticity and Malignant Properties of Epithelial-Mesenchymal Transition in Cancer. Oncogene 2021, 40, 2884–2897. [Google Scholar] [CrossRef]
- Smith, S.C.; Baras, A.S.; Owens, C.R.; Dancik, G.; Theodorescu, D. Transcriptional Signatures of Ral GTPase Are Associated with Aggressive Clinicopathologic Characteristics in Human Cancer. Cancer Res. 2012, 72, 3480–3491. [Google Scholar] [CrossRef]
- Parihar, S.P.; Guler, R.; Brombacher, F. Statins: A Viable Candidate for Host-Directed Therapy against Infectious Diseases. Nat. Rev. Immunol. 2019, 19, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; He, Y.; Zhao, P.; Hu, Y.; Tao, J.; Chen, J.; Huang, Y. Targeting Lipid Metabolism to Overcome EMT-Associated Drug Resistance via Integrin Β3/FAK Pathway and Tumor-Associated Macrophage Repolarization Using Legumain-Activatable Delivery. Theranostics 2019, 9, 265–278. [Google Scholar] [CrossRef]
- van der Horst, G.; Bos, L.; van der Mark, M.; Cheung, H.; Heckmann, B.; Clément-Lacroix, P.; Lorenzon, G.; Pelger, R.C.M.; Bevers, R.F.M.; van der Pluijm, G. Targeting of Alpha-v Integrins Reduces Malignancy of Bladder Carcinoma. PLoS ONE 2014, 9, e108464. [Google Scholar] [CrossRef]
- Slack, R.J.; Macdonald, S.J.F.; Roper, J.A.; Jenkins, R.G.; Hatley, R.J.D. Emerging Therapeutic Opportunities for Integrin Inhibitors. Nat. Rev. Drug Discov. 2022, 21, 60–78. [Google Scholar] [CrossRef]
- Vannini, A.; Leoni, V.; Barboni, C.; Sanapo, M.; Zaghini, A.; Malatesta, P.; Campadelli-Fiume, G.; Gianni, T. Avβ3-Integrin Regulates PD-L1 Expression and Is Involved in Cancer Immune Evasion. Proc. Natl. Acad. Sci. USA 2019, 116, 20141–20150. [Google Scholar] [CrossRef]
- Buckwalter, J.M.; Chan, W.; Shuman, L.; Wildermuth, T.; Ellis-Mohl, J.; Walter, V.; Warrick, J.I.; Wu, X.-R.; Kaag, M.; Raman, J.D.; et al. Characterization of Histone Deacetylase Expression Within In Vitro and In Vivo Bladder Cancer Model Systems. Int. J. Mol. Sci. 2019, 20, E2599. [Google Scholar] [CrossRef]
- El-Chaar, N.N.; Smith, T.E.; Shrestha, G.; Piccolo, S.R.; Harper, M.K.; Van Wagoner, R.M.; Lu, Z.; Venancio, A.R.; Ireland, C.M.; Bild, A.H.; et al. Topsentinol L Trisulfate, a Marine Natural Product That Targets Basal-like and Claudin-Low Breast Cancers. Mar. Drugs 2021, 19, 41. [Google Scholar] [CrossRef] [PubMed]
- Göder, A.; Emmerich, C.; Nikolova, T.; Kiweler, N.; Schreiber, M.; Kühl, T.; Imhof, D.; Christmann, M.; Heinzel, T.; Schneider, G.; et al. HDAC1 and HDAC2 Integrate Checkpoint Kinase Phosphorylation and Cell Fate through the Phosphatase-2A Subunit PR130. Nat. Commun. 2018, 9, 764. [Google Scholar] [CrossRef] [PubMed]
- Huan, J.; Grivas, P.; Birch, J.; Hansel, D.E. Emerging Roles for Mammalian Target of Rapamycin (MTOR) Complexes in Bladder Cancer Progression and Therapy. Cancers 2022, 14, 1555. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.; Rinaldetti, S.; Cheikh, B.B.; Zhou, Q.; Hass, E.P.; Jones, R.T.; Joshi, M.; LaBarbera, D.V.; Knott, S.R.V.; Cech, T.R.; et al. TRIM28 Is a Transcriptional Activator of the Mutant TERT Promoter in Human Bladder Cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2102423118. [Google Scholar] [CrossRef]
- Hau, A.M.; Nakasaki, M.; Nakashima, K.; Krish, G.; Hansel, D.E. Differential MTOR Pathway Profiles in Bladder Cancer Cell Line Subtypes to Predict Sensitivity to MTOR Inhibition. Urol. Oncol. 2017, 35, 593–599. [Google Scholar] [CrossRef]
- Hayashi, T.; Nishiyama, K.; Shirahama, T. Inhibition of 5-Lipoxygenase Pathway Suppresses the Growth of Bladder Cancer Cells. Int. J. Urol. 2006, 13, 1086–1091. [Google Scholar] [CrossRef]
- Casadei, C.; Dizman, N.; Schepisi, G.; Cursano, M.C.; Basso, U.; Santini, D.; Pal, S.K.; De Giorgi, U. Targeted Therapies for Advanced Bladder Cancer: New Strategies with FGFR Inhibitors. Ther. Adv. Med. Oncol. 2019, 11, 1758835919890285. [Google Scholar] [CrossRef]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef]
- Napolitano, F.; Sirci, F.; Carrella, D.; di Bernardo, D. Drug-Set Enrichment Analysis: A Novel Tool to Investigate Drug Mode of Action. Bioinformatics 2016, 32, 235–241. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional Characterization of Somatic Mutations in Cancer Using Network-Based Inference of Protein Activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Aracne.Networks. Available online: http://bioconductor.org/packages/aracne.networks/ (accessed on 16 May 2022).
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational Correction of Copy Number Effect Improves Specificity of CRISPR-Cas9 Essentiality Screens in Cancer Cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the Anti-Cancer Potential of Non-Oncology Drugs by Systematic Viability Profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Álvarez-Twose, I.; Shoumariyeh, K.; Naumann, N.; Lübke, J.; Perkins, C.; Muñoz-González, J.I.; Meggendorfer, M.; Kennedy, V.; et al. MARS: Mutation-Adjusted Risk Score for Advanced Systemic Mastocytosis. J. Clin. Oncol. 2019, 37, 2846–2856. [Google Scholar] [CrossRef]
- Abbott, J.M.; Zhou, Q.; Esquer, H.; Pike, L.; Broneske, T.P.; Rinaldetti, S.; Abraham, A.D.; Ramirez, D.A.; Lunghofer, P.J.; Pitts, T.M.; et al. First-in-Class Inhibitors of Oncogenic CHD1L with Preclinical Activity against Colorectal Cancer. Mol. Cancer Ther. 2020, 19, 1598–1612. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rinaldetti, S.; Zhou, Q.; Abbott, J.M.; de Jong, F.C.; Esquer, H.; Costello, J.C.; Theodorescu, D.; LaBarbera, D.V. High-Content Drug Discovery Targeting Molecular Bladder Cancer Subtypes. Int. J. Mol. Sci. 2022, 23, 10605. https://doi.org/10.3390/ijms231810605
Rinaldetti S, Zhou Q, Abbott JM, de Jong FC, Esquer H, Costello JC, Theodorescu D, LaBarbera DV. High-Content Drug Discovery Targeting Molecular Bladder Cancer Subtypes. International Journal of Molecular Sciences. 2022; 23(18):10605. https://doi.org/10.3390/ijms231810605
Chicago/Turabian StyleRinaldetti, Sébastien, Qiong Zhou, Joshua M. Abbott, Florus C. de Jong, Hector Esquer, James C. Costello, Dan Theodorescu, and Daniel V. LaBarbera. 2022. "High-Content Drug Discovery Targeting Molecular Bladder Cancer Subtypes" International Journal of Molecular Sciences 23, no. 18: 10605. https://doi.org/10.3390/ijms231810605
APA StyleRinaldetti, S., Zhou, Q., Abbott, J. M., de Jong, F. C., Esquer, H., Costello, J. C., Theodorescu, D., & LaBarbera, D. V. (2022). High-Content Drug Discovery Targeting Molecular Bladder Cancer Subtypes. International Journal of Molecular Sciences, 23(18), 10605. https://doi.org/10.3390/ijms231810605