

Integrated Analysis of Transcriptome and Small RNAome Reveals the Regulatory Network for Rapid Growth in Mikania micrantha


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
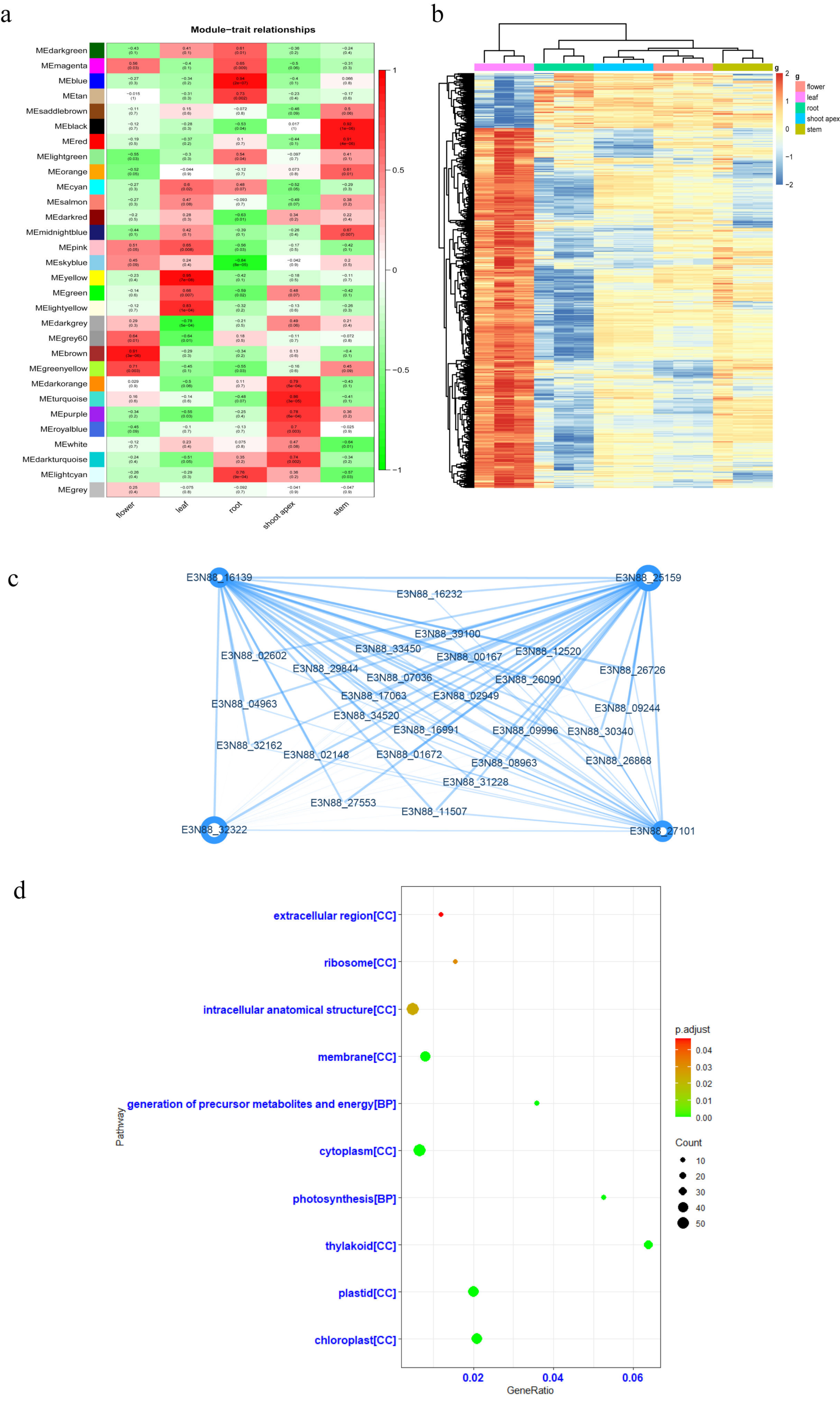
2.1. Weighted Gene Co-Expression Network Analysis (WGCNA) of the Transcriptome
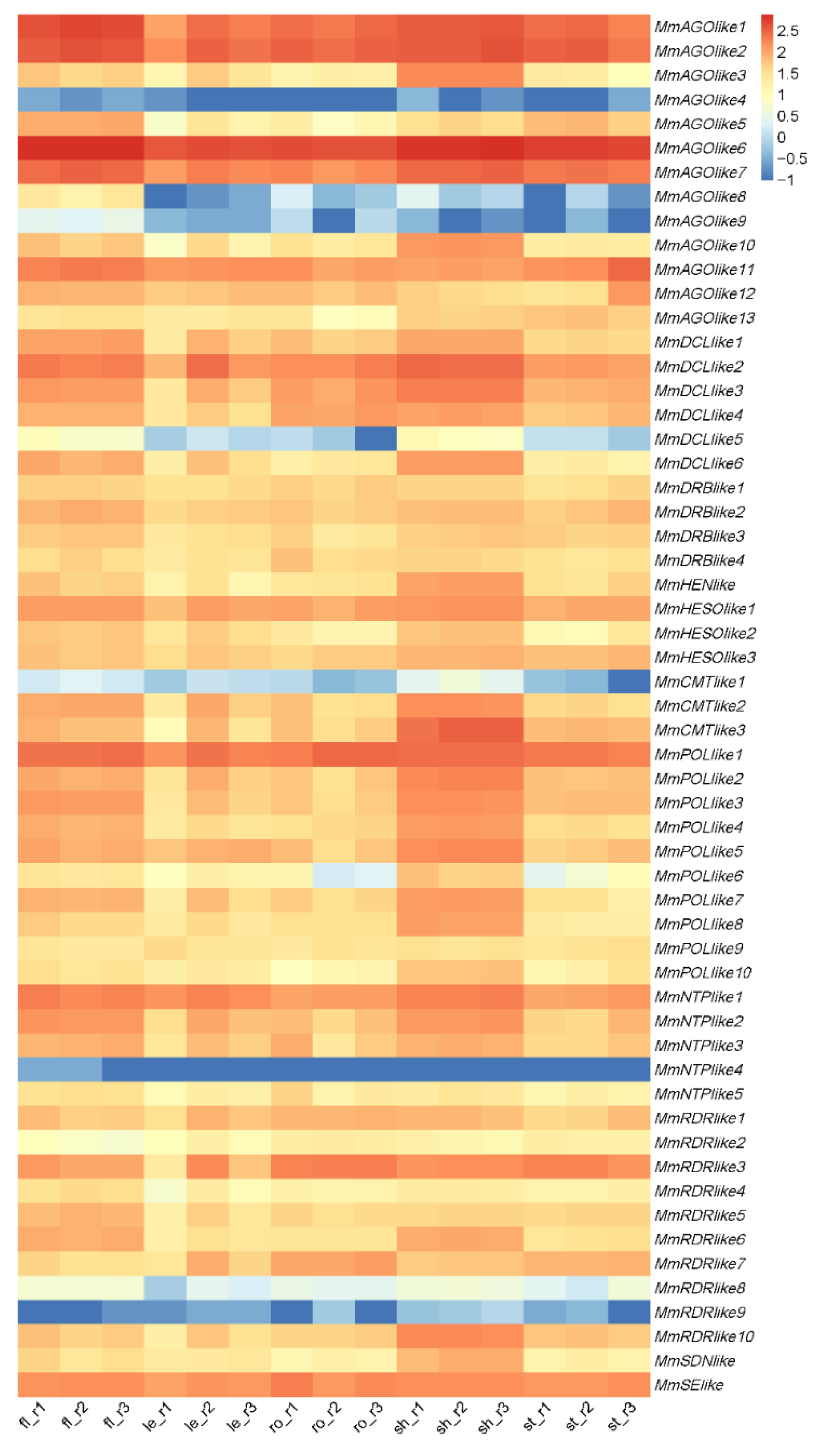
2.2. Small RNA Regulatory Pathways Exist in M. micrantha
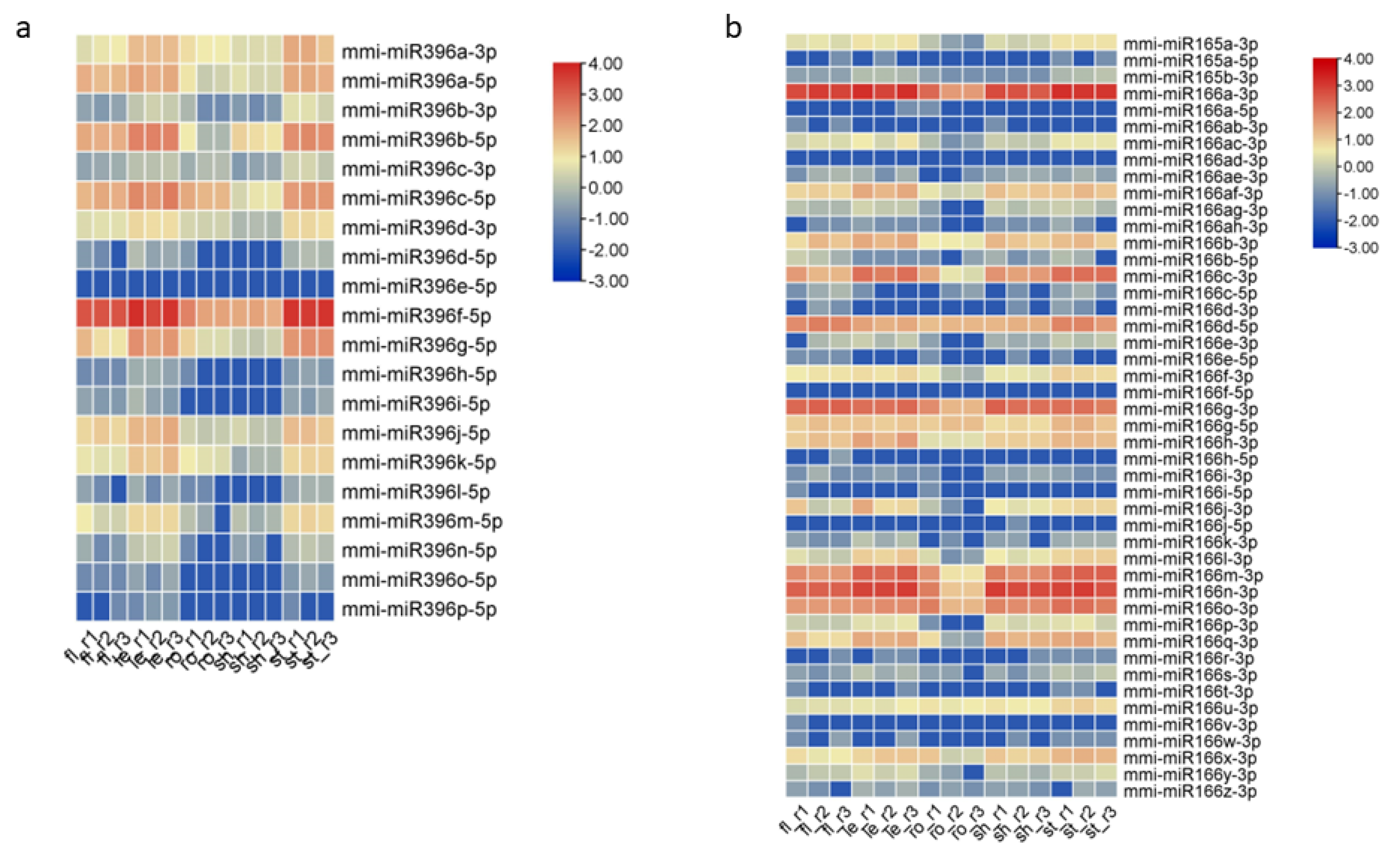
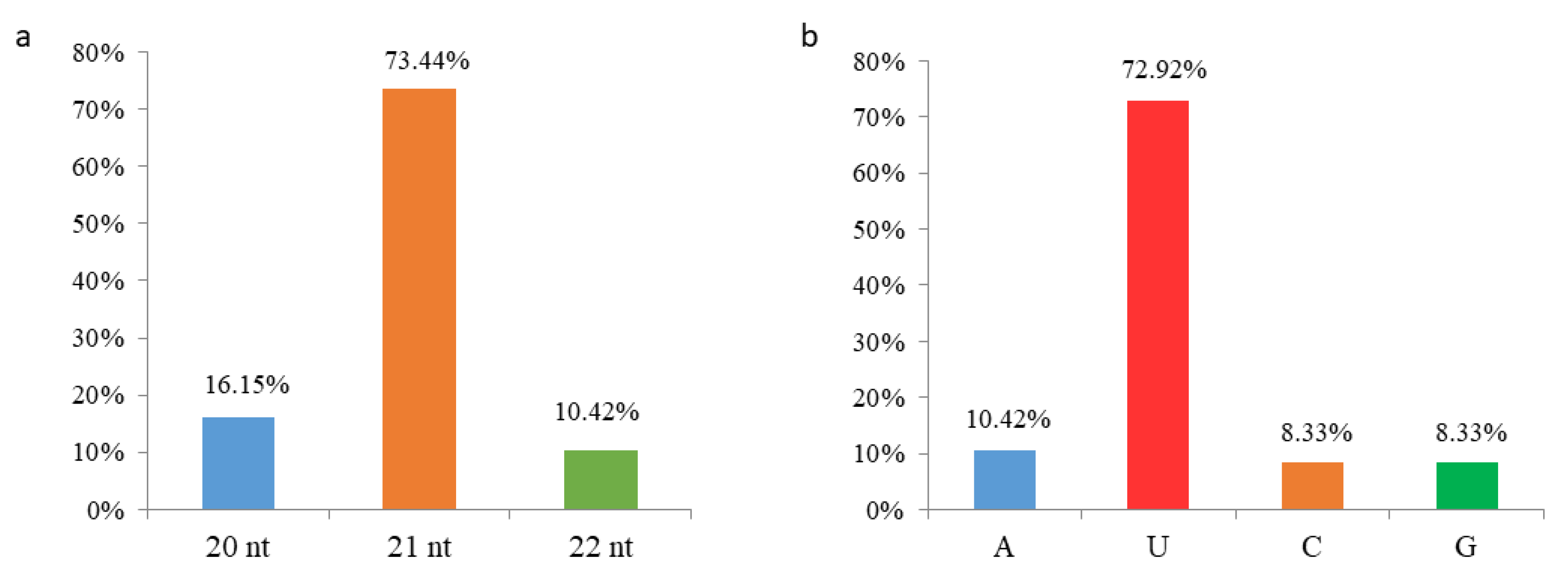
2.3. Identification of Conserved and Novel miRNAs in M. micrantha
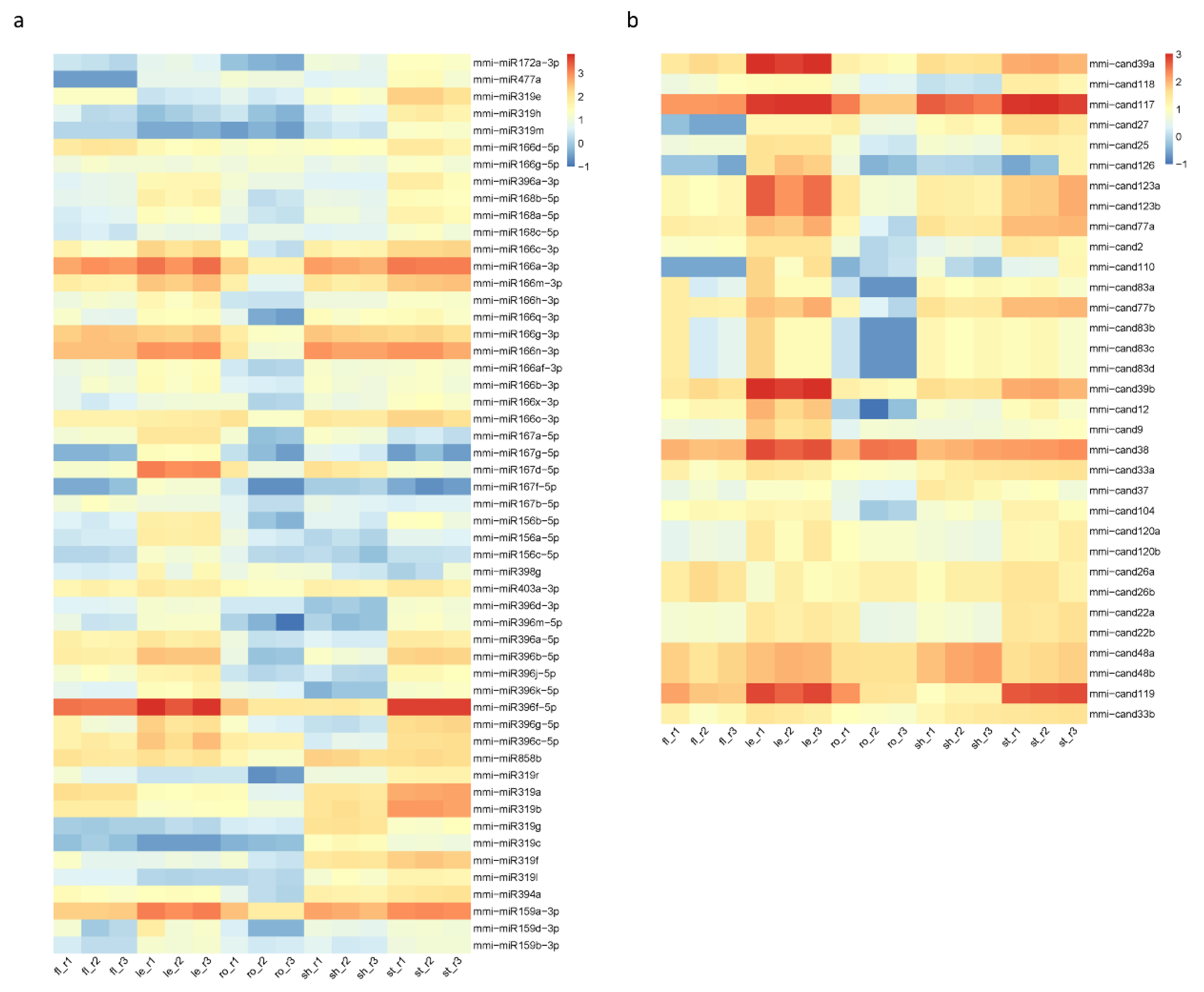
2.4. Differential Expression of miRNAs (DEmiRs) in Five Tissues of M. micrantha
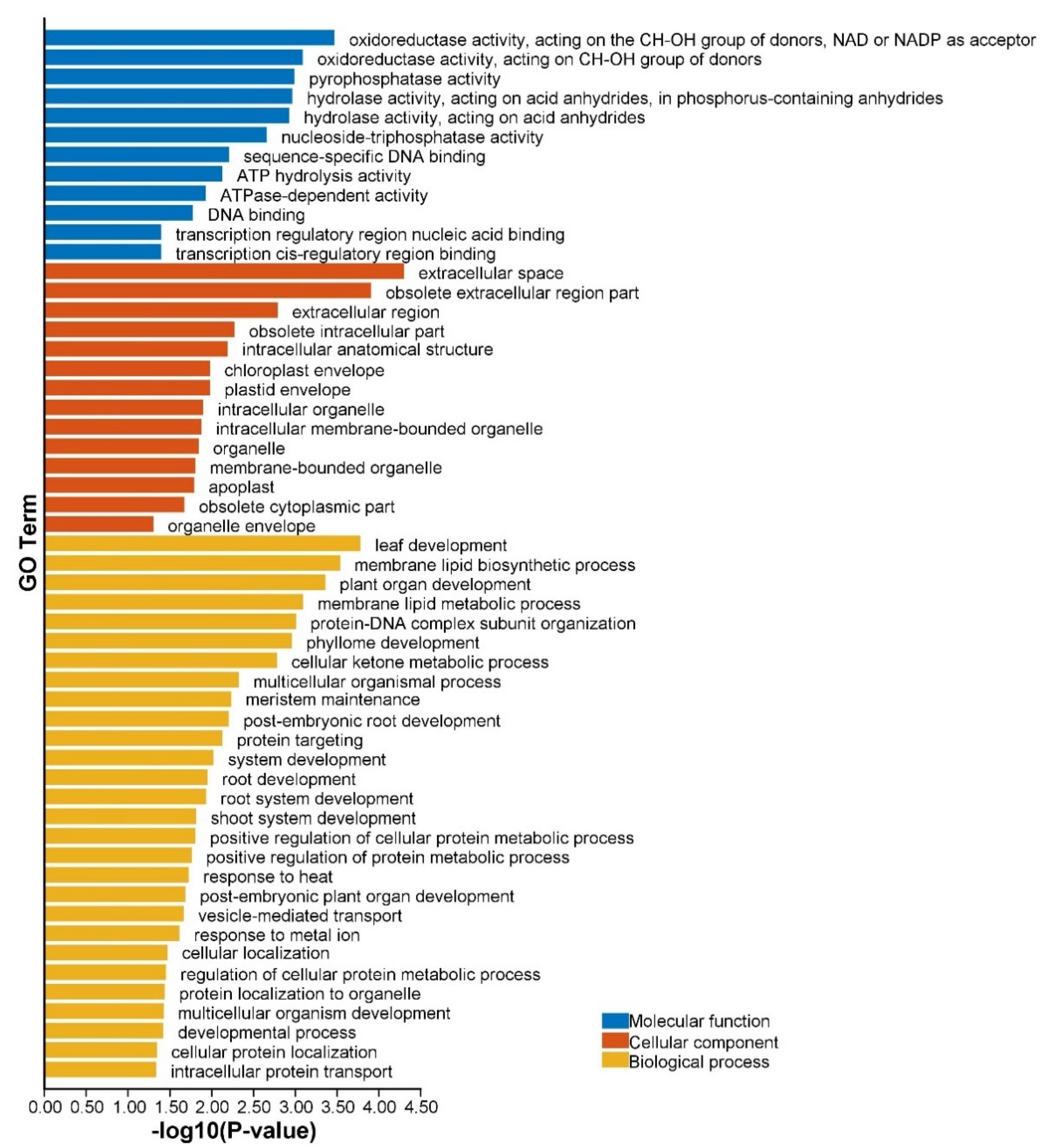
2.5. Prediction of miRNA Target Genes
2.6. Potential Roles of miRNAs in Regulating M. micrantha Development
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction, Library Construction and Sequencing
4.3. WGCNA of Transcriptome
4.4. Identification and Validation of Differential Expression Analysis of Conserved and Novel miRNAs
4.5. Prediction of miRNA Target Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lowe, S.; Browne, M.; Boudjelas, S.; De Poorter, M. 100 of the World’s Worst Invasive Alien Species: A Selection from the Global Invasive Species Database; IUCN/SSC Invasive Species Specialist Group: Auckland, New Zealand, 2000. [Google Scholar]
- Holm, L.G.; Plucknett, D.L.; Pancho, J.V.; Herberger, J.P. The World’s Worst Weeds: Distribution and Biology; University Press of Hawaii: Honolulu, HI, USA, 1977. [Google Scholar]
- Du, F.; Yang, Y.M.; Li, J.Q.; Yin, W.Y. A Review of Mikania and the Impact of M. micrantha (Asteraceae) in Yunnan. Acta Bot. Yunnanica 2006, 28, 505–508. [Google Scholar]
- Waterhouse, D.F. Biological Control of Weeds: Southeast Asian Prospects; ACIAR Monograph No. 26; Australian Center for International Agricultural Research (ACIAR): Canberra, Australia, 1994.
- Zhang, L.Y.; Ye, W.H.; Cao, H.L.; Feng, H.L. Mikania micrantha HBK in China—An overview. Weed Res. 2004, 44, 42–49. [Google Scholar] [CrossRef]
- Day, M.D.; Clements, D.R.; Gile, C.; Senaratne, W.K.A.D.; Shen, S.C.; Weston, L.A.; Zhang, F.D. Biology and Impacts of Pacific Islands Invasive Species. 13. Mikania micrantha Kunth (Asteraceae). Pac. Sci. 2016, 70, 257–285. [Google Scholar] [CrossRef]
- Huang, Z.L.; Cao, H.L.; Liang, X.D.; Ye, W.H.; Feng, H.L.; Cai, C.X. The growth and damaging effect of Mikania micrantha in different habitats. J. Trop. Subtrop. Bot. 2000, 8, 131–138. [Google Scholar]
- Li, J.M.; Dong, M. Fine-scale clonal structure and diversity of invasive plant Mikania micrantha HBK and its plant parasite Cuscuta campestris Yunker. Biol. Invasions 2009, 11, 687–695. [Google Scholar] [CrossRef]
- Li, M.G.; Lu, E.B.; Guo, Q.; Zan, Q.J.; Wei, P.P.; Jiang, L.; Xu, H.L.; Zhong, T.K. Evaluation of the controlling methods and strategies for Mikania micrantha H. B. K. Acta Ecol. Sin. 2012, 32, 3240–3251. [Google Scholar]
- Yang, M.; He, Z.W.; Huang, Y.L.; Lu, L.; Yan, Y.B.; Hong, L.; Shen, H.; Liu, Y.; Guo, Q.; Jiang, L.; et al. The emergence of the hyperinvasive vine, Mikania micrantha (Asteraceae), via admixture and founder events inferred from population transcriptomics. Mol. Ecol. 2017, 26, 3405–3423. [Google Scholar] [CrossRef]
- Li, J.M.; Dong, M.; Zhong, Z.C. Population genetic differentiations in the invasive plant Mikania micrantha in China. J. Plant Ecol. 2007, 31, 680–688. [Google Scholar]
- Wang, T.; Su, Y.J.; Chen, G.P. Population genetic variation and structure of the invasive weed Mikania micrantha in southern China: Consequences of rapid range expansion. J. Hered. 2008, 99, 22–33. [Google Scholar] [CrossRef]
- Yan, Y.B.; Huang, Y.L.; Fang, X.T.; Lu, L.; Zhou, R.C.; Ge, X.J.; Shi, S.H. Development and Characterization of Est-Ssr Markers in the Invasive Weed Mikania micrantha (Asteraceae). Am. J. Bot. 2011, 98, E1–E3. [Google Scholar] [CrossRef]
- Wang, T.; Chen, G.P.; Zan, Q.J.; Wang, C.B.; Su, Y.J. AFLP Genome Scan to Detect Genetic Structure and Candidate Loci under Selection for Local Adaptation of the Invasive Weed Mikania micrantha. PLoS ONE 2012, 7, e41310. [Google Scholar] [CrossRef]
- Bravo-Monzón, Á.E.; González-Rodríguez, A.; Espinosa-García, F.J. Spatial structure of genetic and chemical variation in native populations of the mile-a-minute weed Mikania micrantha. Biochem. Syst. Ecol. 2018, 76, 23–31. [Google Scholar] [CrossRef]
- Zhou, S.H.; Liu, N.N.; Sun, Y.; Wu, D.; Hou, Y.X. MmZFP1 Response to Abiotic Stress in the Invasive Plant Mikania micrantha. Nat. Environ. Polution Technol. 2015, 14, 251–258. [Google Scholar]
- Huang, Y.L.; Fang, X.T.; Lu, L.; Yan, Y.B.; Chen, S.F.; Hu, L.; Zhu, C.C.; Ge, X.J.; Shi, S.H. Transcriptome analysis of an invasive weed Mikania micrantha. Biol. Plant 2012, 56, 111–116. [Google Scholar] [CrossRef]
- Mai, J.T.; Liao, L.L.; Ling, R.S.; Guo, X.L.; Lin, J.Y.; Mo, B.X.; Chen, W.Z.; Yu, Y. Study on RNAi-based herbicide for Mikania micrantha. Synth. Syst. Biotechnol. 2021, 6, 437–445. [Google Scholar] [CrossRef]
- Liu, B.; Yan, J.; Li, W.H.; Yin, L.J.; Li, P.; Yu, H.X.; Xing, L.S.; Cai, M.L.; Wang, H.C.; Zhao, M.X.; et al. Mikania micrantha genome provides insights into the molecular mechanism of rapid growth. Nat. Commun. 2020, 11, 340. [Google Scholar] [CrossRef]
- Chen, X.M.; Rechavi, O. Plant and animal small RNA communications between cells and organisms. Nat. Rev. Mol. Cell Biol. 2022, 23, 185–203. [Google Scholar] [CrossRef]
- Chung, P.J.; Park, B.S.; Wang, H.; Liu, J.; Jang, I.C.; Chua, N.H. Light-Inducible MiR163 Targets PXMT1 Transcripts to Promote Seed Germination and Primary Root Elongation in Arabidopsis. Plant Physiol. 2016, 170, 1772–1782. [Google Scholar] [CrossRef]
- Lee, B.H.; Ko, J.H.; Lee, S.; Lee, Y.; Pak, J.H.; Kim, J.H. The Arabidopsis GRF-INTERACTING FACTOR Gene Family Performs an Overlapping Function in Determining Organ Size as Well as Multiple Developmental Properties. Plant Physiol. 2009, 151, 655–668. [Google Scholar] [CrossRef]
- Bao, M.L.; Bian, H.W.; Zha, Y.L.; Li, F.Y.; Sun, Y.Z.; Bai, B.; Chen, Z.H.; Wang, J.H.; Zhu, M.Y.; Han, N. miR396a-Mediated Basic Helix-Loop-Helix Transcription Factor bHLH74 Repression Acts as a Regulator for Root Growth in Arabidopsis Seedlings. Plant Cell Physiol. 2014, 55, 1343–1353. [Google Scholar] [CrossRef]
- Sharma, D.; Tiwari, M.; Pandey, A.; Bhatia, C.; Sharma, A.; Trivedi, P.K. MicroRNA858 Is a Potential Regulator of Phenylpropanoid Pathway and Plant Development. Plant Physiol. 2016, 171, 944–959. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Ke, L.L.; Wu, G.Z.; Xu, Y.T.; Wu, X.M.; Xia, R.; Deng, X.X.; Xu, Q. miR3954 is a trigger of phasiRNAs that affects flowering time in citrus. Plant J. 2017, 92, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Finotello, F.; Di Camillo, B. Measuring differential gene expression with RNA-seq: Challenges and strategies for data analysis. Brief. Funct. Genom. 2015, 14, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.S.; Gan, L.; Chen, D.J.; Zhang, Y.C.; Zhang, Y.J.; Liu, X.L.; Chen, S.; Wei, Z.S.; Tong, L.Q.; Song, Z.J.; et al. Integration of small RNA, degradome and proteome sequencing in Oryza sativa reveals a delayed senescence network in tetraploid rice seed. PLoS ONE 2020, 15, e0242260. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.P.; Able, A.J.; Able, J.A. Integrated Analysis of Small RNA, Transcriptome, and Degradome Sequencing Reveals the Water-Deficit and Heat Stress Response Network in Durum Wheat. Int. J. Mol. Sci. 2020, 21, 6017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Gong, H.H.; Li, D.H.; Zhou, R.; Zhao, F.T.; Zhang, X.R.; You, J. Integrated small RNA and Degradome sequencing provide insights into salt tolerance in sesame (Sesamum indicum L.). BMC Genom. 2020, 21, 494. [Google Scholar] [CrossRef]
- Cai, C.P.; Li, C.; Sun, R.R.; Zhang, B.H.; Nichols, R.L.; Hake, K.D.; Pan, X.P. Small RNA and degradome deep sequencing reveals important roles of microRNAs in cotton (Gossypium hirsutum L.) response to root-knot nematode Meloidogyne incognita infection. Genomics 2021, 113, 1146–1156. [Google Scholar] [CrossRef]
- Tong, B.; Shi, Y.S.; Ntambiyukuri, A.; Li, X.; Zhan, J.; Wang, A.Q.; Xiao, D.; He, L.F. Integration of Small RNA and Degradome Sequencing Reveals the Regulatory Network of Al-Induced Programmed Cell Death in Peanut. Int. J. Mol. Sci. 2022, 23, 246. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Li, Z.C.; Li, W.Q.; Guo, M.X.; Liu, S.M.; Liu, L.; Yu, Y.; Mo, B.X.; Chen, X.M.; Gao, L. Origin, evolution and diversification of plant ARGONAUTE proteins. Plant J. 2022, 109, 1086–1097. [Google Scholar] [CrossRef]
- Axtell, M.J.; Meyers, B.C. Revisiting Criteria for Plant MicroRNA Annotation in the Era of Big Data. Plant Cell 2018, 30, 272–284. [Google Scholar] [CrossRef]
- Chen, X.M. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 2004, 303, 2022–2025. [Google Scholar] [CrossRef]
- Van der Knaap, E.; Kim, J.H.; Kende, H. A novel gibberellin-induced gene from rice and its potential regulatory role in stem growth. Plant Physiol. 2000, 122, 695–704. [Google Scholar] [CrossRef]
- Yu, Y.; Ji, L.J.; Le, B.H.; Zhai, J.X.; Chen, J.Y.; Luscher, E.; Gao, L.; Liu, C.Y.; Cao, X.F.; Mo, B.X.; et al. ARGONAUTE10 promotes the degradation of miR165/6 through the SDN1 and SDN2 exonucleases in Arabidopsis. PLoS Biol. 2017, 15, e2001272. [Google Scholar] [CrossRef]
- Fan, L.S.; Zhang, C.; Gao, B.; Zhang, Y.; Stewart, E.; Jez, J.; Nakajima, K.; Chen, X.M. Microtubules promote the non-cell autonomous action of microRNAs by inhibiting their cytoplasmic loading onto ARGONAUTE1 in Arabidopsis. Dev. Cell 2022, 57, 995–1008. [Google Scholar] [CrossRef]
- Clepet, C.; Devani, R.S.; Boumlik, R.; Hao, Y.W.; Morin, H.; Marcel, F.; Verdenaud, M.; Mania, B.; Brisou, G.; Citerne, S.; et al. The miR166-SlHB15A regulatory module controls ovule development and parthenocarpic fruit set under adverse temperatures in tomato. Mol. Plant 2021, 14, 1185–1198. [Google Scholar] [CrossRef]
- Koyama, T.; Sato, F.; Ohme-Takagi, M. Roles of miR319 and TCP Transcription Factors in Leaf Development. Plant Physiol. 2017, 175, 874–885. [Google Scholar] [CrossRef]
- Lopez, J.A.; Sun, Y.; Blair, P.B.; Mukhtar, M.S. TCP three-way handshake: Linking developmental processes with plant immunity. Trends Plant Sci. 2015, 20, 238–245. [Google Scholar] [CrossRef]
- Nicolas, M.; Cubas, P. TCP factors: New kids on the signaling block. Curr. Opin. Plant Biol. 2016, 33, 33–41. [Google Scholar] [CrossRef]
- Yu, S.W.; Li, P.H.; Zhao, X.C.; Tan, M.M.; Ahmad, M.Z.; Xu, Y.J.; Tadege, M.; Zhao, J. CsTCPs regulate shoot tip development and catechin biosynthesis in tea plant (Camellia sinensis). Hortic. Res. Engl. 2021, 8, 104. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, D.S.; Kende, H. The AtGRF family of putative transcription factors is involved in leaf and cotyledon growth in Arabidopsis. Plant J. 2003, 36, 94–104. [Google Scholar] [CrossRef]
- Kim, J.S.; Mizoi, J.; Kidokoro, S.; Maruyama, K.; Nakajima, J.; Nakashima, K.; Mitsuda, N.; Takiguchi, Y.; Ohme-Takagi, M.; Kondou, Y.; et al. Arabidopsis GROWTH-REGULATING FACTOR7 Functions as a Transcriptional Repressor of Abscisic Acid- and Osmotic Stress-Responsive Genes, Including DREB2A. Plant Cell 2012, 24, 3393–3405. [Google Scholar] [CrossRef]
- Hewezi, T.; Maier, T.R.; Nettleton, D.; Baum, T.J. The Arabidopsis MicroRNA396-GRF1/GRF3 Regulatory Module Acts as a Developmental Regulator in the Reprogramming of Root Cells during Cyst Nematode Infection. Plant Physiol. 2012, 159, 321–335. [Google Scholar] [CrossRef]
- Borges, F.; Parent, J.S.; van Ex, F.; Wolff, P.; Martinez, G.; Kohler, C.; Martienssen, R.A. Transposon-derived small RNAs triggered by miR845 mediate genome dosage response in Arabidopsis. Nat. Genet. 2018, 50, 186–192. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C-Elegans Heterochronic Gene Lin-4 Encodes Small RNAs with Antisense Complementarity to Lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Park, W.; Li, J.J.; Song, R.T.; Messing, J.; Chen, X.M. CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr. Biol. 2002, 12, 1484–1495. [Google Scholar] [CrossRef]
- Llave, C.; Kasschau, K.D.; Rector, M.A.; Carrington, J.C. Endogenous and silencing-associated small RNAs in plants. Plant Cell 2002, 14, 1605–1619. [Google Scholar] [CrossRef]
- Long, J.A.; Moan, E.I.; Medford, J.I.; Barton, M.K. A member of the KNOTTED class of homeodomain proteins encoded by the STM gene of Arabidopsis. Nature 1996, 379, 66–69. [Google Scholar] [CrossRef]
- Vollbrecht, E.; Reiser, L.; Hake, S. Shoot meristem size is dependent on inbred background and presence of the maize homeobox gene, knotted1. Development 2000, 127, 3161–3172. [Google Scholar] [CrossRef]
- Luo, L.L.; Yang, X.Y.; Guo, M.X.; Lan, T.; Yu, Y.; Mo, B.X.; Chen, X.M.; Gao, L.; Liu, L. TRANS-ACTING SIRNA3-derived short interfering RNAs confer cleavage of mRNAs in rice. Plant Physiol. 2022, 188, 347–362. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Ren, Y.J.; Zhang, D.Y.; Chen, X.W.; Huang, J.Z.; Xu, Y.; Aucapina, C.B.; Zhang, Y.; Miao, Y. Transcriptome-Based WGCNA Analysis Reveals Regulated Metabolite Fluxes between Floral Color and Scent in Narcissus tazetta Flower. Int. J. Mol. Sci. 2021, 22, 8249. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wu, C.; Liu, L. AUSPP: A universal short-read pre-processing package. J. Bioinform. Comput. Biol. 2019, 17, 1950037. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Kuang, Z.; Wang, Y.; Li, L.; Yang, X.Z. miRDeep-P2: Accurate and fast analysis of the microRNA transcriptome in plants. Bioinformatics 2019, 35, 2521–2522. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Dai, X.B.; Zhuang, Z.H.; Zhao, P.X.C. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mo, X.; Chen, H.; Yang, X.; Mo, B.; Gao, L.; Yu, Y. Integrated Analysis of Transcriptome and Small RNAome Reveals the Regulatory Network for Rapid Growth in Mikania micrantha. Int. J. Mol. Sci. 2022, 23, 10596. https://doi.org/10.3390/ijms231810596
Mo X, Chen H, Yang X, Mo B, Gao L, Yu Y. Integrated Analysis of Transcriptome and Small RNAome Reveals the Regulatory Network for Rapid Growth in Mikania micrantha. International Journal of Molecular Sciences. 2022; 23(18):10596. https://doi.org/10.3390/ijms231810596
Chicago/Turabian StyleMo, Xiaowei, Haolang Chen, Xiaolan Yang, Beixin Mo, Lei Gao, and Yu Yu. 2022. "Integrated Analysis of Transcriptome and Small RNAome Reveals the Regulatory Network for Rapid Growth in Mikania micrantha" International Journal of Molecular Sciences 23, no. 18: 10596. https://doi.org/10.3390/ijms231810596
APA StyleMo, X., Chen, H., Yang, X., Mo, B., Gao, L., & Yu, Y. (2022). Integrated Analysis of Transcriptome and Small RNAome Reveals the Regulatory Network for Rapid Growth in Mikania micrantha. International Journal of Molecular Sciences, 23(18), 10596. https://doi.org/10.3390/ijms231810596