
Modeling pKa of the Brønsted Bases as an Approach to the Gibbs Energy of the Proton in Acetonitrile



Abstract
:
1. Introduction
2. Results and Discussion
3. Calculation of pKa
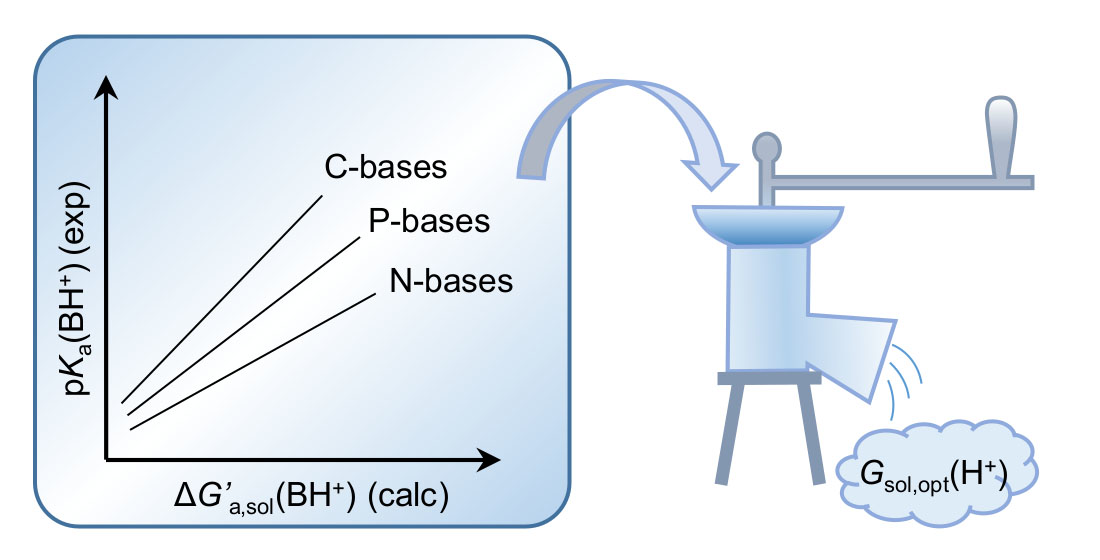
4. Estimation of ΔGsol(H+)
5. Computational Details
- M1 = B3LYP/6-311+G(2df,p)//B3LYP/6-31G(d),
- M2 = B3LYP(GD3)/6-311+G(2df,p)//B3LYP(GD3)/6-31+G(d),
- M3 = M06-2X/aug-cc-pvtz//M06-2X/cc- pVDZ
- M4 = M06-2X(GD3)/6-311++G(3df,2pd)//M06-2X/cc- pVDZ,
- M5 = MP2(fc)/6-311+G(2df,p)//M06-2X/cc-pVDZ.
- M6 = MP2(fc)/6-311+G(2df,p)//B3LYP/6-31G(d).
- M7 = B2PLYP/6-311+G(2df,p)//B3LYP/6-31G(d)
- IPCM = IPCM(ACN)/B3LYP/6-311+G(d,p)
- CPCM = CPCM(ACN)/B3LYP/6-31G(d)
- SMD1 = SMD(ACN)/M06-2X/6-31G(d)
- SMD2 = SMD(ACN)/M06-2X/6-31+G(d,p)
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ishikawa, T.; Margetić, D. Superbases for organic synthesis: Guanidines, amidines, phosphaznes and related organocatalysts. In Perspectives; Ishikawa, T., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2009; pp. 315–320. [Google Scholar]
- Puleo, T.R.; Sujansky, S.J.; Wright, S.E.; Bandar, J.S. Organic Superbases in Recent Synthetic Methodology Research. Chem. A Eur. J. 2021, 27, 4216–4229. [Google Scholar] [CrossRef] [PubMed]
- Schwesinger, R. Tricyclic 2,4-Diaminovinamidines– Readily Accessible, Very Strong CHN Bases. Angew. Chem. Int. Ed. Engl. 1987, 26, 1164–1165. [Google Scholar] [CrossRef]
- Schwesinger, R.; Missfeldt, M.; Peters, K.; von Schnering, H.G. Novel, Very Strongly Basic, Pentacyclic “Proton Sponges” with Vinamidine Structure. Angew. Chem. Int. Ed. Engl. 1987, 26, 1165–1167. [Google Scholar] [CrossRef]
- Schwesinger, R.; Schlemper, H. Peralkylated Polyaminophosphazenes—Extremely Strong, Neutral Nitrogen Bases. Angew. Chem. Int. Ed. Engl. 1987, 26, 1167–1169. [Google Scholar] [CrossRef]
- Schwesinger, R. Starke ungeladene Stickstoffbasen. Nachr. Chem. Tech. Lab. 1990, 38, 1214–1226. [Google Scholar] [CrossRef]
- Schwesinger, R.; Willaredt, J.; Schlemper, H.; Keller, M.; Schmitt, D.; Fritz, H. Novel, Very Strong, Uncharged Auxiliary Bases; Design and Synthesis of Monomeric and Polymer-Bound Triaminoiminophosphorane Bases of Broadly Varied Steric Demand. Chem. Ber. 1994, 127, 2435–2454. [Google Scholar] [CrossRef]
- Schwesinger, R.; Schlemper, H.; Hasenfratz, C.; Willaredt, J.; Dambacher, T.; Breuer, T.; Ottaway, C.; Fletschinger, M.; Boele, H.; Fritz, D.; et al. Extremly Strong, Uncharged Auxiliary Bases; Monomeric and polymer-Supported Polyaminophosphazenes (P2–P5). Liebigs Ann. 1996, 1996, 1055–1081. [Google Scholar] [CrossRef]
- Kaljurand, I.; Rodima, T.; Leito, I.; Koppel, I.A.; Schwesinger, R. Self-Consistent Spectrophotometric Basicity Scale in Acetonitrile Covering the Range between Pyridine and DBU. J. Org. Chem. 2000, 65, 6202–6208. [Google Scholar] [CrossRef]
- Štrukil, V.; Glasovac, Z.; Đilović, I.; Matković-Čalogović, D.; Šuman, L.; Kralj, M.; Eckert-Maksić, M. Triguanide Derivatives: Synthesis, Crystal Structure and Evaluation of the Proliferation Effect on Some Tumor Cell Lines. Eur. J. Org. Chem. 2012, 2012, 6785–6797. [Google Scholar] [CrossRef]
- Vazdar, K.; Kunetskiy, R.; Saame, J.; Kaupmees, K.; Leito, I.; Jahn, U. Very Strong Organosuperbases Formed by Combining Imidazole and Guanidine Bases: Synthesis, Structure, and Basicity. Angew. Chem. Int. Ed. 2014, 53, 1435–1438. [Google Scholar] [CrossRef]
- Petuškova, J.; Bruns, H.; Alcarazo, M. Cyclopropenylylidene-Stabilized Diaryl and Dialkyl Phosphenium Cations: Applications in Homogeneous Gold Catalysis. Angew. Chem. Int. Ed. 2011, 50, 3799–3802. [Google Scholar] [CrossRef]
- Bandar, J.S.; Lambert, T.H. Enantioselective Brønsted Base Catalysis with Chiral Cyclopropenimines. J. Am. Chem. Soc. 2012, 134, 5552–5555. [Google Scholar] [CrossRef]
- Bandar, J.S.; Lambert, T.H. Aminocyclopropenium Ions: Synthesis, Properties, and Applications. Synthesis 2013, 45, 2485–2498. [Google Scholar]
- Kögel, J.F.; Oelkers, B.; Kovačević, B.; Sundermeyer, J. A New Synthetic Pathway to the Second and Third Generation of Superbasic Bisphosphazene Proton Sponges: The Run for the Best Chelating Ligand for a Proton. J. Am. Chem. Soc. 2013, 35, 17768–17774. [Google Scholar] [CrossRef]
- Kögel, J.F.; Xie, X.; Baal, E.; Gesevičius, D.; Oelkers, B.; Kovačević, B.; Sundermeyer, J. Superbasic Alkyl-Substituted Bisphosphazene Proton Sponges: Synthesis, Structural Features, Thermodynamic and Kinetic Basicity, Nucleophilicity and Coordination Chemistry. Chem. Eur. J. 2014, 20, 7670–7685. [Google Scholar] [CrossRef]
- Belding, L.; Dudding, T. Synthesis and Theoretical Investigation of a 1,8-Bis(bis(diisopropylamino)cyclopropeniminyl)naphthalene Proton Sponge Derivative. Chem. Eur. J. 2014, 20, 1032–1037. [Google Scholar] [CrossRef]
- Belding, L.; Stoyanov, L.; Dudding, T. Synthesis, Theoretical Analysis, and Experimental pKa Determination of a Fluorescent, Nonsymmetric, In–Out Proton Sponge. J. Org. Chem. 2016, 81, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Angelici, R.J. Basicities of Transition Metal Complexes from Studies of Their Heats of Protonation: A Guide to Complex Reactivity. Acc. Chem. Res. 1995, 28, 51–60. [Google Scholar] [CrossRef]
- Vazdar, K.; Margetić, D.; Kovačević, B.; Sundermeyer, J.; Leito, I.; Jahn, U. Design of Novel Uncharged Organic Superbases: Merging Basicity and Functionality. Acc. Chem. Res. 2021, 54, 3108–3123. [Google Scholar] [CrossRef]
- Weitkamp, R.F.; Neumann, B.; Stammler, H.-G.; Hoge, B. Phosphorus-Containing Superbases: Recent Progress in the Chemistry of Electron-Abundant Phosphines and Phosphazenes. Chem. Eur. J. 2021, 27, 10807–10825. [Google Scholar] [CrossRef]
- Tang, J.; Dopke, J.; Verkade, J.G. Synthesis of New Exceedingly Strong Non-ionic Bases: RN:P(MeNCH2CH2)3N. J. Am. Chem. Soc. 1993, 115, 5015–5020. [Google Scholar] [CrossRef]
- Verkade, J.G. Atranes: New Examples with Unexpected Properties. Acc. Chem. Res. 1993, 26, 483–489. [Google Scholar] [CrossRef]
- Verkade, J.G. Main Group Atranes: Chemical and Structural Features. Coord. Chem. Rev. 1994, 137, 233–295. [Google Scholar] [CrossRef]
- Wroblewski, A.E.; Pinkas, J.; Verkade, J.G. Strongly Basic Proazaphosphatranes: P(EtNCH2CH2)3N and P(iso-PrNCH2CH2)3N. Main Group Chem. 1995, 1, 69–79. [Google Scholar] [CrossRef]
- Verkade, J.G. P(RNCH2CH2)3N as Very Strong Non-Ionic Bases and Catalysts: Recent Advances and Applications. Phosphorus Sulfur Silicon Relat. Elem. 2002, 177, 1621–1631. [Google Scholar] [CrossRef]
- Verkade, J.G. P(RNCH2CH2)3N: Very Strong Non-Ionic Bases Useful in Organic Synthesis. Top. Curr. Chem. 2003, 223, 1–44. [Google Scholar]
- Verkade, J.G.; Kisanga, P.B. Recent Applications of Proazaphosphatranes in Organic Synthesis. Aldrichimica. Acta 2004, 37, 3–14. [Google Scholar] [CrossRef]
- Münchenberg, J.; Böge, O.; Fischer, A.K.; Jones, P.G.; Schmutzler, R. Synthesis and Structure of Tetraorganoguanidinyl–Substituted-Phosphorus–Halogen Compounds and of Tris–N–(N′,N′,N′′,N′′–Tetramethyl)–Guanidinyl Phosphonium Salts. Phosphorus Sulfur Silicon Relat. Elem. 1994, 86, 103–121. [Google Scholar] [CrossRef]
- Plack, V.; Münchenberg, J.; Thönnessen, H.; Jones, P.G.; Schmutzler, R. Synthesis of Stable N′,N′,N′′,N′′-Tetramethylguanidine-Substituted σ4(P)- and σ3(P)-Organophosphorus Compounds with N-Protonated P–N Bonds - The First σ3-Phosphorus-Substituted Ammonium Salts. Eur. J. Inorg. Chem. 1998, 1998, 865–875. [Google Scholar] [CrossRef]
- Freytag, M.; Plack, V.; Jones, P.G.; Schmutzler, R. Tris–N–(N′,N′,N″,N″–tetramethylguanidyl)phosphine Oxide – Synthesis and Reaction with Acids. Z. Naturforsch. B 2004, 59, 499–502. [Google Scholar]
- Buß, F.; Röthel, M.B.; Werra, J.A.; Rotering, P.; Wilm, L.F.B.; Daniliuc, C.G.; Löwe, P.; Dielmann, F. Tris(tetramethylguanidinyl)phosphine: The Simplest Non-ionic Phosphorus Superbase and Strongly Donating Phosphine Ligand. Chem. Eur. J. 2022, 28, e202104021. [Google Scholar] [CrossRef]
- Marchenko, A.P.; Koidan, G.N.; Pinchuk, A.M.; Kirsanov, A.V. Phosphorylated Phosphorimidic Triamides. Zh. Obshch. Khim. 1984, 54, 1774–1782. [Google Scholar]
- Ullrich, S.; Kovačević, B.; Xie, X.; Sundermeyer, J. Phosphazenyl Phosphines: The Most Electron-Rich Uncharged Phosphorus Brønsted and Lewis Bases. Angew. Chem. Int. Ed. 2019, 58, 10335–10339. [Google Scholar] [CrossRef]
- Mehlmann, P.; Dielmann, F. Switching the Electron-Donating Ability of Phosphines through Proton-Responsive Imidazolin-2-ylidenamino Substituents. Chem. Eur. J. 2019, 25, 2352–2357. [Google Scholar] [CrossRef]
- Kisanga, P.B.; Verkade, J.G.; Schwesinger, R. pKa Measurements of P(RNCH2CH3)3N. J. Org. Chem. 2000, 65, 5431–5432. [Google Scholar] [CrossRef]
- Kaljurand, I.; Koppel, I.A.; Kütt, A.; Rõõm, E.-I.; Rodima, T.; Koppel, I.; Mishima, M.; Leito, I. Experimental Gas-Phase Basicity Scale of Superbasic Phosphazenes. J. Phys. Chem. A 2007, 111, 1245–1250. [Google Scholar] [CrossRef]
- Leito, I.; Koppel, I.A.; Koppel, I.; Kaupmees, K.; Tshepelevitsh, S.; Saame, J. Basicity Limits of Neutral Organic Superbases. Angew. Chem. Int. Ed. 2015, 54, 9262–9265. [Google Scholar] [CrossRef]
- Saame, J.; Rodima, T.; Tshepelevitsh, S.; Kütt, A.; Kaljurand, I.; Haljasorg, T.; Koppel, I.A.; Leito, I. Experimental Basicities of Superbasic Phosphonium Ylides and Phosphazenes. J. Org. Chem. 2016, 81, 7349–7361. [Google Scholar] [CrossRef]
- Kögel, J.F.; Margetić, D.; Xie, X.; Finger, L.H.; Sundermeyer, J. A Phosphorus Bisylide: Exploring a New Class of Superbases with Two Interacting Carbon Atoms as Basicity Centers. Angew. Chem. Int. Ed. 2017, 56, 3090–3093. [Google Scholar] [CrossRef]
- Kovačević, B.; Maksić, Z.B. High Basicity of Phosphorus–Proton Affinity of Tris-(Tetramethylguanidinyl)phosphine and Tris-(Hexamethyltriaminophosphazenyl)phosphine by DFT Calculations. Chem. Commun. 2006, 2006, 1524–1526. [Google Scholar] [CrossRef]
- pKa(PP3H+) Was Measured in THF and Its Value in Acetonitrile Was Estimated to 43.4 Using the Correlation for the “Transfer between the Solvents”. Available online: https://analytical.chem.ut.ee/HA_UT/ (accessed on 27 August 2022).
- Liu, M.; Yang, I.; Buckley, B.; Lee, J.K. Proton Affinities of Phosphines versus N-Heterocyclic Carbenes. Org. Lett. 2010, 12, 4764–4767. [Google Scholar] [CrossRef]
- Liptak, M.D.; Shields, G.C. Accurate pKa Calculations for Carboxylic Acids Using Complete Basis Set and Gaussian-n Models Combined with CPCM Continuum Solvation Methods. J. Am. Chem. Soc. 2001, 123, 7314–7319. [Google Scholar] [CrossRef]
- Glasovac, Z.; Eckert-Maksić, M.; Maksić, Z.B. Basicity of Organic Bases and Superbases in Acetonitrile by the Polarized Continuum Model and DFT Calculations. New J. Chem. 2009, 33, 588–597. [Google Scholar] [CrossRef]
- Klamt, A.; Eckert, F.; Diedenhofen, M.; Beck, M.E. First Principles Calculations of Aqueous pKa Values for Organic and Inorganic Acids Using COSMO-RS Reveal an Inconsistency in the Slope of the pKa Scale. J. Phys. Chem. A 2003, 107, 9380–9386. [Google Scholar] [CrossRef] [Green Version]
- Eckert, F.; Leito, I.; Kaljurand, I.; Kütt, A.; Klamt, A.; Diedenhofen, M. Prediction of acidity in acetonitrile solution with COSMO-RS. J. Comput. Chem. 2009, 30, 799–810. [Google Scholar] [CrossRef]
- Peräkyllä, M. Ab Initio Quantum Mechanical Study on the Origin of the pK(a) Differences of the Proton Sponges 1,8-Bis(dimethylamino)naphthalene, 1,8-Bis(dimethylamino)-2,7-dimethoxynaphthalene, 1,6-Dimethyl-1,6-diazacyclodecane, and 1,6-Diazabicyclo[4.4.4]tetradecane. J. Org. Chem. 1996, 61, 7420–7425. [Google Scholar] [CrossRef]
- Kovačević, B.; Maksić, Z.B. Basicity of Some Organic Superbases in Acetonitrile. Org. Lett. 2001, 3, 1523–1526. [Google Scholar] [CrossRef]
- Busch, M.; Ahlberg, E.; Ahlberg, E.; Laasonen, K. How to Predict the pKa of Any Compound in Any Solvent. ACS Omega 2022, 7, 17369–17383. [Google Scholar] [CrossRef]
- Malloum, A.; Fifen, J.J.; Conradie, J. Determination of the Absolute Solvation Free Energy and Enthalpy of the Proton in Solutions. J. Mol. Liq. 2021, 322, 114919. [Google Scholar] [CrossRef]
- Matsui, T.; Baba, T.; Kamiyad, K.; Shigeta, Y. An Accurate Density Functional Theory Based Estimation of pKa Values of Polar Residues Combined with Experimental Data: From Amino acids to Minimal Proteins. Phys. Chem. Chem. Phys. 2012, 14, 4181–4187. [Google Scholar] [CrossRef]
- Fujiki, R.; Matsui, T.; Shigeta, Y.; Nakano, H.; Yoshida, N. Recent Developments of Computational Methods for pKa Prediction Based on Electronic Structure Theory with Solvation Models. J 2021, 4, 849–864. [Google Scholar] [CrossRef]
- Lias, S.G.; Bartmess, J.E.; Liebman, J.F.; Holmes, J.L.; Levin, R.D.; Mallard, W.G. “Ion Energetics Data” in NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom, P.J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2022.
- Cooks, R.G.; Kruger, T.L. Intrinsic Basicity Determination Using Metastable Ions. J. Am. Chem. Soc. 1977, 99, 1279–1281. [Google Scholar] [CrossRef]
- Bartmess, J.E. The bracketing method for determining the thermochemistry of ion-molecule reactions. In Encyclopedia of Mass Spectrometry; Elsevier: Amsterdam, The Netherlands, 2003; pp. 315–319. [Google Scholar]
- Cerda, B.A.; Wesdemiotis, C. Li+, Na+, and K+ Binding to the DNA and RNA Nucleobases. Bond Energies and Attachment Sites from the Dissociation of Metal Ion-Bound Heterodimers. J. Am. Chem. Soc. 1996, 118, 11884–11892. [Google Scholar] [CrossRef]
- Glasovac, Z.; Pavošević, F.; Štrukil, V.; Eckert-Maksić, M.; Schlangen, M.; Kretschmer, R. Toward Extension of the Gas-phase Basicity Scale by Novel Pyridine Containing Guanidines. Int. J. Mass Spectrom. 2013, 354–355, 113–122. [Google Scholar] [CrossRef]
- Glasovac, Z.; Eckert-Maksić, M.; Kaljurand, I.; Saame, J.; Leito, I. Gas phase Basicity of Biguanides – Comparison of the Equilibrium and the Kinetic Methods. Int. J. Mass Spectrom. 2019, 435, 61–68. [Google Scholar] [CrossRef]
- Haav, K.; Saame, J.; Kütt, A.; Leito, I. Basicity of Phosphanes and Diphosphanes in Acetonitrile. Eur. J. Org. Chem. 2012, 2012, 2167–2172. [Google Scholar] [CrossRef]
- Li, J.-N.; Fu, Y.; Liu, L.; Guo, Q.-X. First-Principle Predictions of Basicity of Organic Amines and Phosphines in Acetonitrile. Tetrahedron 2006, 62, 11801–11813. [Google Scholar] [CrossRef]
- Kaljurand, I.; Kütt, A.; Sooväli, L.; Rodima, T.; Mäemets, V.; Leito, I.; Koppel, I.A. Extension of the Self-Consistent Spectrophotometric Basicity Scale in Acetonitrile to a Full Span of 28 pKa Units: Unification of Different Basicity Scales. J. Org. Chem. 2005, 70, 1019–1028. [Google Scholar] [CrossRef]
- Abdur-Rashid, K.; Fong, T.P.; Greaves, B.; Gusev, D.G.; Hinman, J.G.; Landau, S.E.; Lough, A.J.; Morris, R.H. An Acidity Scale for Phosphorus-Containing Compounds Including Metal Hydrides and Dihydrogen Complexes in THF: Toward the Unification of Acidity Scales. J. Am. Chem. Soc. 2000, 122, 9155–9171. [Google Scholar] [CrossRef]
- Streuli, C.A. Determination of Basicity of Substituted Phosphines by Nonaqueous Titrimetry. Anal. Chem. 1960, 32, 985–987. [Google Scholar] [CrossRef]
- Greb, L.; Quack, S.; Schirmer, B.; Oña-Burgos, P.; Kaupmees, K.; Lõkov, M.; Leito, I.; Grimme, S.; Paradies, J. Electronic effects of triarylphosphines in metal-free hydrogen activation: A kinetic and computational study. Chem. Sci. 2013, 4, 2788–2796. [Google Scholar] [CrossRef]
- Tshepelevitsh, S.; Kütt, A.; Lõkov, M.; Kaljurand, I.; Saame, J.; Heering, A.; Plieger, P.; Vianello, R.; Leito, I. On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem. 2019, 2019, 6735–6748. [Google Scholar] [CrossRef]
- Mehlmann, P.; Mück-Lichtenfeld, C.; Tan, T.T.Y.; Dielmann, F. Tris(imidazolin-2-ylidenamino)phosphine: A Crystalline Phosphorus(III) Superbase That Splits Carbon Dioxide. Chem. Eur. J. 2017, 23, 5929–5933. [Google Scholar] [CrossRef]
- Himmel, D.; Goll, S.K.; Leito, I.; Krossing, I. Anchor Points for the Unified Brønsted Acidity Scale: The rCCC Model for the Calculation of Standard Gibbs Energies of Proton Solvation in Eleven Representative Liquid Media. Chem. Eur. J. 2011, 17, 5808–5826. [Google Scholar] [CrossRef]
- Carvalho, N.F.; Pliego, J.R. Cluster-continuum Quasichemical Theory Calculation of the Lithium Ion Solvation in Water, Acetonitrile and Dimethyl Sulfoxide: An Absolute Single-ion Solvation Free Energy Scale. Phys. Chem. Chem. Phys. 2015, 17, 26745–26755. [Google Scholar] [CrossRef]
- Rossini, E.; Knapp, E.-W. Proton Solvation in Protic and Aprotic Solvents. J. Comput. Chem. 2016, 37, 1082–1091. [Google Scholar] [CrossRef]
- Fawcett, W.R. The Ionic Work Function and its Role in Estimating Absolute Electrode Potentials. Langmuir 2008, 24, 9868–9875. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09. In Rev D. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results Obtained with the Correlation Energy Density Functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [PubMed]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 0618–0622. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. Semi-direct Algorithms for the MP2 Energy and Gradient. Chem. Phys. Lett. 1990, 166, 281–289. [Google Scholar] [CrossRef]
- Foresman, J.B.; Keith, T.A.; Wiberg, K.B.; Snoonian, J.; Frisch, M.J. Solvent Effects. 5. Influence of Cavity Shape, Truncation of Electrostatics, and Electron Correlation on ab Initio Reaction Field Calculations. J. Phys. Chem. 1996, 100, 16098–16104. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comp. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Schaftenaar, G.; Vlieg, E.; Vriend, G. Molden 2.0: Quantum chemistry meets proteins. J. Comp. Aided. Mol. Des. 2017, 31, 789–800. [Google Scholar] [CrossRef]
- Schaftenaar, G.; Nordik, J.H. Molden: A Pre- and Post-processing Program for Molecular and Electronic Structures. J. Comput. Aided Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef]
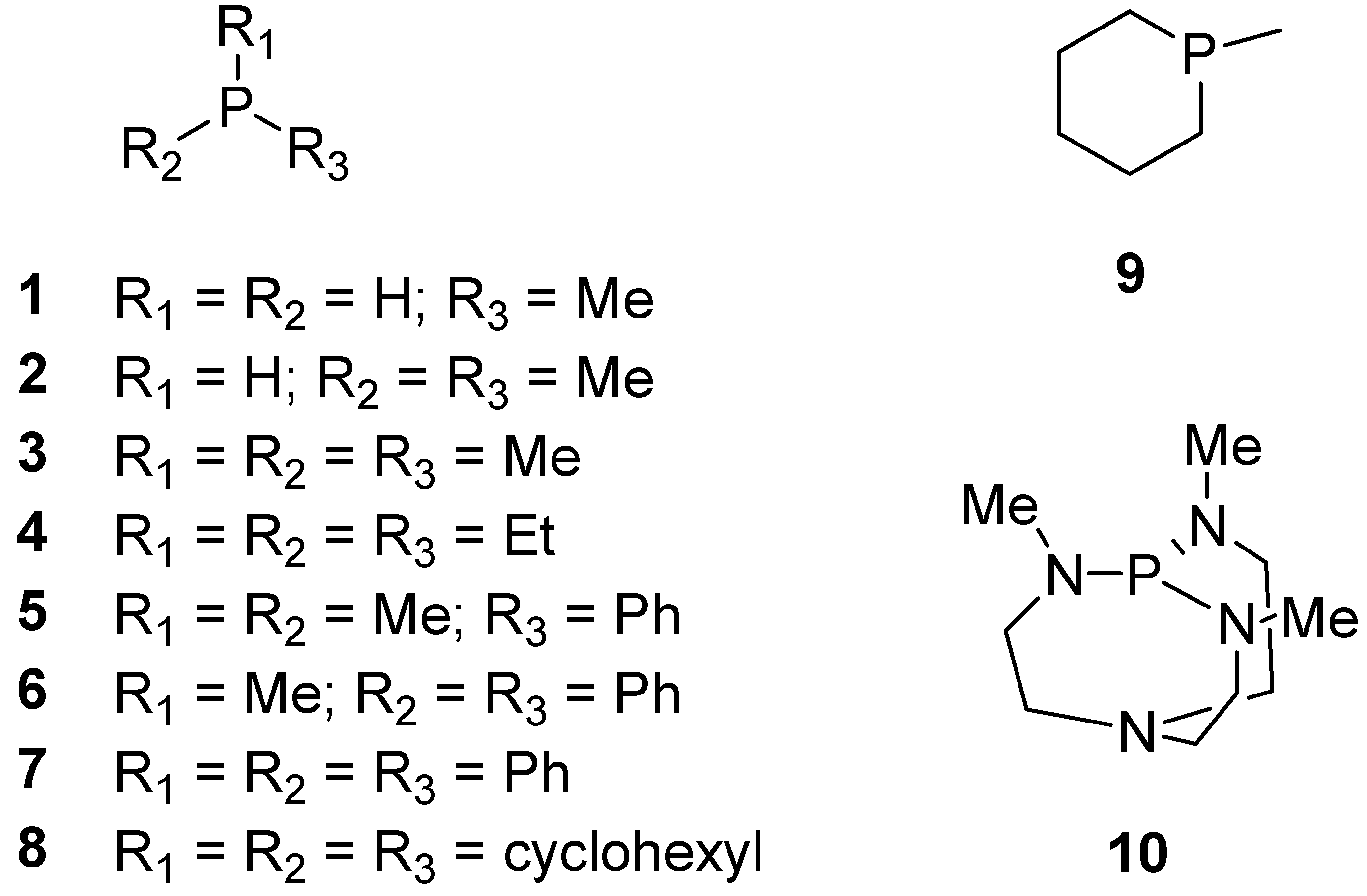


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Base | GB | |||||||
|---|---|---|---|---|---|---|---|---|
| exp | M1 | M2 | M3 | M4 | M5 | M6 | M7 | |
| 1 | 195.4 | 196.9 | 196.9 | 194.5 | 195.2 | 196.6 | 196.5 | 199.7 |
| 2 | 209.8 | 210.6 | 210.5 | 207.5 | 208.3 | 209.2 | 209.1 | 213.3 |
| 3 | 221.4 | 221.5 | 221.8 | 217.4 | 218.4 | 218.7 | 218.9 | 224.1 |
| 4 | 227.5 | 227.4 | 227.2 | 223.3 | 224.2 | 223.7 | 223.9 | 229.7 |
| 5 | 223.9 | 225.3 | 225.7 | 220.3 | 221.2 | 221.3 | 221.1 | 227.6 |
| 6 | 224.6 | 227.0 | 227.8 | 221.9 | 222.7 | 221.9 | 221.8 | 229.9 |
| 7 | 224.8 | 229.2 | 230.4 | 223.5 | 224.3 | 222.5 | 222.8 | 232.0 |
| 8 | 236.4 (241) b | 239.4 | 240.3 | 235.3 | 236.2 | 234.7 | 234.3 | 242.6 |
| 9 | 223.9 | 225.8 | 225.6 | 221.8 | 222.7 | 222.3 | 222.4 | 228.5 |
| 10 | 259.0 | 252.8 | 251.1 | 249.2 | 251.6 | 258.8 | 258.1 | 257.0 |
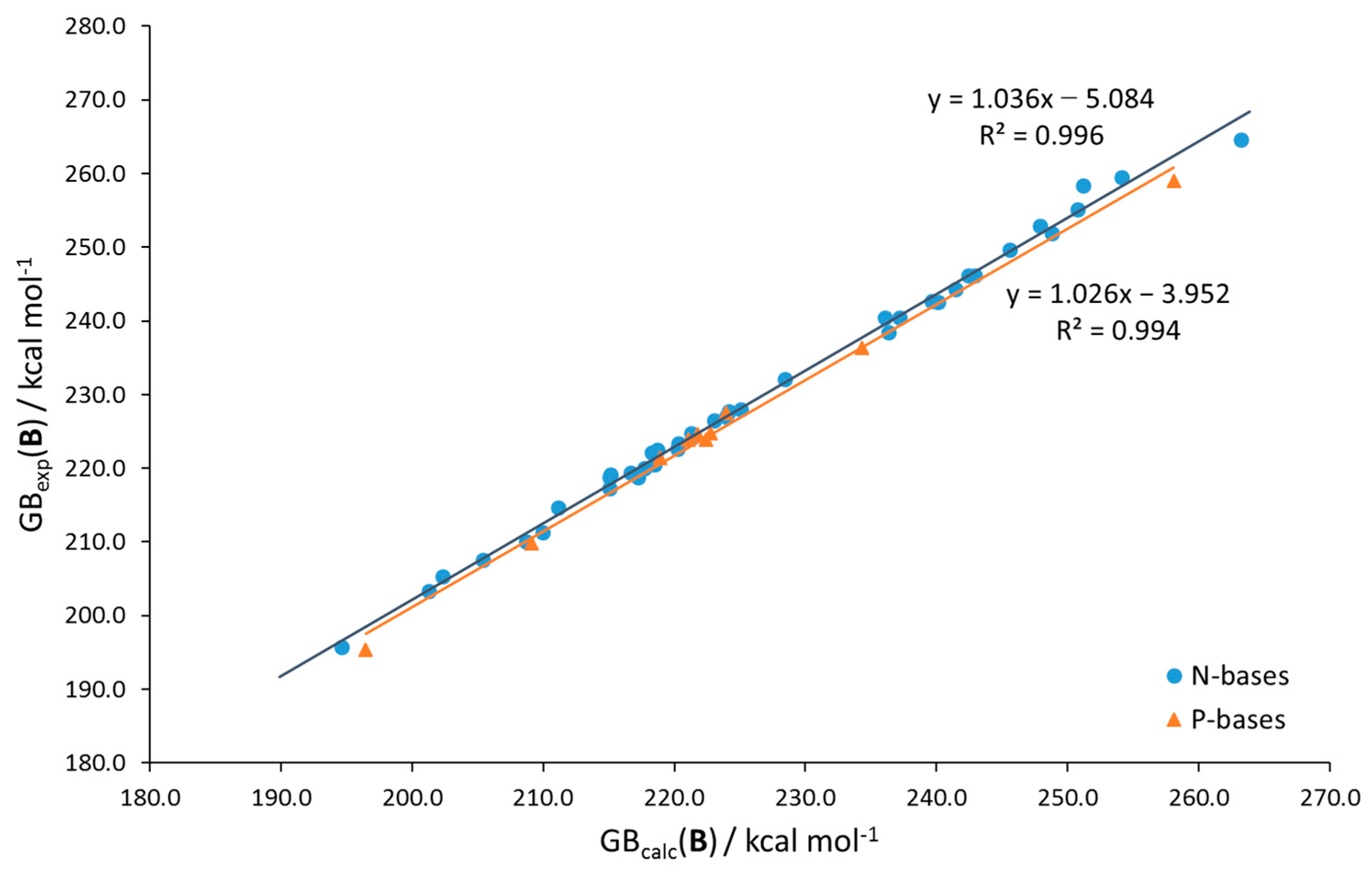
| slope intercept R2 | 1.081 −19.1 0.975 | 1.084 −20.1 0.958 | 1.117 −22.8 0.986 | 1.091 −18.2 0.990 | 1.014 −1.5 0.992 | 1.026 −4.0 0.994 | 1.059 −17.3 0.979 |
| Base B | ACNpKa (BH+) | //M6 | Base B | ACNpKa (BH+) | //M6 | ||||
|---|---|---|---|---|---|---|---|---|---|
| exp. | CPCM | IPCM | SMD1 | exp. | CPCM | IPCM | SMD1 | ||
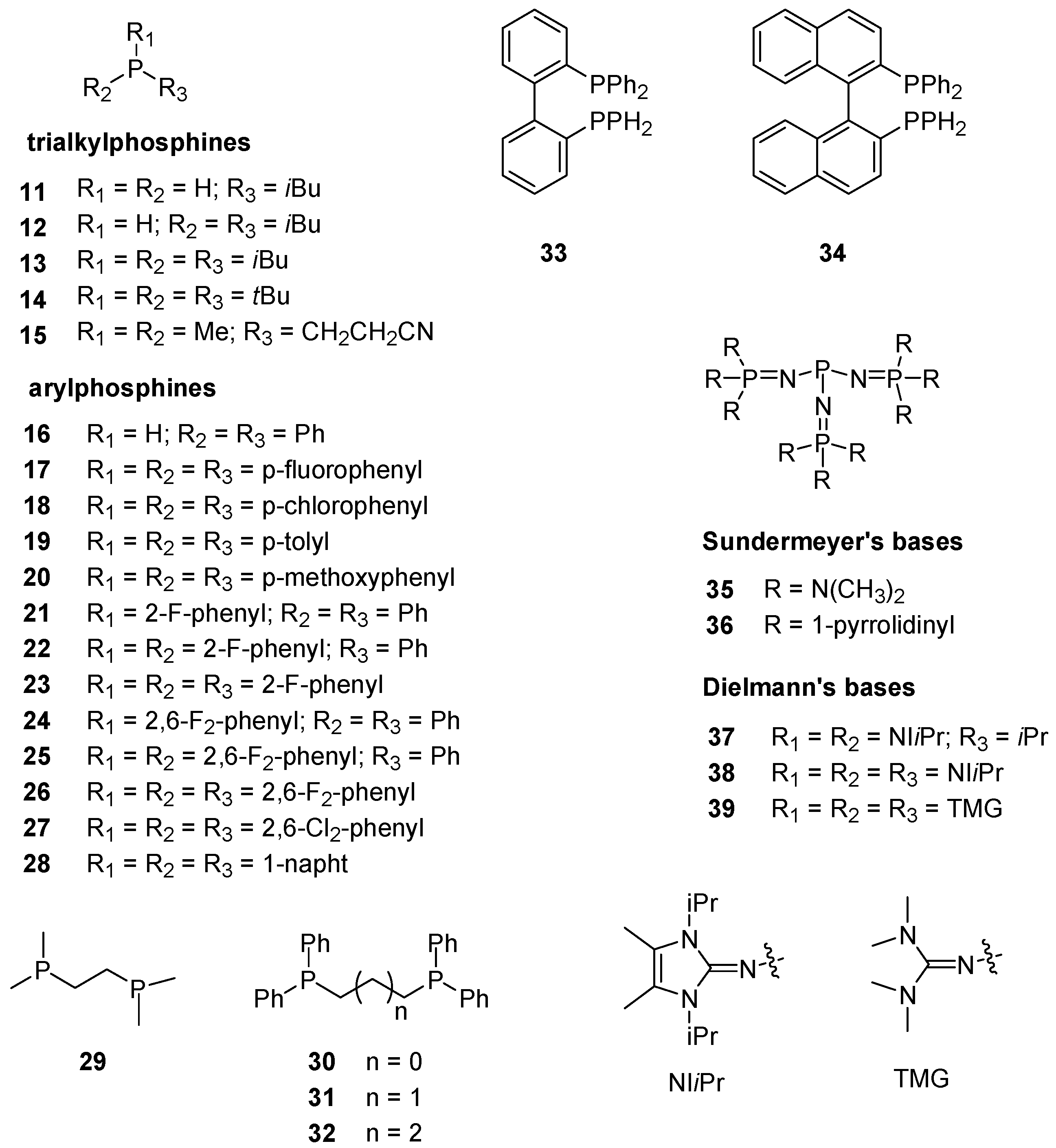
| 3 | 15.5 b | 16.4 | 15.7 | 17.4 | 22 | 4.6 j | 4.1 | 4.9 | 4.0 |
| 4 | 16.6 c | 16.0 | 15.1 | 17.4 | 23 | 3.0 j | 2.7 | 3.6 | 2.2 |
| 5 | 12.7 b | 13.1 | 12.9 | 14.2 | 24 | 5.2 j | 5.7 | 6.0 | 4.8 |
| 6 | 10.0 d | 9.9 | 9.8 | 10.2 | 25 | 2.5 j | 2.7 | 3.7 | 2.0 |
| 7 | 7.6 b | 7.3 | 7.7 | 7.3 | 26 | 0.7 j | −0.3 | 0.5 | −2.1 |
| 8 | 16.1 e,f | 16.9 | 15.7 | 18.6 | 27 | 1.7 j | 2.1 | 3.1 | −1.4 |
| 10 | 32.9 g | 34.1 | 32.7 | 36.0 | 28 | 6.6 j | 6.6 | 7.0 | 6.1 |
| 11 | 5.2 c | 6.2 | 5.5 | 7.4 | 29 | 15.6 b | 15.2 | 14.0 | 16.5 |
| 12 | 10.6 c | 11.7 | 10.2 | 12.9 | 30 | 9.4 b | 9.4 | 9.0 | 9.6 |
| 13 | 15.7 c | 15.4 | 13.8 | 16.8 | 31 | 9.8 b | 9.7 | 9.0 | 10.1 |
| 14 | 17.0 e | 17.9 | 17.4 | 20.0 | 32 | 10.2 b | 9.3 | 8.2 | 9.5 |
| 15 | 13.5 c | 12.3 | 11.7 | 14.1 | 33 | 8.0 b | 7.7 | 8.6 | 6.2 |
| 16 | 4.2 h | 5.0 | 5.1 | 5.3 | 34 | 7.8 b | 8.3 | 8.8 | 6.9 |
| 17 | 6.6 i | 5.7 | 6.7 | 6.4 | 35 | 43.4 f,k | 41.4 | 42.7 | 38.8 |
| 18 | 5.4 i | 5.5 | 5.0 | 6.2 | 36 | 45.4 f,k | 44.1 | 45.3 | 41.6 |
| 19 | 9.1 i | 8.3 | 8.5 | 8.1 | 37 | 32.7 l | 32.4 | 32.4 | 31.7 |
| 20 | 10.1 b | 9.9 | 10.9 | 10.1 | 38 | 29.5 m | 32.5 | 32.6 | 31.3 |
| 21 | 6.1 j | 5.8 | 6.4 | 5.7 | 39 | 38.8 n | 38.8 | 39.3 | 37.7 |
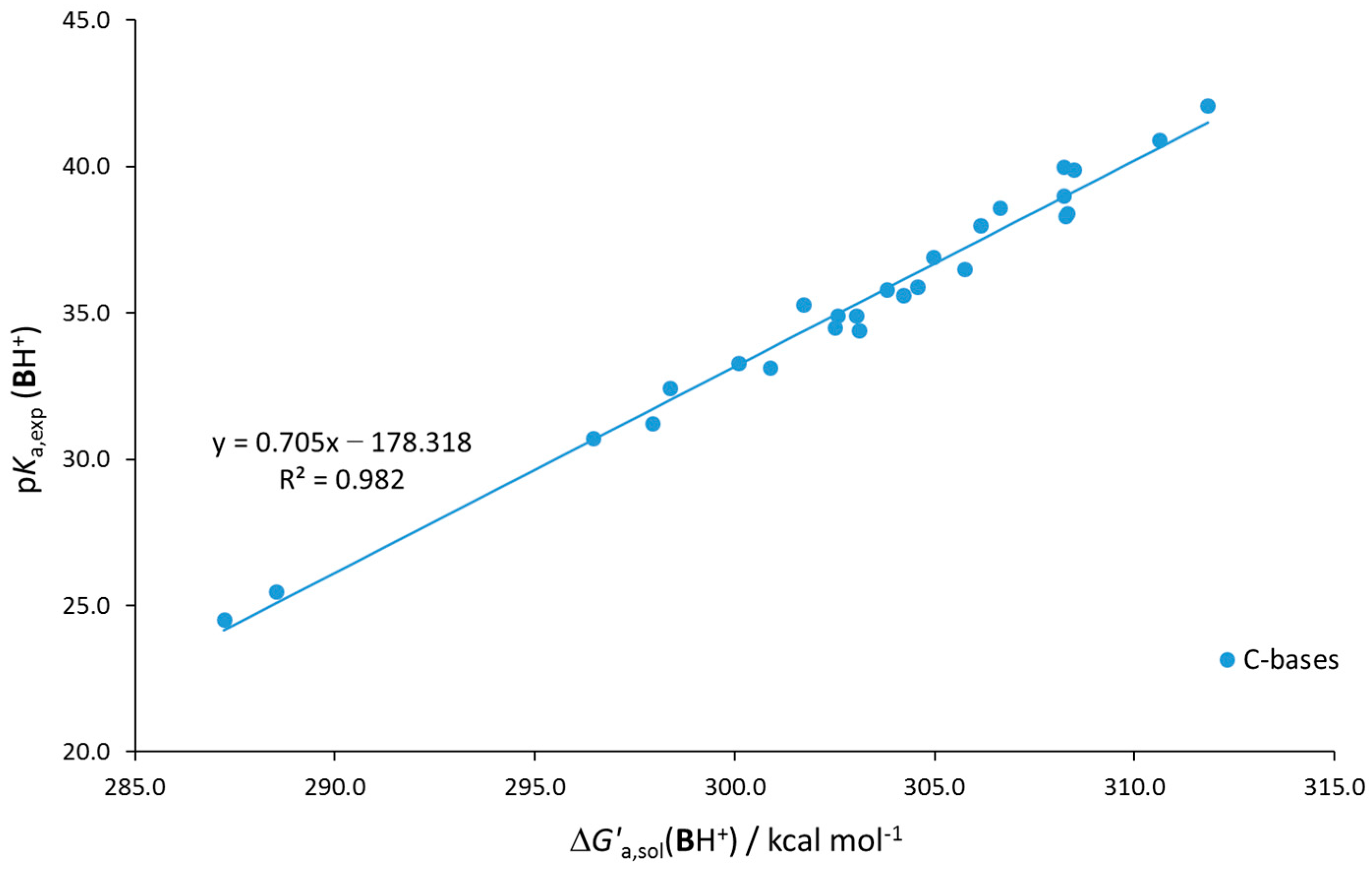
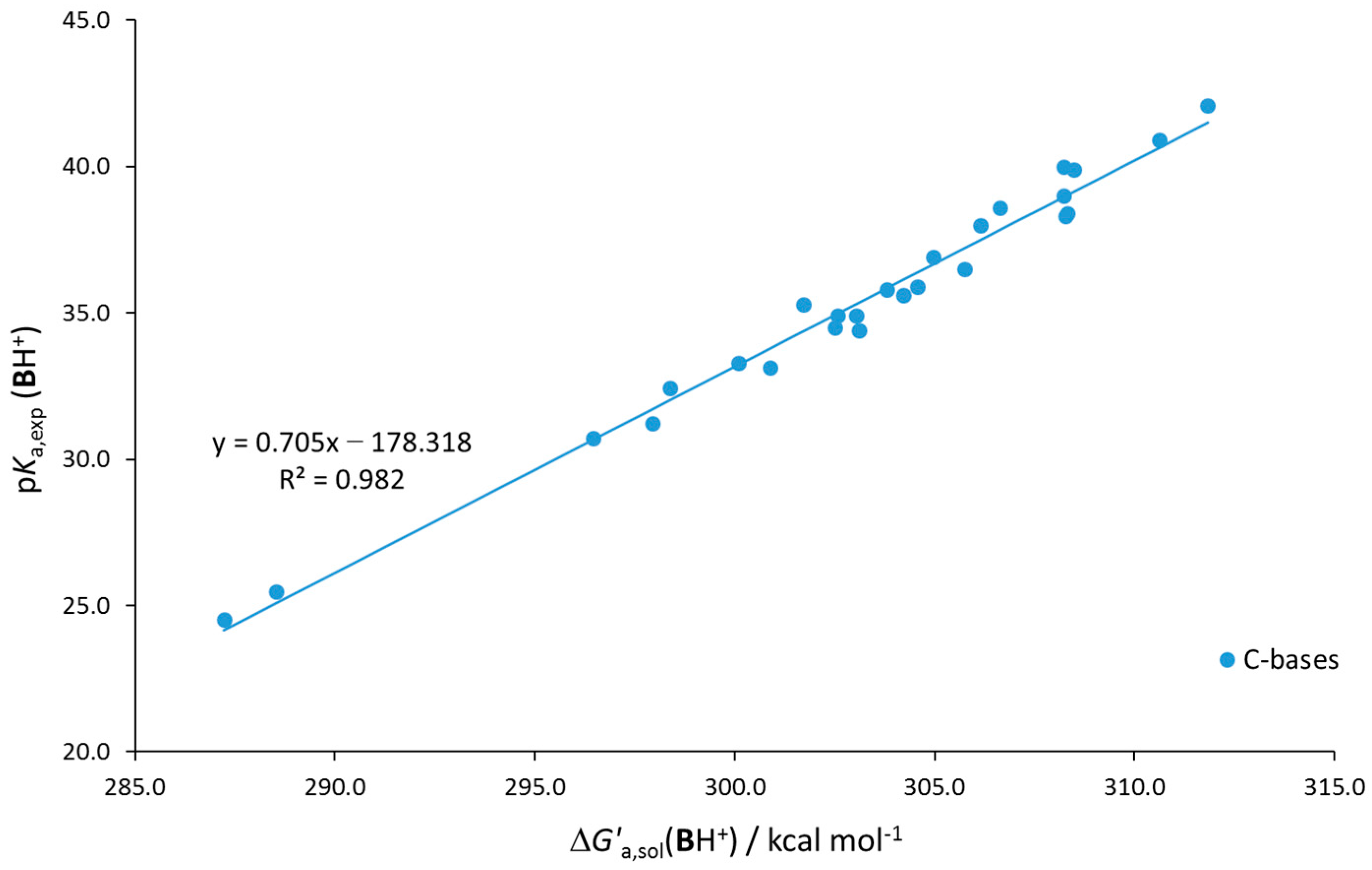
| a b R2 | – | 0.690 −173.2 0.994 | 0.637 −158.8 0.993 | 0.726 −187.6 0.977 | |||||
| MUE RMS | – | 0.7 0.9 | 0.7 1.0 | 1.3 1.8 |
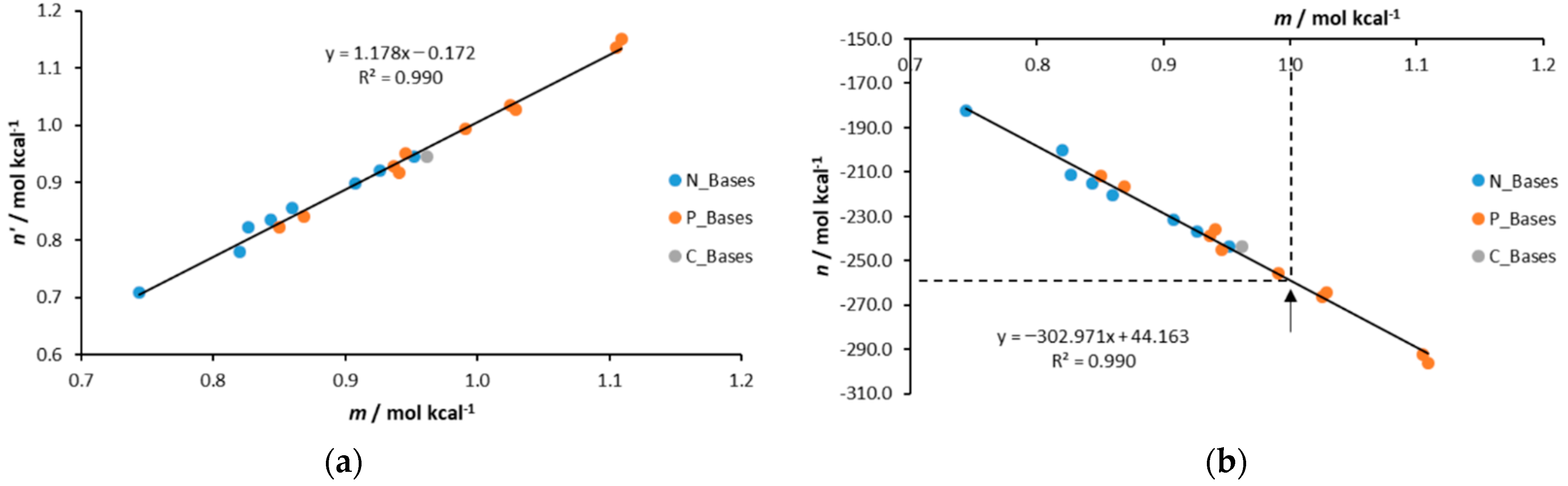
| Method | m | n | n’ | Gsol,calc(H+) b | Δ(Gsol,calc(H+)) | |
|---|---|---|---|---|---|---|
| Lit. data | −257.3 c | 0.0 | ||||
| N_Bases | IPCM//M1 | 0.744 | −182.153 | 0.708 | −244.7 | 12.5 |
| CPCM//M1 | 0.827 | −211.419 | 0.822 | −255.8 | 1.5 | |
| SMD1//M1 | 0.860 | −220.408 | 0.857 | −256.4 | 0.9 | |
| IPCM//M6 | 0.820 | −200.367 | 0.779 | −244.4 | 12.9 | |
| CPCM//M6 | 0.908 | −231.446 | 0.900 | −255.0 | 2.3 | |
| SMD1//M6 | 0.952 | −243.467 | 0.946 | −255.7 | 1.5 | |
| SMD2//M3 | 0.844 | −214.846 | 0.835 | −254.7 | 2.6 | |
| SMD2//M5 | 0.926 | −236.972 | 0.921 | −255.8 | 1.5 | |
| P_Bases | IPCM//M1 | 0.937 | −238.865 | 0.929 | −254.8 | 2.4 |
| CPCM//M1 | 1.029 | −264.296 | 1.027 | −256.9 | 0.4 | |
| SMD1//M1 | 1.105 | −292.404 | 1.137 | −264.6 | 7.3 | |
| IPCM//M6 | 0.869 | −216.304 | 0.841 | −248.9 | 8.4 | |
| CPCM//M6 | 0.941 | −235.924 | 0.917 | −250.6 | 6.7 | |
| SMD1//M6 | 0.991 | −255.585 | 0.993 | −258.0 | 0.8 | |
| SMD2//M3 | 1.025 | −266.374 | 1.035 | −260.0 | 2.7 | |
| SMD2//M5 | 0.946 | −244.794 | 0.952 | −258.9 | 1.6 | |
| IPCM//M5 | 0.850 | −211.562 | 0.822 | −248.9 | 8.4 | |
| SMD2//M2 | 1.109 | −296.051 | 1.151 | −266.9 | 9.6 | |
| C_Bases | IPCM//M6 | 0.962 | −243.305 | 0.946 | −252.9 | 4.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glasovac, Z.; Kovačević, B. Modeling pKa of the Brønsted Bases as an Approach to the Gibbs Energy of the Proton in Acetonitrile. Int. J. Mol. Sci. 2022, 23, 10576. https://doi.org/10.3390/ijms231810576
Glasovac Z, Kovačević B. Modeling pKa of the Brønsted Bases as an Approach to the Gibbs Energy of the Proton in Acetonitrile. International Journal of Molecular Sciences. 2022; 23(18):10576. https://doi.org/10.3390/ijms231810576
Chicago/Turabian StyleGlasovac, Zoran, and Borislav Kovačević. 2022. "Modeling pKa of the Brønsted Bases as an Approach to the Gibbs Energy of the Proton in Acetonitrile" International Journal of Molecular Sciences 23, no. 18: 10576. https://doi.org/10.3390/ijms231810576
APA StyleGlasovac, Z., & Kovačević, B. (2022). Modeling pKa of the Brønsted Bases as an Approach to the Gibbs Energy of the Proton in Acetonitrile. International Journal of Molecular Sciences, 23(18), 10576. https://doi.org/10.3390/ijms231810576