
Glial Cell-Mediated Neuroinflammation in Alzheimer’s Disease




Abstract

1. Introduction
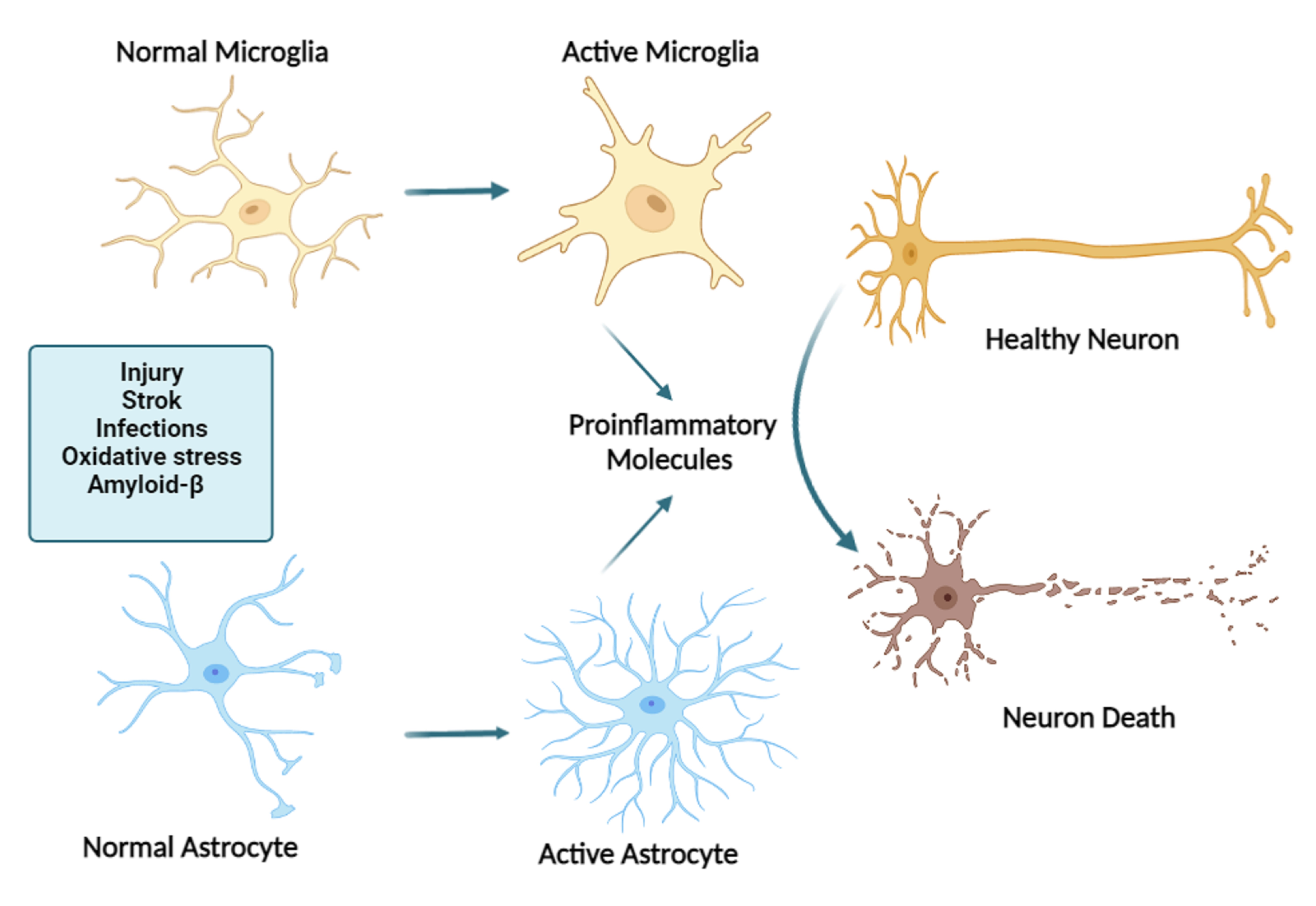
2. Neuroinflammation in Alzheimer’s Disease
Factors Affecting Neuroinflammation in Alzheimer’s Disease
3. Glial Cells Role in Neuroinflammation
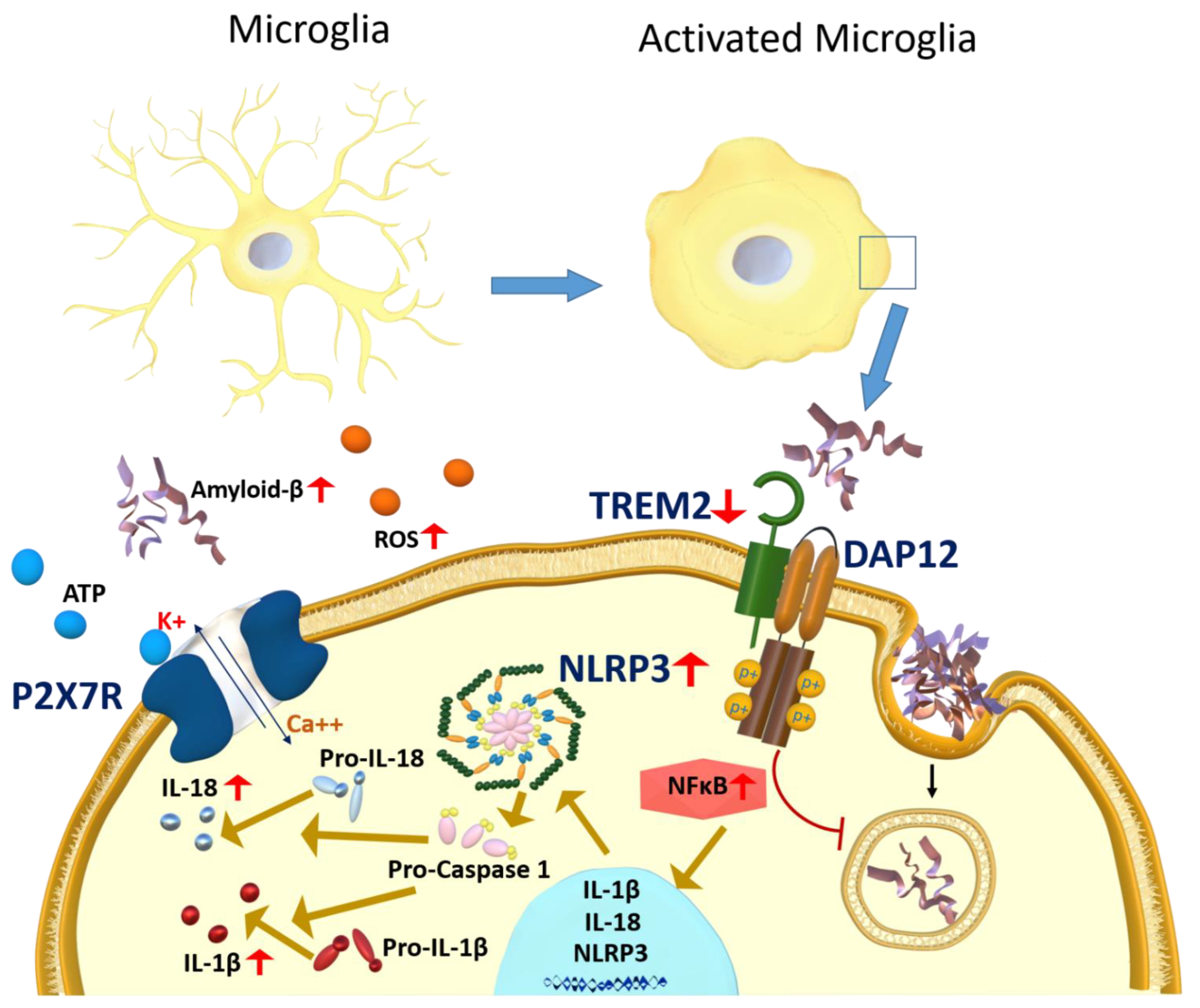
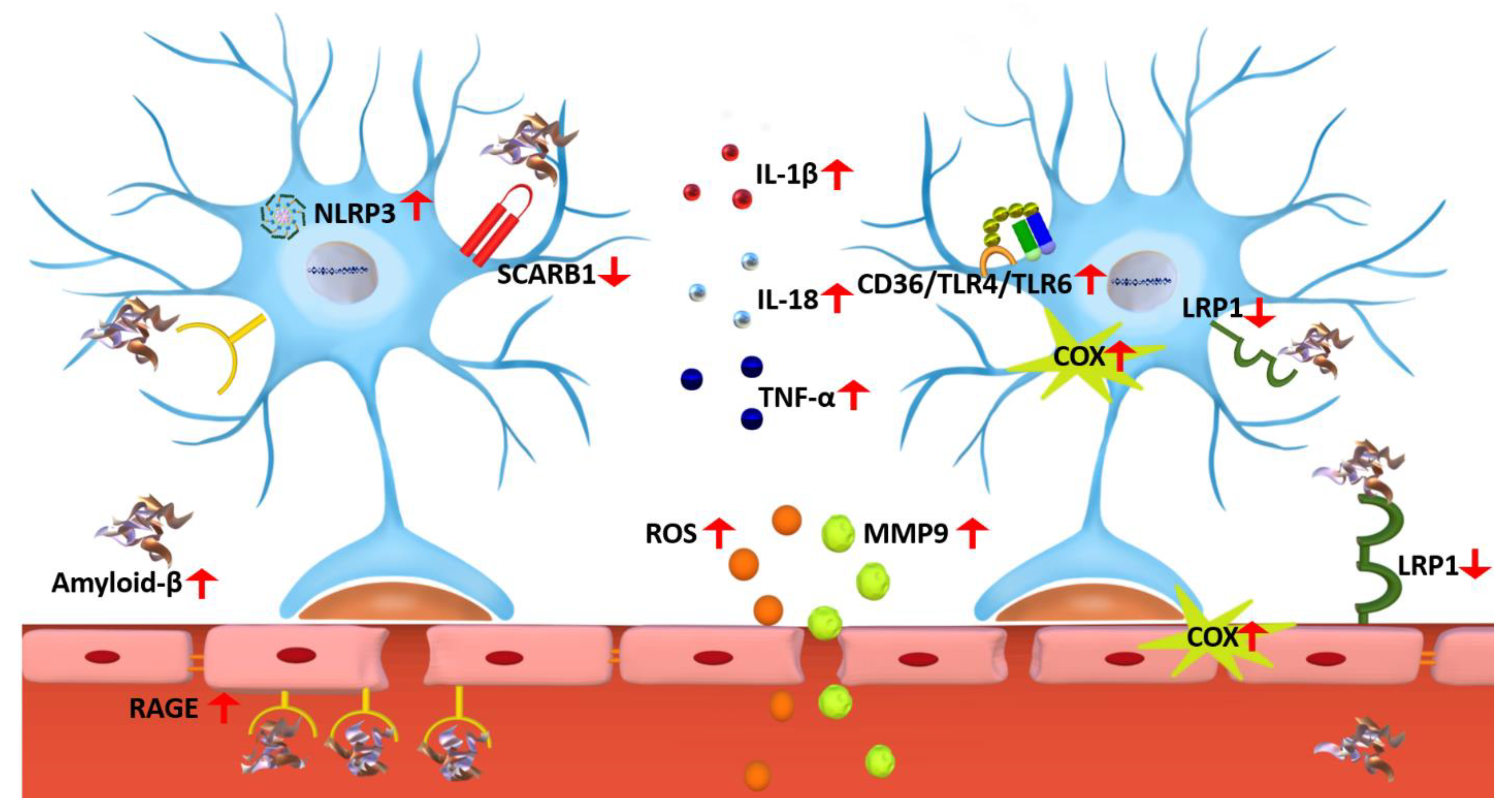
3.1. Microglia
3.1.1. P2X7R Role in Microglia Activation
3.1.2. RAGE Role in Microglia Activation
3.1.3. NLRP3 Inflammasome Activation in Response to Microglia Activation
3.1.4. TREM2 Role in Microglia Activation
3.1.5. Post-Translational Modification in Microglia Activation
3.2. Astrocytes

4. Diagnosis and Biological Markers
4.1. TPSO
4.2. YKL-40
4.3. sTREM
4.4. VICAM-1/ICAM-1
4.5. GFAP
4.6. MCP-1
4.7. S100B

{kind=link}
{kind=link}
{kind=link}
| Biomarkers | Imaging/Biological Sample | Reference |
|---|---|---|
| TSPO | PET | [204,206,235,236] |
| YKL-40 | CSF | [212,213] |
| sTREM2 | CSF | [212,217] |
| VCAM-1 | CSF, Blood | [213,218,237] |
| ICAM-1 | CSF, Blood | [213,218,220] |
| GFAP | CSF, Blood | [223,224,225] |
| MCP-1 | CSF, Blood | [212,227,228,229] |
| S100B | CSF, Blood | [156,224,233,234] |
5. Drug Development Targeting Neuroinflammation for Alzheimer’s Disease
5.1. Immunotherapy
5.1.1. Passive Immunotherapy
5.1.2. Active Immunotherapy or Vaccination
5.2. Small Molecule Compounds
5.3. Other Pharmacological Treatments of AD
5.4. Non-Pharmacological Treatments of AD
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Association. 2018 Alzheimer’s disease facts, and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Adav, S.S.; Sze, S.K. Insight of brain degenerative protein modifications in the pathology of neurodegeneration and dementia by proteomic profiling. Mol. Brain 2016, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann. N. Y. Acad. Sci. 2000, 924, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer disease: An update on pathobiology and treatment strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Guerreiro, R.; Bras, J. The age factor in Alzheimer’s disease. Genome Med. 2015, 7, 106. [Google Scholar] [CrossRef]
- Qiu, C.; Kivipelto, M.; Von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2022, 11, 111–128. [Google Scholar] [CrossRef]
- Patterson, C. Alzheimer’s Disease International (2018) World Alzheimer Report 2018. In Proceedings of the the State of the Art of Dementia Research: New Frontiers, London, UK, 21 September 2018; pp. 1–48. [Google Scholar]
- Guerchet, M.; Prince, M.; Prina, M. Numbers of People with Dementia Worldwide: An Update to the Estimates in the World Alzheimer Report 2015; Alzheimer’s Disease International: London, UK, 2020. [Google Scholar]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Hippius, H.; Neundörfer, G. The discovery of Alzheimer’s disease. Dialogues Clin. Neurosci. 2022, 5, 101–108. [Google Scholar] [CrossRef]
- Nordberg, A. Towards early diagnosis in Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments in Alzheimer disease: An update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration. FDA Grants Accelerated Approval for Alzheimer’s Drug. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug (accessed on 31 August 2022).
- Massoud, F.; Léger, G.C. Pharmacological treatment of Alzheimer disease. Can. J. Psychiatry 2011, 56, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Winslow, B.T.; Onysko, M.; Stob, C.M.; Hazlewood, K.A. Treatment of Alzheimer disease. Am. Fam. Physician 2011, 83, 1403–1412. [Google Scholar]
- Abeysinghe, A.; Deshapriya, R.; Udawatte, C. Alzheimer’s disease; a review of the pathophysiological basis and therapeutic interventions. Life Sci. 2020, 256, 117996. [Google Scholar] [CrossRef] [PubMed]
- Hazan, S. Rapid improvement in Alzheimer’s disease symptoms following fecal microbiota transplantation: A case report. J. Int. Med. Res. 2020, 48, 0300060520925930. [Google Scholar] [CrossRef]
- Frisoni, G.B.; Altomare, D.; Thal, D.R.; Ribaldi, F.; van der Kant, R.; Ossenkoppele, R.; Blennow, K.; Cummings, J.; van Duijn, C.; Nilsson, P.M. The probabilistic model of Alzheimer disease: The amyloid hypothesis revised. Nat. Rev. Neurosci. 2022, 23, 53–66. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Zhong, W.; Wu, A.; Berglund, K.; Gu, X.; Jiang, M.Q.; Talati, J.; Zhao, J.; Wei, L.; Yu, S.P. Pathogenesis of sporadic Alzheimer’s disease by deficiency of NMDA receptor subunit GluN3A. Alzheimer’s Dement. 2022, 18, 222–239. [Google Scholar] [CrossRef]
- Ryman, D.C.; Acosta-Baena, N.; Aisen, P.S.; Bird, T.; Danek, A.; Fox, N.C.; Goate, A.; Frommelt, P.; Ghetti, B.; Langbaum, J.B. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology 2014, 83, 253–260. [Google Scholar] [CrossRef]
- Tanzi, R.E. The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef]
- Edwards, G.A., III; Gamez, N.; Escobedo, G., Jr.; Calderon, O.; Moreno-Gonzalez, I. Modifiable risk factors for Alzheimer’s disease. Front. Aging Neurosci. 2019, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-X.; Tian, Y.; Wang, Z.-T.; Ma, Y.-H.; Tan, L.; Yu, J.-T. The epidemiology of Alzheimer’s disease modifiable risk factors and prevention. J. Prev. Alzheimer’s Dis. 2021, 8, 313–321. [Google Scholar] [CrossRef]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Nicola, D.; Boche, D. Post-mortem analysis of neuroinflammatory changes in human Alzheimer’s disease. Alzheimer’s Res. Ther. 2015, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, D.; Mizrahi, R. Molecular imaging of neuroinflammation in Alzheimer’s disease and mild cognitive impairment. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 80, 123–131. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 276. [Google Scholar] [CrossRef]
- Griciuc, A.; Tanzi, R.E. The role of innate immune genes in Alzheimer’s disease. Curr. Opin. Neurol. 2021, 34, 228. [Google Scholar] [CrossRef]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in neurological diseases: A road map to brain-disease dependent-inflammatory response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Moser, V.A.; Pike, C.J. Obesity and sex interact in the regulation of Alzheimer’s disease. Neurosci. Biobehav. Rev. 2016, 67, 102–118. [Google Scholar] [CrossRef] [PubMed]
- Maioli, S.; Leander, K.; Nilsson, P.; Nalvarte, I. Estrogen receptors and the aging brain. Essays Biochem. 2021, 65, 913–925. [Google Scholar] [CrossRef]
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef]
- Lee, J.H.; Jiang, Y.; Han, D.H.; Shin, S.K.; Choi, W.H.; Lee, M.J. Targeting Estrogen Receptors for the Treatment of Alzheimer’s Disease. Mol. Neurobiol. 2014, 49, 39–49. [Google Scholar] [CrossRef]
- Chakrabarti, M.; Haque, A.; Banik, N.L.; Nagarkatti, P.; Nagarkatti, M.; Ray, S.K. Estrogen receptor agonists for attenuation of neuroinflammation and neurodegeneration. Brain Res. Bull. 2014, 109, 22–31. [Google Scholar] [CrossRef]
- Tecalco-Cruz, A.C.; Zepeda–Cervantes, J.; Ortega-Domínguez, B. Estrogenic hormones receptors in Alzheimer’s disease. Mol. Biol. Rep. 2021, 48, 7517–7526. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Yeo, I.J.; Hwang, C.J.; Choi, D.-Y.; Im, H.-S.; Kim, J.Y.; Choi, W.R.; Jung, M.H.; Han, S.-B.; Hong, J.T. Estrogen deficiency exacerbates Aβ-induced memory impairment through enhancement of neuroinflammation, amyloidogenesis and NF-κB activation in ovariectomized mice. Brain Behav. Immun. 2018, 73, 282–293. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Rozemuller, A.J.M.; van Haastert, E.S.; Eikelenboom, P.; van Gool, W.A. Neuroinflammation in Alzheimer’s disease wanes with age. J. Neuroinflamm. 2011, 8, 171. [Google Scholar] [CrossRef]
- Fuller, S.; Steele, M.; Münch, G. Activated astroglia during chronic inflammation in Alzheimer’s disease—Do they neglect their neurosupportive roles? Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2010, 690, 40–49. [Google Scholar] [CrossRef]
- Kloske, C.M.; Wilcock, D.M. The Important Interface Between Apolipoprotein E and Neuroinflammation in Alzheimer’s Disease. Front. Immunol. 2020, 11, 754. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Yu, W.S.; Lim, L.W. Exploring ER stress response in cellular aging and neuroinflammation in Alzheimer’s disease. Ageing Res. Rev. 2021, 70, 101417. [Google Scholar] [CrossRef] [PubMed]
- Justice, N.J. The relationship between stress and Alzheimer’s disease. Neurobiol. Stress 2018, 8, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Lesuis, S.L.; Weggen, S.; Baches, S.; Lucassen, P.J.; Krugers, H.J. Targeting glucocorticoid receptors prevents the effects of early life stress on amyloid pathology and cognitive performance in APP/PS1 mice. Transl. Psychiatry 2018, 8, 53. [Google Scholar] [CrossRef]
- Carroll, J.C.; Iba, M.; Bangasser, D.A.; Valentino, R.J.; James, M.J.; Brunden, K.R.; Lee, V.M.Y.; Trojanowski, J.Q. Chronic stress exacerbates tau pathology, neurodegeneration, and cognitive performance through a corticotropin-releasing factor receptor-dependent mechanism in a transgenic mouse model of tauopathy. J. Neurosci. 2011, 31, 14436–14449. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Niculescu, A.G.; Lungu, I.I.; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An overview of oxidative stress, neuroinflammation, and neurodegenerative diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef]
- Bondy, S.C. Metal toxicity and neuroinflammation. Curr. Opin. Toxicol. 2021, 26, 8–13. [Google Scholar] [CrossRef]
- Harischandra, D.S.; Ghaisas, S.; Zenitsky, G.; Jin, H.; Kanthasamy, A.; Anantharam, V.; Kanthasamy, A.G. Manganese-Induced Neurotoxicity: New Insights into the Triad of Protein Misfolding, Mitochondrial Impairment, and Neuroinflammation. Front Neurosci. 2019, 13, 654. [Google Scholar] [CrossRef]
- Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal Toxicity Links to Alzheimer’s Disease and Neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868. [Google Scholar] [CrossRef]
- Martins, R.N.; Villemagne, V.; Sohrabi, H.R.; Chatterjee, P.; Shah, T.M.; Verdile, G.; Fraser, P.; Taddei, K.; Gupta, V.B.; Rainey-Smith, S.R. Alzheimer’s disease: A journey from amyloid peptides and oxidative stress, to biomarker technologies and disease prevention strategies—Gains from AIBL and DIAN cohort studies. J. Alzheimer’s Dis. 2018, 62, 965–992. [Google Scholar] [CrossRef]
- Cheignon, C.M.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative stress, amyloid-β peptide, and altered key molecular pathways in the pathogenesis and progression of Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of chronic oxidative stress on neuroinflammatory response mediated by CD4+ T cells in neurodegenerative diseases. Front. Cell. Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef]
- Pawate, S.; Shen, Q.; Fan, F.; Bhat, N.R. Redox regulation of glial inflammatory response to lipopolysaccharide and interferonγ. J. Neurosci. Res. 2004, 77, 540–551. [Google Scholar] [CrossRef]
- Chan, P.H. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 2001, 21, 2–14. [Google Scholar] [CrossRef]
- Hsieh, H.-L.; Yang, C.-M. Role of redox signaling in neuroinflammation and neurodegenerative diseases. BioMed Res. Int. 2013, 2013, 484613. [Google Scholar] [CrossRef]
- Saha, R.N.; Pahan, K. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid. Redox Signal. 2006, 8, 929–947. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, D.; Picone, P.; Baldassano, S.; Caruana, L.; Messina, E.; Marino Gammazza, A.; Cappello, F.; Mule, F.; Di Carlo, M. Insulin Resistance as Common Molecular Denominator Linking Obesity to Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 723–735. [Google Scholar] [CrossRef]
- Cavaliere, G.; Trinchese, G.; Penna, E.; Cimmino, F.; Pirozzi, C.; Lama, A.; Annunziata, C.; Catapano, A.; Mattace Raso, G.; Meli, R.; et al. High-Fat Diet Induces Neuroinflammation and Mitochondrial Impairment in Mice Cerebral Cortex and Synaptic Fraction. Front. Cell. Neurosci. 2019, 13, 509. [Google Scholar] [CrossRef] [PubMed]
- McGrattan, A.M.; McGuinness, B.; McKinley, M.C.; Kee, F.; Passmore, P.; Woodside, J.V.; McEvoy, C.T. Diet and Inflammation in Cognitive Ageing and Alzheimer’s Disease. Curr. Nutr. Rep. 2019, 8, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Więckowska-Gacek, A.; Mietelska-Porowska, A.; Wydrych, M.; Wojda, U. Western diet as a trigger of Alzheimer’s disease: From metabolic syndrome and systemic inflammation to neuroinflammation and neurodegeneration. Ageing Res. Rev. 2021, 70, 101397. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Cechova, K.; Amlerova, J.; Hort, J. Antibiotics, gut microbiota, and Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 108. [Google Scholar] [CrossRef]
- Lorena, F.B.; do Nascimento, B.P.P.; Camargo, E.; Bernardi, M.M.; Fukushima, A.R.; do N. Panizza, J.; de B. Nogueira, P.; Brandão, M.E.S.; Ribeiro, M.O. Long-term obesity is associated with depression and neuroinflammation. Arch. Endocrinol. Metab. 2021, 65, 537–548. [Google Scholar] [CrossRef]
- Noble, E.E.; Hsu, T.M.; Kanoski, S.E. Gut to Brain Dysbiosis: Mechanisms Linking Western Diet Consumption, the Microbiome, and Cognitive Impairment. Front. Behav. Neurosci. 2017, 11, 9. [Google Scholar] [CrossRef]
- Fang, P.; Kazmi, S.A.; Jameson, K.G.; Hsiao, E.Y. The Microbiome as a Modifier of Neurodegenerative Disease Risk. Cell Host Microbe 2020, 28, 201–222. [Google Scholar] [CrossRef]
- Goyal, D.; Ali, S.A.; Singh, R.K. Emerging role of gut microbiota in modulation of neuroinflammation and neurodegeneration with emphasis on Alzheimer’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 106, 110112. [Google Scholar] [CrossRef]
- Chen, C.; Liao, J.; Xia, Y.; Liu, X.; Jones, R.; Haran, J.; McCormick, B.; Sampson, T.R.; Alam, A.; Ye, K. Gut microbiota regulate Alzheimer’s disease pathologies and cognitive disorders via PUFA-associated neuroinflammation. Gut 2022, gutjnl-2021-326269. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.V.; Sadler, R.K.; Llovera, G.; Singh, V.; Roth, S.; Heindl, S.; Sebastian Monasor, L.; Verhoeven, A.; Peters, F.; Parhizkar, S.; et al. Microbiota-derived short chain fatty acids modulate microglia and promote Aβ plaque deposition. eLife 2021, 10, e59826. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Shi, Q.-Q.; Zhang, L.; Yue, C.-P.; He, Z.-J.; Li, X.-X.; He, Q.-J.; Liu, Q.; Du, X.-B. Hydrogen-rich water ameliorates neuropathological impairments in a mouse model of Alzheimer’s disease through reducing neuroinflammation and modulating intestinal microbiota. Neural Regen. Res. 2022, 17, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System during Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Solito, E.; Sastre, M. Microglia function in Alzheimer’s disease. Front. Pharmacol. 2012, 3, 14. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, Z.; Hu, H.; Zhao, M.; Sun, L. Microglia in Alzheimer’s Disease: A Target for Therapeutic Intervention. Front. Cell. Neurosci. 2021, 15, 749587. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Harry, G.J.; Kraft, A.D. Neuroinflammation and microglia: Considerations and approaches for neurotoxicity assessment. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Megur, A.; Baltriukienė, D.; Bukelskienė, V.; Burokas, A. The Microbiota-Gut-Brain Axis and Alzheimer’s Disease: Neuroinflammation Is to Blame? Nutrients 2020, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Francistiová, L.; Bianchi, C.; Di Lauro, C.; Sebastián-Serrano, Á.; de Diego-García, L.; Kobolák, J.; Dinnyés, A.; Díaz-Hernández, M. The Role of P2X7 Receptor in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 94. [Google Scholar] [CrossRef]
- Oliveira-Giacomelli, Á.; Petiz, L.L.; Andrejew, R.; Turrini, N.; Silva, J.B.; Sack, U.; Ulrich, H. Role of P2X7 Receptors in Immune Responses During Neurodegeneration. Front. Cell. Neurosci. 2021, 15, 662935. [Google Scholar] [CrossRef]
- Chen, Y.H.; Lin, R.R.; Tao, Q.Q. The role of P2X7R in neuroinflammation and implications in Alzheimer’s disease. Life Sci. 2021, 271, 119187. [Google Scholar] [CrossRef]
- Sanz, J.M.; Chiozzi, P.; Ferrari, D.; Colaianna, M.; Idzko, M.; Falzoni, S.; Fellin, R.; Trabace, L.; Di Virgilio, F. Activation of Microglia by Amyloid β Requires P2X7 Receptor Expression. J. Immunol. 2009, 182, 4378. [Google Scholar] [CrossRef]
- Martin, E.; Amar, M.; Dalle, C.; Youssef, I.; Boucher, C.; Le Duigou, C.; Brückner, M.; Prigent, A.; Sazdovitch, V.; Halle, A.; et al. New role of P2X7 receptor in an Alzheimer’s disease mouse model. Mol. Psychiatry 2019, 24, 108–125. [Google Scholar] [CrossRef]
- Monif, M.; Burnstock, G.; Williams, D.A. Microglia: Proliferation and activation driven by the P2X7 receptor. Int. J. Biochem. Cell Biol. 2010, 42, 1753–1756. [Google Scholar] [CrossRef]
- Martínez-Frailes, C.; Di Lauro, C.; Bianchi, C.; de Diego-García, L.; Sebastián-Serrano, Á.; Boscá, L.; Díaz-Hernández, M. Amyloid Peptide Induced Neuroinflammation Increases the P2X7 Receptor Expression in Microglial Cells, Impacting on Its Functionality. Front. Cell. Neurosci. 2019, 13, 143. [Google Scholar] [CrossRef] [PubMed]
- Illes, P.; Rubini, P.; Huang, L.; Tang, Y. The P2X7 receptor: A new therapeutic target in Alzheimer’s disease. Expert Opin. Ther. Targets 2019, 23, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): From Risk Factors to Therapeutic Targeting. Cells 2020, 9, 383. [Google Scholar] [CrossRef]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.-J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.P.; Hong, H.; Liao, J.M.; Wang, T.S.; Wu, J.; Chen, S.S.; Li, Y.Q.; Long, Y.; Xia, Y.Z. Upregulation of RAGE at the blood-brain barrier in streptozotocin-induced diabetic mice. Synapse 2009, 63, 636–642. [Google Scholar] [CrossRef]
- Abdallah, I.M.; Al-Shami, K.M.; Yang, E.; Wang, J.; Guillaume, C.; Kaddoumi, A. Oleuropein-Rich Olive Leaf Extract Attenuates Neuroinflammation in the Alzheimer’s Disease Mouse Model. ACS Chem. Neurosci. 2022, 13, 1002–1013. [Google Scholar] [CrossRef]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, L.; Jiang, J.; Li, H.; Wu, Q.; Ooi, K.; Wang, J.; Feng, Y.; Zhu, D.; Xia, C. HMGB1/RAGE axis mediates stress-induced RVLM neuroinflammation in mice via impairing mitophagy flux in microglia. J. Neuroinflamm. 2020, 17, 15. [Google Scholar] [CrossRef]
- Fang, F.; Lue, L.-F.; Yan, S.; Xu, H.; Luddy, J.S.; Chen, D.; Walker, D.G.; Stern, D.M.; Yan, S.; Schmidt, A.M.; et al. RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 2010, 24, 1043–1055. [Google Scholar] [CrossRef]
- Wang, X.; Sun, X.; Niu, M.; Zhang, X.; Wang, J.; Zhou, C.; Xie, A. RAGE Silencing Ameliorates Neuroinflammation by Inhibition of p38-NF-κB Signaling Pathway in Mouse Model of Parkinson’s Disease. Front. Neurosci. 2020, 14, 353. [Google Scholar] [CrossRef]
- Baiguera, S.; Fioravanzo, L.; Grandi, C.; Di Liddo, R.; Parnigotto, P.P.; Folin, M. Involvement of the receptor for advanced glycation-end products (RAGE) in beta-amyloid-induced toxic effects in rat cerebromicrovascular endothelial cells cultured in vitro. Int. J. Mol. Med. 2009, 24, 9–15. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Magna, M.; Pisetsky, D.S. The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Mol. Med. 2014, 20, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.E.; Andersson, U.; Pisetsky, D.S. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Oh, S.; Lee, S.; Byun, K. Ligands of Receptor for Advanced Glycation End-Products Produced by Activated Microglia are Critical in Neurodegenerative Diseases. J. Alzheimer’s Dis. Parkinsonism 2017, 7, 1000318. [Google Scholar] [CrossRef]
- Prasad, K. AGE–RAGE stress: A changing landscape in pathology and treatment of Alzheimer’s disease. Mol. Cell. Biochem. 2019, 459, 95–112. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- de Oliveira, P.; Cella, C.; Locker, N.; Ravindran, K.K.G.; Mendis, A.; Wafford, K.; Gilmour, G.; Dijk, D.J.; Winsky-Sommerer, R. Improved Sleep, Memory, and Cellular Pathological Features of Tauopathy, Including the NLRP3 Inflammasome, after Chronic Administration of Trazodone in rTg4510 Mice. J. Neurosci. 2022, 42, 3494–3509. [Google Scholar] [CrossRef]
- He, X.-F.; Xu, J.-H.; Li, G.; Li, M.-Y.; Li, L.-L.; Pei, Z.; Zhang, L.-Y.; Hu, X.-Q. NLRP3-dependent microglial training impaired the clearance of amyloid-beta and aggravated the cognitive decline in Alzheimer’s disease. Cell Death Dis. 2020, 11, 849. [Google Scholar] [CrossRef]
- Feng, Y.-S.; Tan, Z.-X.; Wu, L.-Y.; Dong, F.; Zhang, F. The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101192. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L., 3rd; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.G.; Jones, R.A.; Zhou, X.Q.; McGinness, J.M.; Van Eldik, L.J.; Mrak, R.E.; Griffin, W.S. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer’s disease: Potential significance for tau protein phosphorylation. Neurochem. Int. 2001, 39, 341–348. [Google Scholar] [CrossRef]
- Tanaka, S.; Ide, M.; Shibutani, T.; Ohtaki, H.; Numazawa, S.; Shioda, S.; Yoshida, T. Lipopolysaccharide-induced microglial activation induces learning and memory deficits without neuronal cell death in rats. J. Neurosci. Res. 2006, 83, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef]
- Mitroulis, I.; Skendros, P.; Ritis, K. Targeting IL-1β in disease; the expanding role of NLRP3 inflammasome. Eur. J. Intern. Med. 2010, 21, 157–163. [Google Scholar] [CrossRef]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef]
- Sharma, B.; Satija, G.; Madan, A.; Garg, M.; Alam, M.M.; Shaquiquzzaman, M.; Khanna, S.; Tiwari, P.; Parvez, S.; Iqubal, A.; et al. Role of NLRP3 Inflammasome and Its Inhibitors as Emerging Therapeutic Drug Candidate for Alzheimer’s Disease: A Review of Mechanism of Activation, Regulation, and Inhibition. Inflammation 2022, 1–32. [Google Scholar] [CrossRef]
- Barczuk, J.; Siwecka, N.; Lusa, W.; Rozpędek-Kamińska, W.; Kucharska, E.; Majsterek, I. Targeting NLRP3-Mediated Neuroinflammation in Alzheimer’s Disease Treatment. Int. J. Mol. Sci. 2022, 23, 8979. [Google Scholar] [CrossRef]
- Haure-Mirande, J.V.; Audrain, M.; Ehrlich, M.E.; Gandy, S. Microglial TYROBP/DAP12 in Alzheimer’s disease: Transduction of physiological and pathological signals across TREM2. Mol. Neurodegener. 2022, 17, 55. [Google Scholar] [CrossRef]
- Yang, J.; Fu, Z.; Zhang, X.; Xiong, M.; Meng, L.; Zhang, Z. TREM2 ectodomain and its soluble form in Alzheimer’s disease. J. Neuroinflamm. 2020, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- Ellwanger, D.C.; Wang, S.; Brioschi, S.; Shao, Z.; Green, L.; Case, R.; Yoo, D.; Weishuhn, D.; Rathanaswami, P.; Bradley, J. Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2017742118. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Tan, M.-S.; Yu, J.-T.; Sun, L.; Tan, L.; Wang, Y.-L.; Jiang, T.; Tan, L. Increased expression of TREM2 in peripheral blood of Alzheimer’s disease patients. J. Alzheimer’s Dis. 2014, 38, 497–501. [Google Scholar] [CrossRef]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Karanfilian, L.; Tosto, M.G.; Malki, K. The role of TREM2 in Alzheimer’s disease; evidence from transgenic mouse models. Neurobiol. Aging 2020, 86, 39–53. [Google Scholar] [CrossRef]
- Ewers, M.; Biechele, G.; Suárez-Calvet, M.; Sacher, C.; Blume, T.; Morenas-Rodriguez, E.; Deming, Y.; Piccio, L.; Cruchaga, C.; Kleinberger, G.; et al. Higher CSF sTREM2 and microglia activation are associated with slower rates of beta-amyloid accumulation. EMBO Mol. Med. 2020, 12, e12308. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Meilandt, W.J.; Xie, L.; Gandham, V.D.; Ngu, H.; Barck, K.H.; Rezzonico, M.G.; Imperio, J.; Lalehzadeh, G.; Huntley, M.A.; et al. Trem2 restrains the enhancement of tau accumulation and neurodegeneration by β-amyloid pathology. Neuron 2021, 109, 1283–1301.e6. [Google Scholar] [CrossRef]
- Jairaman, A.; McQuade, A.; Granzotto, A.; Kang, Y.J.; Chadarevian, J.P.; Gandhi, S.; Parker, I.; Smith, I.; Cho, H.; Sensi, S.L.; et al. TREM2 regulates purinergic receptor-mediated calcium signaling and motility in human iPSC-derived microglia. eLife 2022, 11, e73021. [Google Scholar] [CrossRef] [PubMed]
- Ruganzu, J.B.; Peng, X.; He, Y.; Wu, X.; Zheng, Q.; Ding, B.; Lin, C.; Guo, H.; Yang, Z.; Zhang, X.; et al. Downregulation of TREM2 expression exacerbates neuroinflammatory responses through TLR4-mediated MAPK signaling pathway in a transgenic mouse model of Alzheimer’s disease. Mol. Immunol. 2022, 142, 22–36. [Google Scholar] [CrossRef]
- Long, H.; Zhong, G.; Wang, C.; Zhang, J.; Zhang, Y.; Luo, J.; Shi, S. TREM2 Attenuates Aβ1-42-Mediated Neuroinflammation in BV-2 Cells by Downregulating TLR Signaling. Neurochem. Res. 2019, 44, 1830–1839. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, Y.; Wang, L.; Zhan, H.; Luo, X.; Zeng, Y.; Wu, W.; Zhang, X.; Wang, F. TREM2 ameliorates neuroinflammatory response and cognitive impairment via PI3K/AKT/FoxO3a signaling pathway in Alzheimer’s disease mice. Aging 2020, 12, 20862–20879. [Google Scholar] [CrossRef] [PubMed]
- Gallart-Palau, X.; Serra, A.; Sze, S.K. Uncovering Neurodegenerative Protein Modifications via Proteomic Profiling. Int. Rev. Neurobiol. 2015, 121, 87–116. [Google Scholar] [CrossRef] [PubMed]
- Tilvawala, R.; Nguyen, S.H.; Maurais, A.J.; Nemmara, V.V.; Nagar, M.; Salinger, A.J.; Nagpal, S.; Weerapana, E.; Thompson, P.R. The Rheumatoid Arthritis-Associated Citrullinome. Cell Chem. Biol. 2018, 25, 691–704.e6. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Perez, K.A.; Dubois, C.; Nisbet, R.M.; Li, Q.-X.; Varghese, S.; Jin, L.; Birchall, I.; Streltsov, V.A.; Vella, L.J.; et al. Citrullination of Amyloid-β Peptides in Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 3719–3732. [Google Scholar] [CrossRef] [PubMed]
- Gallart-Palau, X.; Serra, A.; Lee, B.S.T.; Guo, X.; Sze, S.K. Brain ureido degenerative protein modifications are associated with neuroinflammation and proteinopathy in Alzheimer’s disease with cerebrovascular disease. J. Neuroinflamm. 2017, 14, 175. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, A.; Masutomi, H.; Handa, S.; Nakamura, M.; Nakaya, S.; Uchida, Y.; Saito, Y.; Murayama, S.; Jang, B.; Jeon, Y.C.; et al. Mass spectrometric identification of citrullination sites and immunohistochemical detection of citrullinated glial fibrillary acidic protein in Alzheimer’s disease brains. J. Neurosci. Res. 2015, 93, 1664–1674. [Google Scholar] [CrossRef]
- Joshi, P.; Riffel, F.; Satoh, K.; Enomoto, M.; Qamar, S.; Scheiblich, H.; Villacampa, N.; Kumar, S.; Theil, S.; Parhizkar, S.; et al. Differential interaction with TREM2 modulates microglial uptake of modified Aβ species. Glia 2021, 69, 2917–2932. [Google Scholar] [CrossRef]
- Kumar, S.; Kapadia, A.; Theil, S.; Joshi, P.; Riffel, F.; Heneka, M.T.; Walter, J. Novel Phosphorylation-State Specific Antibodies Reveal Differential Deposition of Ser26 Phosphorylated Aβ Species in a Mouse Model of Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 619639. [Google Scholar] [CrossRef]
- Schilling, S.; Zeitschel, U.; Hoffmann, T.; Heiser, U.; Francke, M.; Kehlen, A.; Holzer, M.; Hutter-Paier, B.; Prokesch, M.; Windisch, M.; et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer’s disease-like pathology. Nat. Med. 2008, 14, 1106–1111. [Google Scholar] [CrossRef]
- Becker, A.; Eichentopf, R.; Sedlmeier, R.; Waniek, A.; Cynis, H.; Koch, B.; Stephan, A.; Bäuscher, C.; Kohlmann, S.; Hoffmann, T.; et al. IsoQC (QPCTL) knock-out mice suggest differential substrate conversion by glutaminyl cyclase isoenzymes. Biol. Chem. 2016, 397, 45–55. [Google Scholar] [CrossRef]
- Hinojosa, A.E.; Garcia-Bueno, B.; Leza, J.C.; Madrigal, J.L.M. CCL2/MCP-1 modulation of microglial activation and proliferation. J. Neuroinflamm. 2011, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Kiyota, T.; Yamamoto, M.; Xiong, H.; Lambert, M.P.; Klein, W.L.; Gendelman, H.E.; Ransohoff, R.M.; Ikezu, T. CCL2 accelerates microglia-mediated Abeta oligomer formation and progression of neurocognitive dysfunction. PLoS ONE 2009, 4, e6197. [Google Scholar] [CrossRef] [PubMed]
- Hartlage-Rübsamen, M.; Waniek, A.; Meißner, J.; Morawski, M.; Schilling, S.; Jäger, C.; Kleinschmidt, M.; Cynis, H.; Kehlen, A.; Arendt, T.; et al. Isoglutaminyl cyclase contributes to CCL2-driven neuroinflammation in Alzheimer’s disease. Acta Neuropathol. 2015, 129, 565–583. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the astrocyte reaction in Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, R.; Fusco, R.; Cuzzocrea, S. Astrocytes: Role and functions in brain pathologies. Front. Pharmacol. 2019, 10, 1114. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T.; Al Mamun, A.; Barreto, G.E.; Rashid, M.; Perveen, A.; Ashraf, G.M. Pharmacological approaches to mitigate neuroinflammation in Alzheimer’s disease. Int. Immunopharmacol. 2020, 84, 106479. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive astrocytes in neurodegenerative diseases. Aging Dis. 2019, 10, 664. [Google Scholar] [CrossRef]
- Carter, S.F.; Herholz, K.; Rosa-Neto, P.; Pellerin, L.; Nordberg, A.; Zimmer, E.R. Astrocyte biomarkers in Alzheimer’s disease. Trends Mol. Med. 2019, 25, 77–95. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- Leng, K.; Rooney, B.; Kim, H.; Xia, W.; Koontz, M.; Krawczyk, M.; Zhang, Y.; Ullian, E.M.; Fancy, S.P.; Schrag, M.S. CRISPRi screens in human astrocytes elucidate regulators of distinct inflammatory reactive states. BioRxiv 2021. [Google Scholar] [CrossRef]
- Lv, J.; Ma, S.; Zhang, X.; Zheng, L.; Ma, Y.; Zhao, X.; Lai, W.; Shen, H.; Wang, Q.; Ji, J. Quantitative proteomics reveals that PEA15 regulates astroglial Aβ phagocytosis in an Alzheimer’s disease mouse model. J. Proteom. 2014, 110, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cheng, D.; Cheng, R.; Zhu, X.; Wan, T.; Liu, J.; Zhang, R. Mechanisms of U87 astrocytoma cell uptake and trafficking of monomeric versus protofibril Alzheimer’s disease amyloid-β proteins. PLoS ONE 2014, 9, e99939. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.; Bogerts, B.; Schroeter, M.L.; Bernstein, H.-G. S100B protein in neurodegenerative disorders. Clin. Chem. Lab. Med. 2011, 49, 409–424. [Google Scholar] [CrossRef]
- Son, S.M.; Kang, S.; Choi, H.; Mook-Jung, I. Statins induce insulin-degrading enzyme secretion from astrocytes via an autophagy-based unconventional secretory pathway. Mol. Neurodegener. 2015, 10, 56. [Google Scholar] [CrossRef]
- Yamamoto, N.; Tanida, M.; Ono, Y.; Kasahara, R.; Fujii, Y.; Ohora, K.; Suzuki, K.; Sobue, K. Leptin inhibits amyloid β-protein degradation through decrease of neprilysin expression in primary cultured astrocytes. Biochem. Biophys. Res. Commun. 2014, 445, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Hu, X.; Song, H.; Yin, K.; Bateman, R.J.; Cirrito, J.R.; Xiao, Q.; Hsu, F.F.; Turk, J.W.; Xu, J. Matrix metalloproteinase-9 degrades amyloid-β fibrils in vitro and compact plaques in situ. J. Biol. Chem. 2006, 281, 24566–24574. [Google Scholar] [CrossRef]
- Nagele, R.G.; Wegiel, J.; Venkataraman, V.; Imaki, H.; Wang, K.-C.; Wegiel, J. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 663–674. [Google Scholar] [CrossRef]
- Johnston, J.M.; Burnett, P.; Thomas, A.P.; Tezapsidis, N. Calcium oscillations in type-1 astrocytes, the effect of a presenilin 1 (PS1) mutation. Neurosci. Lett. 2006, 395, 159–164. [Google Scholar] [CrossRef]
- Batarseh, Y.S.; Duong, Q.-V.; Mousa, Y.M.; Al Rihani, S.B.; Elfakhri, K.; Kaddoumi, A. Amyloid-β and astrocytes interplay in amyloid-β related disorders. Int. J. Mol. Sci. 2016, 17, 338. [Google Scholar] [CrossRef]
- Oksanen, M.; Hyötyläinen, I.; Trontti, K.; Rolova, T.; Wojciechowski, S.; Koskuvi, M.; Viitanen, M.; Levonen, A.L.; Hovatta, I.; Roybon, L. NF-E2-related factor 2 activation boosts antioxidant defenses and ameliorates inflammatory and amyloid properties in human Presenilin-1 mutated Alzheimer’s disease astrocytes. Glia 2020, 68, 589–599. [Google Scholar] [CrossRef]
- Jeong, E.M.; Yoon, J.-H.; Lim, J.; Shin, J.-W.; Cho, A.Y.; Heo, J.; Lee, K.B.; Lee, J.-H.; Lee, W.J.; Kim, H.-J. Real-time monitoring of glutathione in living cells reveals that high glutathione levels are required to maintain stem cell function. Stem Cell Rep. 2018, 10, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Nakaki, T. Impaired glutathione synthesis in neurodegeneration. Int. J. Mol. Sci. 2013, 14, 21021–21044. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Thiyagarajah, M.; Song, J.; Chen, C.; Herrmann, N.; Gallagher, D.; Rapoport, M.J.; Black, S.E.; Ramirez, J.; Andreazza, A.C. Altered central and blood glutathione in Alzheimer’s disease and mild cognitive impairment: A meta-analysis. Alzheimer’s Res. Ther. 2022, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, M.Y.; Markesbery, W.R. Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci. Lett. 2001, 302, 141–145. [Google Scholar] [CrossRef]
- Gu, F.; Chauhan, V.; Chauhan, A. Glutathione redox imbalance in brain disorders. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 89–95. [Google Scholar] [CrossRef]
- Pocernich, C.B.; Butterfield, D.A. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 625–630. [Google Scholar] [CrossRef]
- Lee, M.; Cho, T.; Jantaratnotai, N.; Wang, Y.T.; McGeer, E.; McGeer, P.L. Depletion of GSH in glial cells induces neurotoxicity: Relevance to aging and degenerative neurological diseases. FASEB J. 2010, 24, 2533–2545. [Google Scholar] [CrossRef]
- Takahashi, S. Neuroprotective Function of High Glycolytic Activity in Astrocytes: Common Roles in Stroke and Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 6568. [Google Scholar] [CrossRef]
- Baxter, P.S.; Hardingham, G.E. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef]
- Chen, Y.; Vartiainen, N.E.; Ying, W.; Chan, P.H.; Koistinaho, J.; Swanson, R.A. Astrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanism. J. Neurochem. 2001, 77, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef] [PubMed]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte dysfunction in Alzheimer disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef]
- Simpson, J.; Ince, P.; Lace, G.; Forster, G.; Shaw, P.; Matthews, F.; Savva, G.; Brayne, C.; Wharton, S. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol. Aging 2010, 31, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Steele, M.L.; Robinson, S.R. Reactive astrocytes give neurons less support: Implications for Alzheimer’s disease. Neurobiol. Aging 2012, 33, 423.e1–423.e13. [Google Scholar] [CrossRef]
- Bernacki, J.; Dobrowolska, A.; Nierwińska, K.; Małecki, A. Physiology and pharmacological role of the blood-brain barrier. Pharmacol. Rep. PR 2008, 60, 600–622. [Google Scholar]
- Hawkins, B.T.; Egleton, R.D. Pathophysiology of the blood-brain barrier: Animal models and methods. Curr. Top. Dev. Biol. 2008, 80, 277–309. [Google Scholar] [CrossRef]
- Sharma, C.; Woo, H.; Kim, S.R. Addressing Blood–Brain Barrier Impairment in Alzheimer’s Disease. Biomedicines 2022, 10, 742. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Ju Hwang, C.; Choi, D.Y.; Park, M.H.; Hong, J.T. NF-κB as a Key Mediator of Brain Inflammation in Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2019, 18, 3–10. [Google Scholar] [CrossRef]
- Yang, S.; Magnutzki, A.; Alami, N.O.; Lattke, M.; Hein, T.M.; Scheller, J.S.; Kröger, C.; Oswald, F.; Yilmazer-Hanke, D.; Wirth, T.; et al. IKK2/NF-κB Activation in Astrocytes Reduces amyloid β Deposition: A Process Associated with Specific Microglia Polarization. Cells 2021, 10, 2669. [Google Scholar] [CrossRef] [PubMed]
- Spampinato, S.F.; Merlo, S.; Sano, Y.; Kanda, T.; Sortino, M.A. Astrocytes contribute to Aβ-induced blood-brain barrier damage through activation of endothelial MMP9. J. Neurochem. 2017, 142, 464–477. [Google Scholar] [CrossRef] [PubMed]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like receptors activation, signaling, and targeting: An overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef]
- Frederiksen, H.R.; Haukedal, H.; Freude, K. Cell Type Specific Expression of Toll-Like Receptors in Human Brains and Implications in Alzheimer’s Disease. BioMed Res. Int. 2019, 2019, 7420189. [Google Scholar] [CrossRef]
- Walker, D.G.; Tang, T.M.; Lue, L.-F. Increased expression of toll-like receptor 3, an anti-viral signaling molecule, and related genes in Alzheimer’s disease brains. Exp. Neurol. 2018, 309, 91–106. [Google Scholar] [CrossRef]
- Leitner, G.R.; Wenzel, T.J.; Marshall, N.; Gates, E.J.; Klegeris, A. Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert Opin. Ther. Targets 2019, 23, 865–882. [Google Scholar] [CrossRef]
- Zhou, C.; Sun, X.; Hu, Y.; Song, J.; Dong, S.; Kong, D.; Wang, Y.; Hua, X.; Han, J.; Zhou, Y.; et al. Genomic deletion of TLR2 induces aggravated white matter damage and deteriorated neurobehavioral functions in mouse models of Alzheimer’s disease. Aging 2019, 11, 7257–7273. [Google Scholar] [CrossRef]
- Dallas, M.L.; Widera, D. TLR2 and TLR4-mediated inflammation in Alzheimer’s disease: Self-defense or sabotage? Neural Regen. Res. 2021, 16, 1552–1553. [Google Scholar] [CrossRef]
- Ioghen, O.; Chițoiu, L.; Gherghiceanu, M.; Ceafalan, L.C.; Hinescu, M.E. CD36—A novel molecular target in the neurovascular unit. Eur. J. Neurosci. 2021, 53, 2500–2510. [Google Scholar] [CrossRef]
- Sankar, S.B.; Donegan, R.K.; Shah, K.J.; Reddi, A.R.; Wood, L.B. Heme and hemoglobin suppress amyloid β-mediated inflammatory activation of mouse astrocytes. J. Biol. Chem. 2018, 293, 11358–11373. [Google Scholar] [CrossRef]
- Shmuel-Galia, L.; Klug, Y.; Porat, Z.; Charni, M.; Zarmi, B.; Shai, Y. Intramembrane attenuation of the TLR4-TLR6 dimer impairs receptor assembly and reduces microglia-mediated neurodegeneration. J. Biol. Chem. 2017, 292, 13415–13427. [Google Scholar] [CrossRef] [PubMed]
- Uekawa, K.; Park, L.; Hattori, Y.; Zhou, P.; Murphy, M.; Anrather, J.; Iadecola, C. Abstract 149: CD36 in Perivascular Macrophages Contributes to Neurovascular and Cognitive Dysfunction and Amyloid Angiopathy in Mice Overexpressing the Alzheimer Aβ Peptide. Stroke 2018, 49, A149. [Google Scholar] [CrossRef]
- Aymerich, M.S.; Aso, E.; Abellanas, M.A.; Tolon, R.M.; Ramos, J.A.; Ferrer, I.; Romero, J.; Fernández-Ruiz, J. Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem. Pharmacol. 2018, 157, 67–84. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Carrera-Silva, E.A.; Bosurgi, L.; Ghosh, S. TAM Receptor Signaling in Immune Homeostasis. Annu. Rev. Immunol. 2015, 33, 355–391. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.N.; Andreasson, K.I. TAM-ping down amyloid in Alzheimer’s disease. Nat. Immunol. 2021, 22, 543–544. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.A.; Lemke, G. TAM Receptors Are Pleiotropic Inhibitors of the Innate Immune Response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Varley, J.; Brooks, D.J.; Edison, P. Imaging neuroinflammation in Alzheimer’s disease and other dementias: Recent advances and future directions. Alzheimer’s Dement. 2015, 11, 1110–1120. [Google Scholar] [CrossRef]
- Gaetani, L.; Paolini Paoletti, F.; Bellomo, G.; Mancini, A.; Simoni, S.; Di Filippo, M.; Parnetti, L. CSF and Blood Biomarkers in Neuroinflammatory and Neurodegenerative Diseases: Implications for Treatment. Trends Pharmacol. Sci. 2020, 41, 1023–1037. [Google Scholar] [CrossRef]
- Park, J.-C.; Han, S.-H.; Mook-Jung, I. Peripheral inflammatory biomarkers in Alzheimer’s disease: A brief review. BMB Rep. 2020, 53, 10–19. [Google Scholar] [CrossRef]
- López-Picón, F.R.; Snellman, A.; Eskola, O.; Helin, S.; Solin, O.; Haaparanta-Solin, M.; Rinne, J.O. Neuroinflammation Appears Early on PET Imaging and Then Plateaus in a Mouse Model of Alzheimer Disease. J. Nucl. Med. 2018, 59, 509. [Google Scholar] [CrossRef]
- Lagarde, J.; Sarazin, M.; Bottlaender, M. In vivo PET imaging of neuroinflammation in Alzheimer’s disease. J. Neural Transm. 2018, 125, 847–867. [Google Scholar] [CrossRef] [PubMed]
- Tournier, B.B.; Tsartsalis, S.; Ceyzériat, K.; Garibotto, V.; Millet, P. In Vivo TSPO Signal and Neuroinflammation in Alzheimer’s Disease. Cells 2020, 9, 1941. [Google Scholar] [CrossRef] [PubMed]
- Werry, E.L.; Bright, F.M.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kril, J.J.; Kassiou, M. Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders. Int. J. Mol. Sci. 2019, 20, 3161. [Google Scholar] [CrossRef]
- Tournier, B.B.; Tsartsalis, S.; Ceyzériat, K.; Fraser, B.H.; Grégoire, M.-C.; Kövari, E.; Millet, P. Astrocytic TSPO Upregulation Appears Before Microglial TSPO in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 77, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Tournier, B.B.; Tsartsalis, S.; Rigaud, D.; Fossey, C.; Cailly, T.; Fabis, F.; Pham, T.; Grégoire, M.-C.; Kövari, E.; Moulin-Sallanon, M.; et al. TSPO and amyloid deposits in sub-regions of the hippocampus in the 3xTgAD mouse model of Alzheimer’s disease. Neurobiol. Dis. 2019, 121, 95–105. [Google Scholar] [CrossRef]
- Zhou, R.; Ji, B.; Kong, Y.; Qin, L.; Ren, W.; Guan, Y.; Ni, R. PET Imaging of Neuroinflammation in Alzheimer’s Disease. Front. Immunol. 2021, 12, 739130. [Google Scholar] [CrossRef]
- Boche, D.; Gerhard, A.; Rodriguez-Vieitez, E. Prospects and challenges of imaging neuroinflammation beyond TSPO in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2831–2847. [Google Scholar] [CrossRef]
- Park, S.A.; Han, S.M.; Kim, C.E. New fluid biomarkers tracking non-amyloid-β and non-tau pathology in Alzheimer’s disease. Exp. Mol. Med. 2020, 52, 556–568. [Google Scholar] [CrossRef]
- Nordengen, K.; Kirsebom, B.-E.; Henjum, K.; Selnes, P.; Gísladóttir, B.; Wettergreen, M.; Torsetnes, S.B.; Grøntvedt, G.R.; Waterloo, K.K.; Aarsland, D.; et al. Glial activation and inflammation along the Alzheimer’s disease continuum. J. Neuroinflamm. 2019, 16, 46. [Google Scholar] [CrossRef]
- Janelidze, S.; Mattsson, N.; Stomrud, E.; Lindberg, O.; Palmqvist, S.; Zetterberg, H.; Blennow, K.; Hansson, O. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology 2018, 91, e867–e877. [Google Scholar] [CrossRef]
- Mavroudis, I.; Chowdhury, R.; Petridis, F.; Karantali, E.; Chatzikonstantinou, S.; Balmus, I.M.; Luca, I.S.; Ciobica, A.; Kazis, D. YKL-40 as a Potential Biomarker for the Differential Diagnosis of Alzheimer’s Disease. Medicina 2022, 58, 60. [Google Scholar] [CrossRef] [PubMed]
- Hok-A-Hin, Y.S.; Hoozemans, J.J.M.; Hu, W.T.; Wouters, D.; Howell, J.C.; Rábano, A.; van der Flier, W.M.; Pijnenburg, Y.A.L.; Teunissen, C.E.; del Campo, M. YKL-40 changes are not detected in post-mortem brain of patients with Alzheimer’s disease and frontotemporal lobar degeneration. Alzheimer’s Res. Ther. 2022, 14, 100. [Google Scholar] [CrossRef] [PubMed]
- Angiulli, F.; Conti, E.; Zoia, C.P.; Da Re, F.; Appollonio, I.; Ferrarese, C.; Tremolizzo, L. Blood-Based Biomarkers of Neuroinflammation in Alzheimer’s Disease: A Central Role for Periphery? Diagnostics 2021, 11, 1525. [Google Scholar] [CrossRef]
- Bekris, L.M.; Khrestian, M.; Dyne, E.; Shao, Y.; Pillai, J.A.; Rao, S.M.; Bemiller, S.M.; Lamb, B.; Fernandez, H.H.; Leverenz, J.B. Soluble TREM2 and biomarkers of central and peripheral inflammation in neurodegenerative disease. J. Neuroimmunol. 2018, 319, 19–27. [Google Scholar] [CrossRef]
- Klyucherev, T.O.; Olszewski, P.; Shalimova, A.A.; Chubarev, V.N.; Tarasov, V.V.; Attwood, M.M.; Syvänen, S.; Schiöth, H.B. Advances in the development of new biomarkers for Alzheimer’s disease. Transl. Neurodegener. 2022, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Tan, C.-C.; Cao, X.-P.; Tan, L. Neuroinflammation modulates the association of PGRN with cerebral amyloid-β burden. Neurobiol. Aging 2021, 103, 52–59. [Google Scholar] [CrossRef]
- Milà-Alomà, M.; Suárez-Calvet, M.; Molinuevo, J.L. Latest advances in cerebrospinal fluid and blood biomarkers of Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419888819. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.D.; Chambers, A.; Ott, B.R.; Daiello, L.A. Peripheral markers of vascular endothelial dysfunction show independent but additive relationships with brain-based biomarkers in association with functional impairment in Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, e046554. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Verberk, I.M.W.; Thijssen, E.H.; Vermunt, L.; Hansson, O.; Zetterberg, H.; van der Flier, W.M.; Mielke, M.M.; del Campo, M. Blood-based biomarkers for Alzheimer’s disease: Towards clinical implementation. Lancet Neurol. 2022, 21, 66–77. [Google Scholar] [CrossRef]
- Abdelhak, A.; Foschi, M.; Abu-Rumeileh, S.; Yue, J.K.; D’Anna, L.; Huss, A.; Oeckl, P.; Ludolph, A.C.; Kuhle, J.; Petzold, A.; et al. Blood GFAP as an emerging biomarker in brain and spinal cord disorders. Nat. Rev. Neurol. 2022, 18, 158–172. [Google Scholar] [CrossRef]
- Bellaver, B.; Ferrari-Souza, J.P.; Uglione da Ros, L.; Carter, S.F.; Rodriguez-Vieitez, E.; Nordberg, A.; Pellerin, L.; Rosa-Neto, P.; Leffa, D.T.; Zimmer, E.R. Astrocyte Biomarkers in Alzheimer Disease. A Systematic Review and Meta-Analysis. Neurology 2021, 96, e2944–e2955. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-T.; Benedet, A.L.; Tissot, C.; Lussier, F.Z.; Bezgin, G.; Therriault, J.; Servaes, S.; Arias, J.F.; Gauthier, S.; Rosa-Neto, P. The neuroinflammation signatures in different stages of the Alzheimer’s disease continuum. Alzheimer’s Dement. 2021, 17, e055183. [Google Scholar] [CrossRef]
- Westin, K.; Buchhave, P.; Nielsen, H.; Minthon, L.; Janciauskiene, S.; Hansson, O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s disease. PLoS ONE 2012, 7, e30525. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-N.; Niu, L.-D.; Wang, Y.-J.; Cao, X.-P.; Liu, Q.; Tan, L.; Zhang, C.; Yu, J.-T. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: A meta-analysis and systematic review of 170 studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-J.; Liao, Y.-C.; Wang, Y.-F.; Lin, I.F.; Wang, S.-J.; Fuh, J.-L. Plasma MCP-1 and Cognitive Decline in Patients with Alzheimer’s Disease and Mild Cognitive Impairment: A Two-year Follow-up Study. Sci. Rep. 2018, 8, 1280. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Singh, S.; Anshita, D.; Ravichandiran, V. MCP-1: Function, regulation, and involvement in disease. Int. Immunopharmacol. 2021, 101, 107598. [Google Scholar] [CrossRef]
- Cristóvão, J.S.; Gomes, C.M. S100 Proteins in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 463. [Google Scholar] [CrossRef]
- Fong, T.G.; Vasunilashorn, S.M.; Libermann, T.; Marcantonio, E.R.; Inouye, S.K. Delirium and Alzheimer disease: A proposed model for shared pathophysiology. Int. J. Geriatr. Psychiatry 2019, 34, 781–789. [Google Scholar] [CrossRef]
- Peskind, E.R.; Griffin, W.S.T.; Akama, K.T.; Raskind, M.A.; Van Eldik, L.J. Cerebrospinal fluid S100B is elevated in the earlier stages of Alzheimer’s disease. Neurochem. Int. 2001, 39, 409–413. [Google Scholar] [CrossRef]
- Petzold, A.; Jenkins, R.; Watt, H.C.; Green, A.J.E.; Thompson, E.J.; Keir, G.; Fox, N.C.; Rossor, M.N. Cerebrospinal fluid S100B correlates with brain atrophy in Alzheimer’s disease. Neurosci. Lett. 2003, 336, 167–170. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Lyoo, C.H.; McGwier, M.; Snow, J.; Jenko, K.J.; Kimura, N.; Corona, W.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 2013, 136, 2228–2238. [Google Scholar] [CrossRef] [PubMed]
- Hamelin, L.; Lagarde, J.; Dorothée, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18 F-DPA-714 PET imaging. Brain 2016, 139, 1252–1264. [Google Scholar] [CrossRef]
- Zuliani, G.; Cavalieri, M.; Galvani, M.; Passaro, A.; Munari, M.R.; Bosi, C.; Zurlo, A.; Fellin, R. Markers of endothelial dysfunction in older subjects with late onset Alzheimer’s disease or vascular dementia. J. Neurol. Sci. 2008, 272, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Janssen, B.; Mach, R.H. Development of brain PET imaging agents: Strategies for imaging neuroinflammation in Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2019, 165, 371–399. [Google Scholar]
- McGrowder, D.A.; Miller, F.; Vaz, K.; Nwokocha, C.; Wilson-Clarke, C.; Anderson-Cross, M.; Brown, J.; Anderson-Jackson, L.; Williams, L.; Latore, L.; et al. Cerebrospinal Fluid Biomarkers of Alzheimer’s Disease: Current Evidence and Future Perspectives. Brain Sci. 2021, 11, 215. [Google Scholar] [CrossRef]
- Mcgeer, P.; Mcgeer, E.; Rogers, J.; Sibley, J. Anti-inflammatory drugs and Alzheimer disease. Lancet 1990, 335, 1037. [Google Scholar] [CrossRef]
- Breitner, J.C. Inflammatory processes and antiinflammatory drugs in Alzheimer’s disease: A current appraisal. Neurobiol. Aging 1996, 17, 789–794. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. The inflammatory response system of brain: Implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Rev. 1995, 21, 195–218. [Google Scholar] [CrossRef]
- Ozben, T.; Ozben, S. Neuro-inflammation and anti-inflammatory treatment options for Alzheimer’s disease. Clin. Biochem. 2019, 72, 87–89. [Google Scholar] [CrossRef]
- Kamboh, M.I. A brief synopsis on the genetics of Alzheimer’s disease. Curr. Genet. Med. Rep. 2018, 6, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Maslyar, D.; Paul, R.; Long, H.; Rhinn, H.; Tassi, I.; Morrison, G.; Yeh, F.; Schwabe, T.; Ward, M. A Phase 1 Study of AL003 in Healthy Volunteers and Participants with Alzheimer’s disease (P5-3.002). Neurology 2022, 98 (Suppl. S18), 3582. [Google Scholar]
- Griciuc, A.; Patel, S.; Federico, A.N.; Choi, S.H.; Innes, B.J.; Oram, M.K.; Cereghetti, G.; McGinty, D.; Anselmo, A.; Sadreyev, R.I. TREM2 acts downstream of CD33 in modulating microglial pathology in Alzheimer’s disease. Neuron 2019, 103, 820–835.e7. [Google Scholar] [CrossRef]
- Park, H.-H.; Lee, K.-Y.; Kim, S.; Lee, J.W.; Choi, N.-Y.; Lee, E.-H.; Lee, Y.J.; Lee, S.-H.; Koh, S.-H. The novel vaccine peptide GV1001 effectively blocks β-amyloid toxicity by mimicking the extra-telomeric functions of human telomerase reverse transcriptase. Neurobiol. Aging 2014, 35, 1255–1274. [Google Scholar] [CrossRef]
- Park, H.-H.; Yu, H.-J.; Kim, S.; Kim, G.; Choi, N.-Y.; Lee, E.-H.; Lee, Y.J.; Yoon, M.-Y.; Lee, K.-Y.; Koh, S.-H. Neural stem cells injured by oxidative stress can be rejuvenated by GV1001, a novel peptide, through scavenging free radicals and enhancing survival signals. Neurotoxicology 2016, 55, 131–141. [Google Scholar] [CrossRef]
- Ko, Y.-J.; Kwon, K.-Y.; Kum, K.-Y.; Lee, W.-C.; Baek, S.-H.; Kang, M.K.; Shon, W.-J. The anti-inflammatory effect of human telomerase-derived peptide on P. gingivalis lipopolysaccharide-induced inflammatory cytokine production and its mechanism in human dental pulp cells. Mediat. Inflamm. 2015, 2015, 385217. [Google Scholar] [CrossRef]
- Koh, S.-H.; Kwon, H.S.; Choi, S.H.; Jeong, J.H.; Na, H.R.; Lee, C.N.; Yang, Y.; Lee, A.Y.; Lee, J.-H.; Park, K.W. Efficacy and safety of GV1001 in patients with moderate-to-severe Alzheimer’s disease already receiving donepezil: A phase 2 randomized, double-blind, placebo-controlled, multicenter clinical trial. Alzheimer’s Res. Ther. 2021, 13, 66. [Google Scholar] [CrossRef]
- Koeken, V.A.; de Bree, L.C.J.; Mourits, V.P.; Moorlag, S.J.; Walk, J.; Cirovic, B.; Arts, R.J.; Jaeger, M.; Dijkstra, H.; Lemmers, H. BCG vaccination in humans inhibits systemic inflammation in a sex-dependent manner. J. Clin. Investig. 2020, 130, 5591–5602. [Google Scholar] [CrossRef] [PubMed]
- Klinger, D.; Hill, B.L.; Barda, N.; Halperin, E.; Gofrit, O.N.; Greenblatt, C.L.; Rappoport, N.; Linial, M.; Bercovier, H. Bladder cancer immunotherapy by BCG is associated with a significantly reduced risk of Alzheimer’s disease and Parkinson’s disease. Vaccines 2021, 9, 491. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S.T. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, J.; Bali, J.; Rajendran, L.; Nitsch, R.; Tackenberg, C. Calcium flux-independent NMDA receptor activity is required for Aβ oligomer-induced synaptic loss. Cell Death Dis. 2015, 6, e1791. [Google Scholar] [CrossRef]
- Koppensteiner, P.; Trinchese, F.; Fà, M.; Puzzo, D.; Gulisano, W.; Yan, S.; Poussin, A.; Liu, S.; Orozco, I.; Dale, E. Time-dependent reversal of synaptic plasticity induced by physiological concentrations of oligomeric Aβ42: An early index of Alzheimer’s disease. Sci. Rep. 2016, 6, 32553. [Google Scholar] [CrossRef]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef]
- Gee, M.S.; Son, S.H.; Jeon, S.H.; Do, J.; Kim, N.; Ju, Y.-J.; Lee, S.J.; Chung, E.K.; Inn, K.-S.; Kim, N.-J. A selective p38α/β MAPK inhibitor alleviates neuropathology and cognitive impairment, and modulates microglia function in 5XFAD mouse. Alzheimer’s Res. Ther. 2020, 12, 45. [Google Scholar] [CrossRef]
- Duffy, J.P.; Harrington, E.M.; Salituro, F.G.; Cochran, J.E.; Green, J.; Gao, H.; Bemis, G.W.; Evindar, G.; Galullo, V.P.; Ford, P.J.; et al. The Discovery of VX-745: A Novel and Selective p38α Kinase Inhibitor. ACS Med. Chem. Lett. 2011, 2, 758–763. [Google Scholar] [CrossRef]
- Alam, J.J. Selective Brain-Targeted Antagonism of p38 MAPKα Reduces Hippocampal IL-1β Levels and Improves Morris Water Maze Performance in Aged Rats. J. Alzheimer’s Dis. JAD 2015, 48, 219–227. [Google Scholar] [CrossRef]
- Lee, Y.B.; Schrader, J.W.; Kim, S.U. p38 MAP kinase regulates TNF-α production in human astrocytes and microglia by multiple mechanisms. Cytokine 2000, 12, 874–880. [Google Scholar] [CrossRef]
- Kim, S.H.; Smith, C.J.; Van Eldik, L.J. Importance of MAPK pathways for microglial pro-inflammatory cytokine IL-1β production. Neurobiol. Aging 2004, 25, 431–439. [Google Scholar] [CrossRef]
- McDonald, D.R.; Bamberger, M.E.; Combs, C.K.; Landreth, G.E. β-Amyloid fibrils activate parallel mitogen-activated protein kinase pathways in microglia and THP1 monocytes. J. Neurosci. 1998, 18, 4451–4460. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.; Pierrat, B.; Mary, J.-L.; Lesslauer, W. Blockade of p38 mitogen-activated protein kinase pathway inhibits inducible nitric-oxide synthase expression in mouse astrocytes. J. Biol. Chem. 1997, 272, 28373–28380. [Google Scholar] [CrossRef] [PubMed]
- Hua, L.L.; Zhao, M.-L.; Cosenza, M.; Kim, M.-O.; Huang, H.; Tanowitz, H.B.; Brosnan, C.F.; Lee, S.C. Role of mitogen-activated protein kinases in inducible nitric oxide synthase and TNFα expression in human fetal astrocytes. J. Neuroimmunol. 2002, 126, 180–189. [Google Scholar] [CrossRef]
- Han, Q.; Lin, Q.; Huang, P.; Chen, M.; Hu, X.; Fu, H.; He, S.; Shen, F.; Zeng, H.; Deng, Y. Microglia-derived IL-1β contributes to axon development disorders and synaptic deficit through p38-MAPK signal pathway in septic neonatal rats. J. Neuroinflamm. 2017, 14, 52. [Google Scholar] [CrossRef]
- Mancuso, R.; Fryatt, G.; Cleal, M.; Obst, J.; Pipi, E.; Monzón-Sandoval, J.; Ribe, E.; Winchester, L.; Webber, C.; Nevado, A. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain 2019, 142, 3243–3264. [Google Scholar] [CrossRef]
- Lim, G.P.; Yang, F.; Chu, T.; Chen, P.; Beech, W.; Teter, B.; Tran, T.; Ubeda, O.; Ashe, K.H.; Frautschy, S. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J. Neurosci. 2000, 20, 5709–5714. [Google Scholar] [CrossRef]
- Wilkinson, B.L.; Cramer, P.E.; Varvel, N.H.; Reed-Geaghan, E.; Jiang, Q.; Szabo, A.; Herrup, K.; Lamb, B.T.; Landreth, G.E. Ibuprofen attenuates oxidative damage through NOX2 inhibition in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 197.e21–197.e32. [Google Scholar] [CrossRef]
- Green, K.N.; Crapser, J.D.; Hohsfield, L.A. To kill a microglia: A case for CSF1R inhibitors. Trends Immunol. 2020, 41, 771–784. [Google Scholar] [CrossRef]
- Hori, Y.; Takeda, S.; Cho, H.; Wegmann, S.; Shoup, T.M.; Takahashi, K.; Irimia, D.; Elmaleh, D.R.; Hyman, B.T.; Hudry, E. A Food and Drug Administration-approved asthma therapeutic agent impacts amyloid β in the brain in a transgenic model of Alzheimer disease. J. Biol. Chem. 2015, 290, 1966–1978. [Google Scholar] [CrossRef]
- Zhang, C.; Griciuc, A.; Hudry, E.; Wan, Y.; Quinti, L.; Ward, J.; Forte, A.M.; Shen, X.; Ran, C.; Elmaleh, D.R. Cromolyn reduces levels of the Alzheimer’s disease-associated amyloid β-protein by promoting microglial phagocytosis. Sci. Rep. 2018, 8, 1144. [Google Scholar] [CrossRef] [PubMed]
- Elfakhri, K.H.; Abdallah, I.M.; Brannen, A.D.; Kaddoumi, A. Multi-faceted therapeutic strategy for treatment of Alzheimer’s disease by concurrent administration of etodolac and α-tocopherol. Neurobiol. Dis. 2019, 125, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Gardener, H.; Caunca, M.R. Mediterranean diet in preventing neurodegenerative diseases. Curr. Nutr. Rep. 2018, 7, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Loued, S.; Berrougui, H.; Componova, P.; Ikhlef, S.; Helal, O.; Khalil, A. Extra-virgin olive oil consumption reduces the age-related decrease in HDL and paraoxonase 1 anti-inflammatory activities. Br. J. Nutr. 2013, 110, 1272–1284. [Google Scholar] [CrossRef]
- Fezai, M.; Senovilla, L.; Jemaà, M.; Ben-Attia, M. Analgesic, anti-inflammatory and anticancer activities of extra virgin olive oil. J. Lipids 2013, 2013, 129736. [Google Scholar] [CrossRef] [PubMed]
- Luque-Sierra, A.; Alvarez-Amor, L.; Kleemann, R.; Martín, F.; Varela, L.M. Extra-virgin olive oil with natural phenolic content exerts an anti-inflammatory effect in adipose tissue and attenuates the severity of atherosclerotic lesions in Ldlr−/−. Leiden mice. Mol. Nutr. Food Res. 2018, 62, 1800295. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, B.; Ebrahim, S.; Kandeil, M. Anti-inflammatory effects of evening primrose and extra virgin olive oil on arthritc rats. Adv. Anim. Vet. Sci. 2021, 9, 1684–1691. [Google Scholar] [CrossRef]
- Serreli, G.; Deiana, M. Extra virgin olive oil polyphenols: Modulation of cellular pathways related to oxidant species and inflammation in aging. Cells 2020, 9, 478. [Google Scholar] [CrossRef]
- Negro, C.; Aprile, A.; Luvisi, A.; Nicolì, F.; Nutricati, E.; Vergine, M.; Miceli, A.; Blando, F.; Sabella, E.; De Bellis, L. Phenolic profile and antioxidant activity of Italian monovarietal extra virgin olive oils. Antioxidants 2019, 8, 161. [Google Scholar] [CrossRef]
- Batarseh, Y.S.; Kaddoumi, A. Oleocanthal-rich extra-virgin olive oil enhances donepezil effect by reducing amyloid-β load and related toxicity in a mouse model of Alzheimer’s disease. J. Nutr. Biochem. 2018, 55, 113–123. [Google Scholar] [CrossRef]
- Qosa, H.; Batarseh, Y.S.; Mohyeldin, M.M.; El Sayed, K.A.; Keller, J.N.; Kaddoumi, A. Oleocanthal enhances amyloid-β clearance from the brains of TgSwDI mice and in vitro across a human blood-brain barrier model. ACS Chem. Neurosci. 2015, 6, 1849–1859. [Google Scholar] [CrossRef] [PubMed]
- Al Rihani, S.B.; Darakjian, L.I.; Kaddoumi, A. Oleocanthal-rich extra-virgin olive oil restores the blood–brain barrier function through NLRP3 inflammasome inhibition simultaneously with autophagy induction in TgSwDI mice. ACS Chem. Neurosci. 2019, 10, 3543–3554. [Google Scholar] [CrossRef] [PubMed]
- Tsolaki, M.; Lazarou, E.; Kozori, M.; Petridou, N.; Tabakis, I.; Lazarou, I.; Karakota, M.; Saoulidis, I.; Melliou, E.; Magiatis, P. A randomized clinical trial of greek high phenolic early harvest extra virgin olive oil in mild cognitive impairment: The MICOIL pilot study. J. Alzheimer’s Dis. 2020, 78, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Tzekaki, E.E.; Papaspyropoulos, A.; Tsolaki, M.; Lazarou, E.; Kozori, M.; Pantazaki, A.A. Restoration of BMI1 levels after the administration of early harvest extra virgin olive oil as a therapeutic strategy against Alzheimer’s disease. Exp. Gerontol. 2021, 144, 111178. [Google Scholar] [CrossRef] [PubMed]
- Borowiec, K.; Michalak, A. Flavonoids from edible fruits as therapeutic agents in neuroinflammation—A comprehensive review and update. Crit. Rev. Food Sci. Nutr. 2021, 62, 6742–6760. [Google Scholar] [CrossRef]
- Khan, M.S.; Muhammad, T.; Ikram, M.; Kim, M.O. Dietary Supplementation of the Antioxidant Curcumin Halts Systemic LPS-Induced Neuroinflammation-Associated Neurodegeneration and Memory/Synaptic Impairment via the JNK/NF-κB/Akt Signaling Pathway in Adult Rats. Oxidative Med. Cell. Longev. 2019, 2019, 7860650. [Google Scholar] [CrossRef]
- Sil, S.; Ghosh, T.; Gupta, P.; Ghosh, R.; Kabir, S.N.; Roy, A. Dual Role of Vitamin C on the Neuroinflammation Mediated Neurodegeneration and Memory Impairments in Colchicine Induced Rat Model of Alzheimer Disease. J. Mol. Neurosci. 2016, 60, 421–435. [Google Scholar] [CrossRef]
- Devassy, J.G.; Leng, S.; Gabbs, M.; Monirujjaman, M.; Aukema, H.M. Omega-3 Polyunsaturated Fatty Acids and Oxylipins in Neuroinflammation and Management of Alzheimer Disease. Adv. Nutr. 2016, 7, 905–916. [Google Scholar] [CrossRef]
- Sun, J.; Xu, J.; Yang, B.; Chen, K.; Kong, Y.; Fang, N.; Gong, T.; Wang, F.; Ling, Z.; Liu, J. Effect of Clostridium butyricum against Microglia-Mediated Neuroinflammation in Alzheimer’s Disease via Regulating Gut Microbiota and Metabolites Butyrate. Mol. Nutr. Food Res. 2020, 64, 1900636. [Google Scholar] [CrossRef]
- Ribarič, S. Physical Exercise, a Potential Non-Pharmacological Intervention for Attenuating Neuroinflammation and Cognitive Decline in Alzheimer’s Disease Patients. Int. J. Mol. Sci. 2022, 23, 3245. [Google Scholar] [CrossRef]
- Song, S.-H.; Jee, Y.-S.; Ko, I.-G.; Lee, S.-W.; Sim, Y.-J.; Kim, D.-Y.; Lee, S.-J.; Cho, Y.S. Treadmill exercise and wheel exercise improve motor function by suppressing apoptotic neuronal cell death in brain inflammation rats. J. Exerc. Rehabil. 2018, 14, 911–919. [Google Scholar] [CrossRef]
- Cao, W.; Lin, J.; Xiang, W.; Liu, J.; Wang, B.; Liao, W.; Jiang, T. Physical Exercise-Induced Astrocytic Neuroprotection and Cognitive Improvement Through Primary Cilia and Mitogen-Activated Protein Kinases Pathway in Rats With Chronic Cerebral Hypoperfusion. Front. Aging Neurosci. 2022, 14, 866336. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chu, J.M.T.; Yan, T.; Zhang, Y.; Chen, Y.; Chang, R.C.C.; Wong, G.T.C. Short-term resistance exercise inhibits neuroinflammation and attenuates neuropathological changes in 3xTg Alzheimer’s disease mice. J. Neuroinflamm. 2020, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Y.; Wang, X.; Wang, D.; Wu, H.; Chen, H.; Chen, J.; Liu, Y. Treadmill exercise improve recognition memory by TREM2 pathway to inhibit hippocampal microglial activation and neuroinflammation in Alzheimer’s disease model. Physiol. Behav. 2022, 251, 113820. [Google Scholar] [CrossRef] [PubMed]

| Drugs | Clinical Stage | Status | Mechanism | Clinical Trial ID |
|---|---|---|---|---|
| AL003 | Phase I | Completed | Anti-CD33 antibody | NCT03822208 |
| JNJ-40346527 | Phase I | Not yet recruiting | Selective inhibitor of the CSF-1R tyrosine kinase | NCT04121208 |
| Salsalate | Phase I | Active, not recruiting | NSAID | NCT03277573 |
| Sirolimus | Phase I | Recruiting | mTOR inhibitor | NCT04629495 |
| XPro1595 | Phase I | Completed | Neutralize sTNF | NCT03943264 |
| AL002 | Phase II | Recruiting | Anti-TREM2 antibody | NCT04592874 |
| ALZT-OP1(cromolyn and ibuprofen) | Phase II | Completed | Combination of a cytokine release suppressor and an NSAID | NCT04570644 |
| Bacillus Calmette-Guerin (BCG) | Phase II | Recruiting | Systematic immune activation | NCT05004688 |
| Daratumumab | Phase II | Recruiting | Anti-CD38 antibody | NCT04070378 |
| GV1001 | Phase II | Not yet recruiting | Systematic immune activation | NCT05189210 |
| Lenalidomide | Phase II | Recruiting | Modulate innate and adaptive immune responses; inhibit BACE1 | NCT04032626 |
| Montelukast | Phase II | Active, not recruiting | Inhibit the CysLT1 receptor | NCT03991988 |
| Tacrolimus | Phase II | Withdrawn (COVID restrictions) | Calcineurin Inhibition | NCT04263519 |
| VX-745 | Phase II | Completed | Selective p38 MAPKα inhibitor | NCT03402659 |
| NE3107 | Phase III | Recruiting | ERK inhibitor | NCT04669028 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Ghraiybah, N.F.; Wang, J.; Alkhalifa, A.E.; Roberts, A.B.; Raj, R.; Yang, E.; Kaddoumi, A. Glial Cell-Mediated Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 10572. https://doi.org/10.3390/ijms231810572
Al-Ghraiybah NF, Wang J, Alkhalifa AE, Roberts AB, Raj R, Yang E, Kaddoumi A. Glial Cell-Mediated Neuroinflammation in Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(18):10572. https://doi.org/10.3390/ijms231810572
Chicago/Turabian StyleAl-Ghraiybah, Nour F., Junwei Wang, Amer E. Alkhalifa, Andrew B. Roberts, Ruchika Raj, Euitaek Yang, and Amal Kaddoumi. 2022. "Glial Cell-Mediated Neuroinflammation in Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 18: 10572. https://doi.org/10.3390/ijms231810572
APA StyleAl-Ghraiybah, N. F., Wang, J., Alkhalifa, A. E., Roberts, A. B., Raj, R., Yang, E., & Kaddoumi, A. (2022). Glial Cell-Mediated Neuroinflammation in Alzheimer’s Disease. International Journal of Molecular Sciences, 23(18), 10572. https://doi.org/10.3390/ijms231810572