
Interaction between Alzheimer’s Disease and Cerebral Small Vessel Disease: A Review Focused on Neuroimaging Markers

Abstract
:1. Introduction
2. Imaging Markers of Alzheimer’s Disease (AD) and Cerebral Small Vessel Disease (CSVD) in Subcortical Vascular Cognitive Impairment (SVCI)
2.1. Frequency of AD Imaging Markers in SVCI
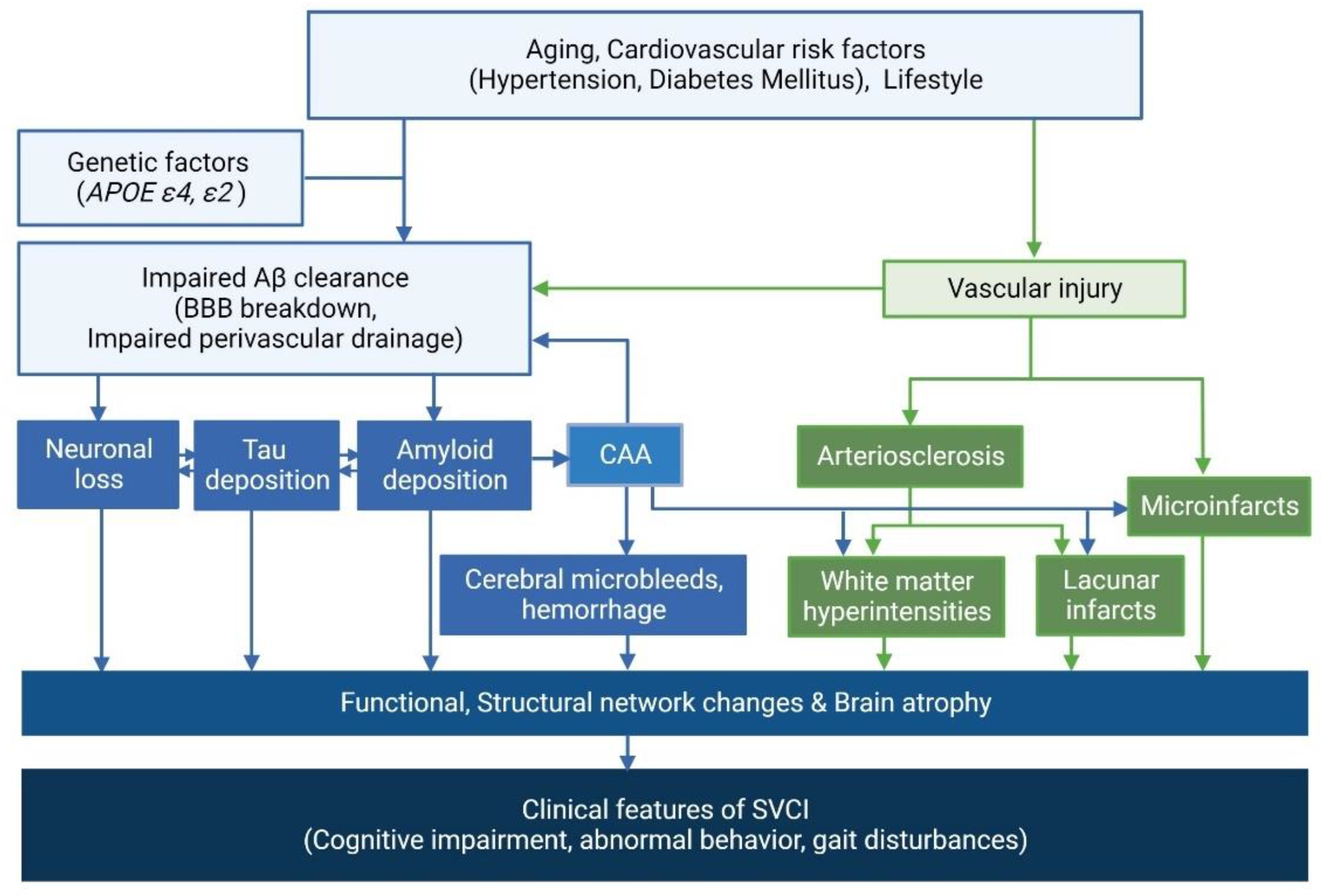
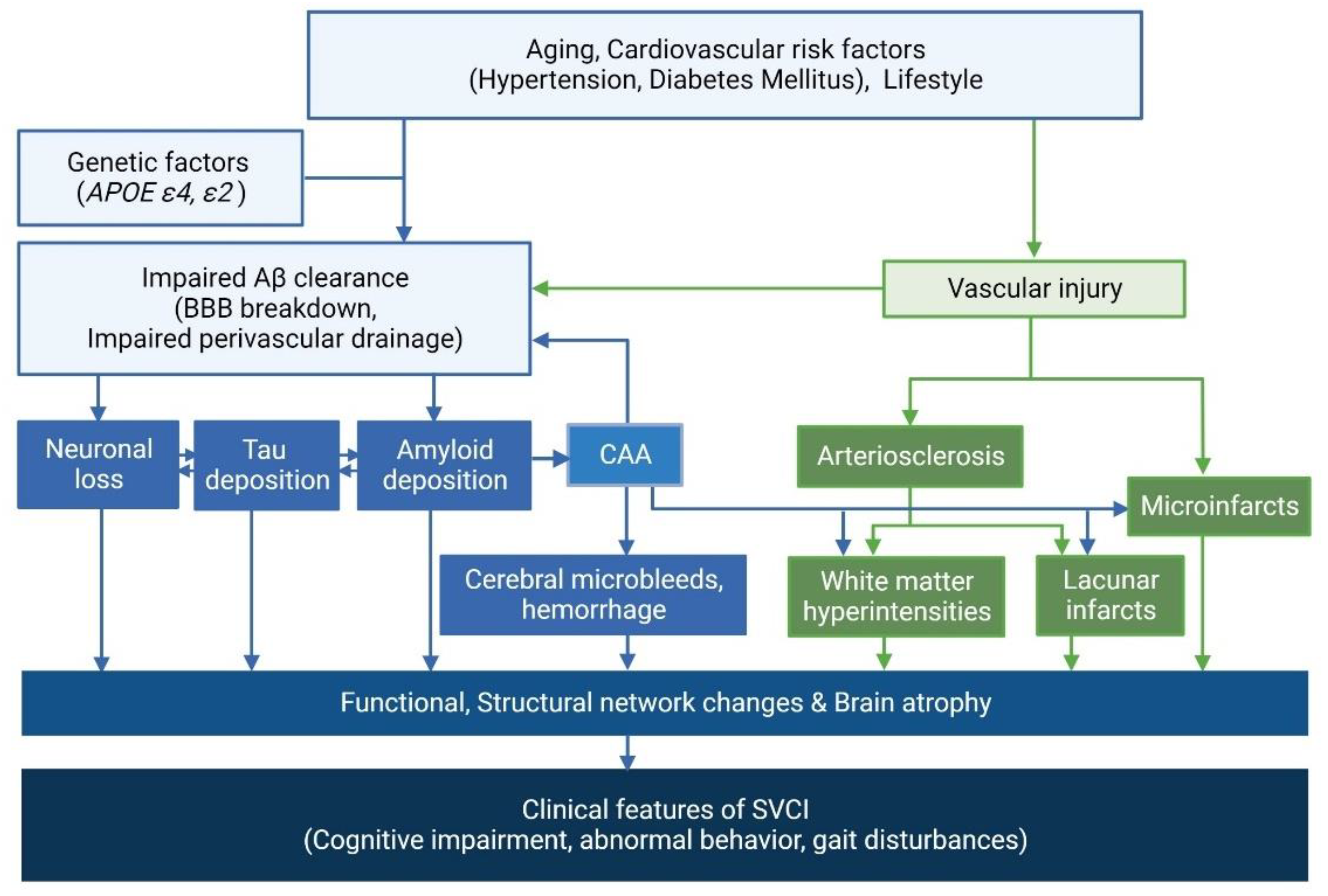
2.2. Correlation between AD and CSVD Imaging Markers
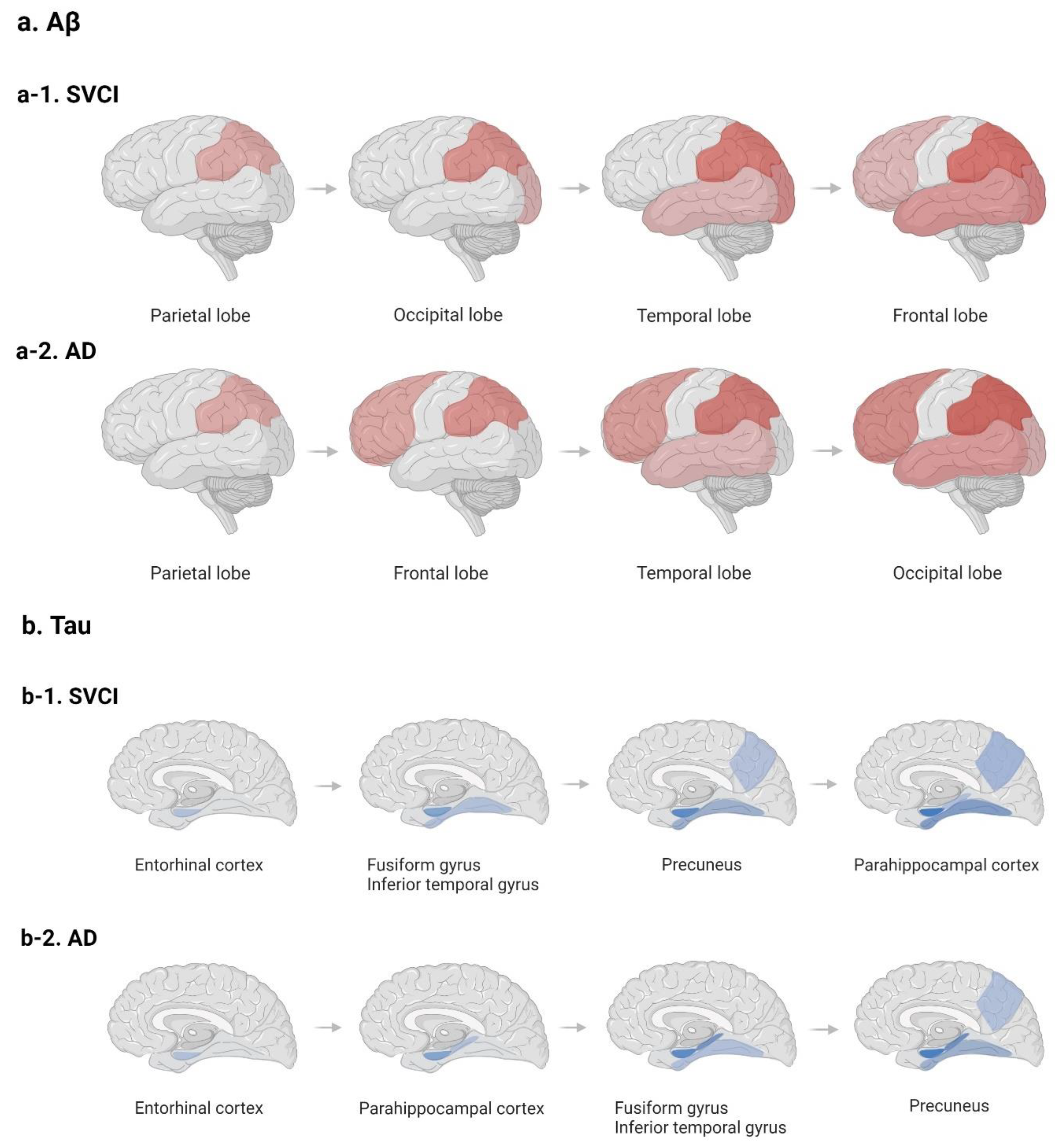
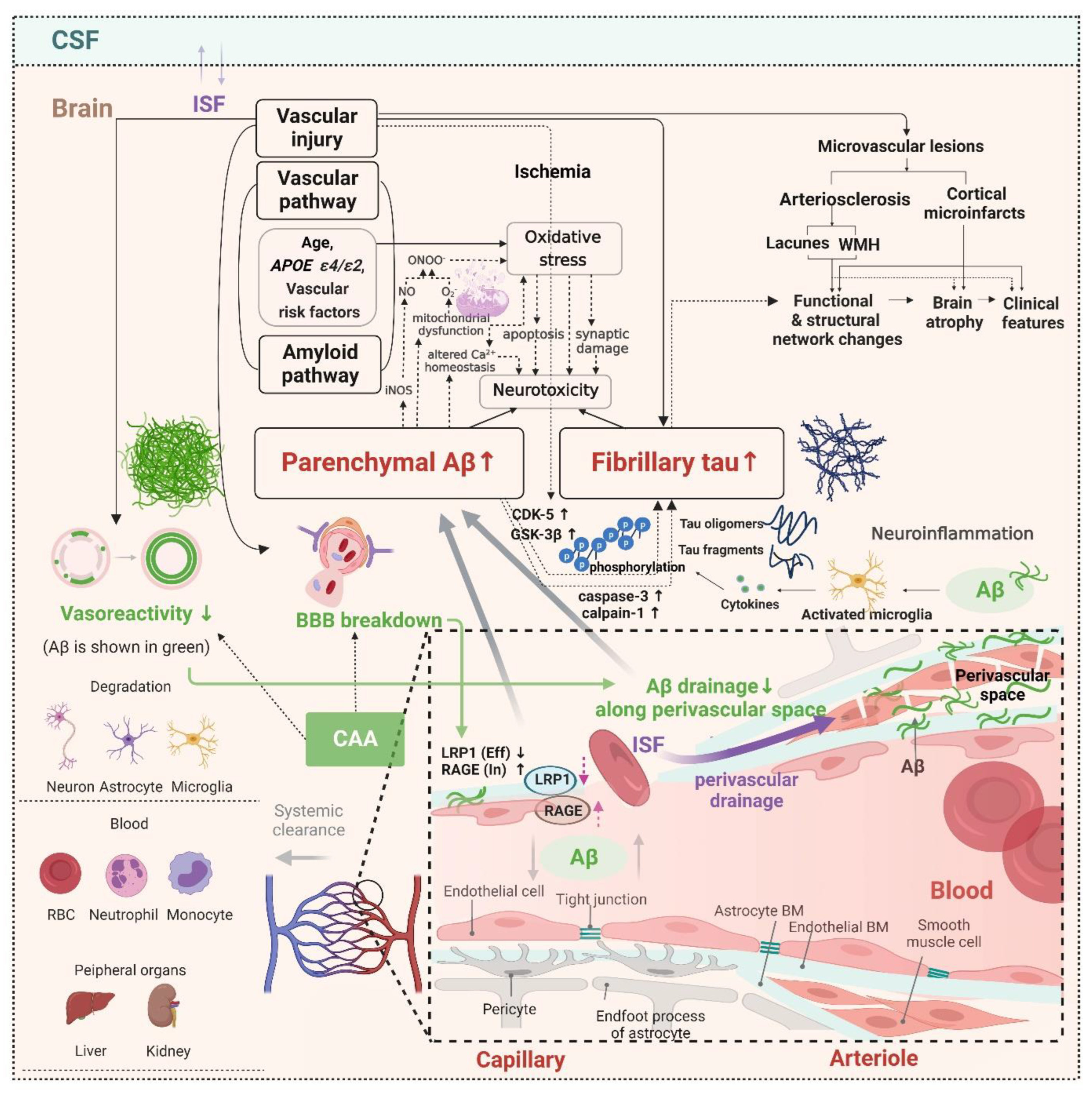
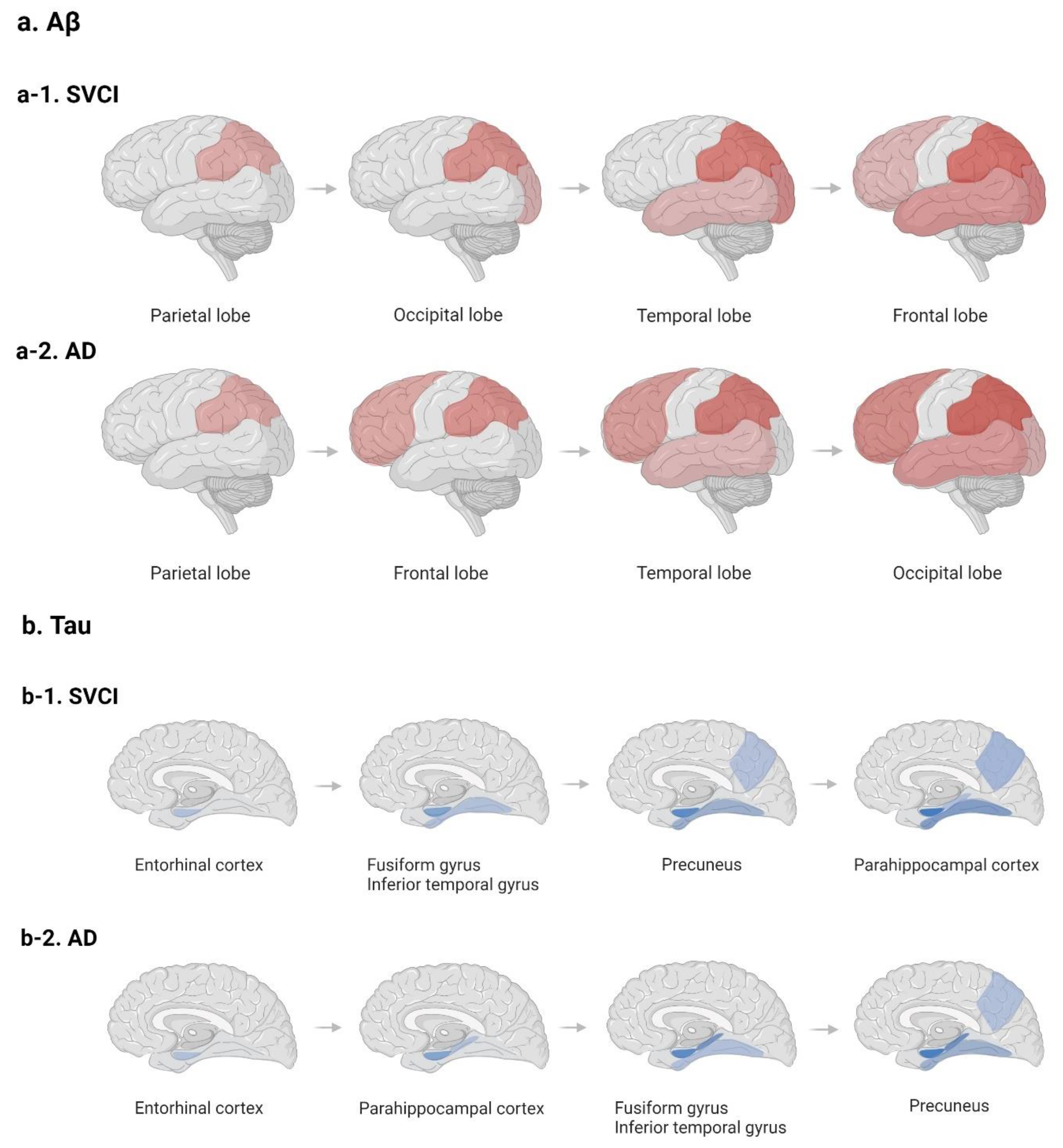
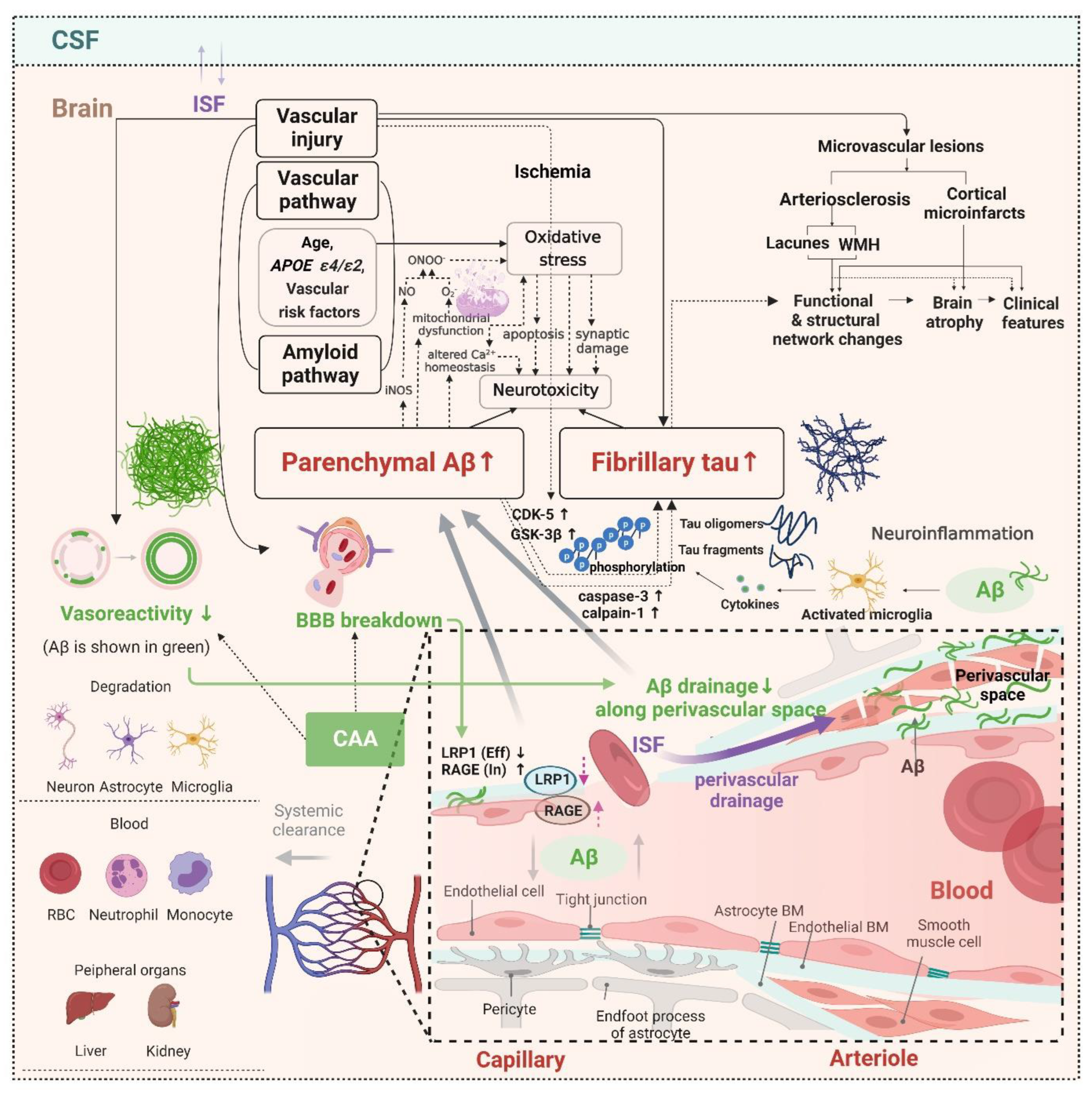
2.3. Potential Distinct Pathobiology of AD Markers in SVCI
2.4. Clinical Effects of AD and CSVD Markers in SVCI Patients
2.5. Hemorrhagic Markers in Cerebral Amyloid Angiopathy (CAA) and the Clinical Effects
3. Current Challenges and Future Perspectives
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scott, K.R.; Barrett, A.M. Dementia syndromes: Evaluation and treatment. Expert Rev. Neurother. 2007, 7, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Roman, G.C. Vascular dementia: Distinguishing characteristics, treatment, and prevention. J. Am. Geriatr. Soc. 2003, 51 (Suppl. S5), S296–S304. [Google Scholar] [CrossRef] [PubMed]
- Wenk, G.L. Neuropathologic changes in Alzheimer’s disease. J. Clin. Psychiatry 2003, 64 (Suppl. 9), 7–10. [Google Scholar] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Wiste, H.J.; Therneau, T.M.; Weigand, S.D.; Knopman, D.S.; Mielke, M.M.; Lowe, V.J.; Vemuri, P.; Machulda, M.M.; Schwarz, C.G.; et al. Associations of Amyloid, Tau, and Neurodegeneration Biomarker Profiles With Rates of Memory Decline Among Individuals Without Dementia. Jama 2019, 321, 2316–2325. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Lerch, J.P.; Pruessner, J.C.; Zijdenbos, A.; Hampel, H.; Teipel, S.J.; Evans, A.C. Focal decline of cortical thickness in Alzheimer’s disease identified by computational neuroanatomy. Cereb. Cortex 2005, 15, 995–1001. [Google Scholar] [CrossRef]
- Márquez, F.; Yassa, M.A. Neuroimaging Biomarkers for Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 21. [Google Scholar] [CrossRef]
- Schneider, J.A.; Arvanitakis, Z.; Bang, W.; Bennett, D.A. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007, 69, 2197–2204. [Google Scholar] [CrossRef]
- Agrawal, S.; Schneider, J.A. Vascular pathology and pathogenesis of cognitive impairment and dementia in older adults. Cereb. Circ.-Cogn. Behav. 2022, 3, 100148. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. The overlap between vascular disease and Alzheimer’s disease--lessons from pathology. BMC Med. 2014, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P.A.; Yu, L.; Nag, S.; Leurgans, S.; Wilson, R.S.; Bennett, D.A.; Schneider, J.A. Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology 2015, 85, 1930–1936. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, P.B.; Scuteri, A.; Black, S.E.; Decarli, C.; Greenberg, S.M.; Iadecola, C.; Launer, L.J.; Laurent, S.; Lopez, O.L.; Nyenhuis, D.; et al. Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the american heart association/american stroke association. Stroke 2011, 42, 2672–2713. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Grinberg, L.T.; Attems, J. Vascular dementia: Different forms of vessel disorders contribute to the development of dementia in the elderly brain. Exp. Gerontol. 2012, 47, 816–824. [Google Scholar] [CrossRef]
- Jorm, A.F.; Jolley, D. The incidence of dementia: A meta-analysis. Neurology 1998, 51, 728–733. [Google Scholar] [CrossRef]
- Lobo, A.; Launer, L.J.; Fratiglioni, L.; Andersen, K.; Di Carlo, A.; Breteler, M.M.; Copeland, J.R.; Dartigues, J.F.; Jagger, C.; Martinez-Lage, J.; et al. Prevalence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000, 54 (Suppl. 5), S4–S9. [Google Scholar]
- Iadecola, C.; Duering, M.; Hachinski, V.; Joutel, A.; Pendlebury, S.T.; Schneider, J.A.; Dichgans, M. Vascular Cognitive Impairment and Dementia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2019, 73, 3326–3344. [Google Scholar] [CrossRef]
- O’Brien, J.T.; Erkinjuntti, T.; Reisberg, B.; Roman, G.; Sawada, T.; Pantoni, L.; Bowler, J.V.; Ballard, C.; DeCarli, C.; Gorelick, P.B.; et al. Vascular cognitive impairment. Lancet Neurol 2003, 2, 89–98. [Google Scholar] [CrossRef]
- Roman, G.C.; Erkinjuntti, T.; Wallin, A.; Pantoni, L.; Chui, H.C. Subcortical ischaemic vascular dementia. Lancet Neurol. 2002, 1, 426–436. [Google Scholar] [CrossRef]
- Seo, S.W.; Cho, S.S.; Park, A.; Chin, J.; Na, D.L. Subcortical vascular versus amnestic mild cognitive impairment: Comparison of cerebral glucose metabolism. J. Neuroimaging 2009, 19, 213–219. [Google Scholar] [CrossRef]
- Seo, S.W.; Ahn, J.; Yoon, U.; Im, K.; Lee, J.M.; Tae Kim, S.; Ahn, H.J.; Chin, J.; Jeong, Y.; Na, D.L. Cortical thinning in vascular mild cognitive impairment and vascular dementia of subcortical type. J. Neuroimaging 2010, 20, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.W.; Im, K.; Lee, J.M.; Kim, Y.H.; Kim, S.T.; Kim, S.Y.; Yang, D.W.; Kim, S.I.; Cho, Y.S.; Na, D.L. Cortical thickness in single- versus multiple-domain amnestic mild cognitive impairment. Neuroimage 2007, 36, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Seo, S.W.; Kim, C.; Kim, G.H.; Noh, H.J.; Kim, S.T.; Kwak, K.C.; Yoon, U.; Lee, J.M.; Lee, J.W.; et al. Pathogenesis of cerebral microbleeds: In vivo imaging of amyloid and subcortical ischemic small vessel disease in 226 individuals with cognitive impairment. Ann. Neurol. 2013, 73, 584–593. [Google Scholar] [CrossRef]
- Dubois, M.F.; Hebert, R. The incidence of vascular dementia in Canada: A comparison with Europe and East Asia. Neuroepidemiology 2001, 20, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Kim, M.E.; Jang, H.; Kwon, H.; Lee, H.; Kim, H.J.; Seo, S.W.; Na, D.L. Amyloid Positivity in the Alzheimer/Subcortical-Vascular Spectrum. Neurology 2021, 96, e2201–e2211. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, P.B. Risk factors for vascular dementia and Alzheimer disease. Stroke 2004, 35 (Suppl. 1), 2620–2622. [Google Scholar] [CrossRef]
- Javanshiri, K.; Waldo, M.L.; Friberg, N.; Sjovall, F.; Wickerstrom, K.; Haglund, M.; Englund, E. Atherosclerosis, Hypertension, and Diabetes in Alzheimer’s Disease, Vascular Dementia, and Mixed Dementia: Prevalence and Presentation. J. Alzheimers Dis. 2018, 65, 1247–1258. [Google Scholar] [CrossRef]
- Saridin, F.N.; Hilal, S.; Villaraza, S.G.; Reilhac, A.; Gyanwali, B.; Tanaka, T.; Stephenson, M.C.; Ng, S.L.; Vrooman, H.; van der Flier, W.M.; et al. Brain amyloid beta, cerebral small vessel disease, and cognition: A memory clinic study. Neurology 2020, 95, e2845–e2853. [Google Scholar] [CrossRef]
- Lee, M.J.; Seo, S.W.; Na, D.L.; Kim, C.; Park, J.H.; Kim, G.H.; Kim, C.H.; Noh, Y.; Cho, H.; Kim, H.J.; et al. Synergistic effects of ischemia and beta-amyloid burden on cognitive decline in patients with subcortical vascular mild cognitive impairment. JAMA Psychiatry 2014, 71, 412–422. [Google Scholar] [CrossRef]
- Toledo, J.B.; Arnold, S.E.; Raible, K.; Brettschneider, J.; Xie, S.X.; Grossman, M.; Monsell, S.E.; Kukull, W.A.; Trojanowski, J.Q. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain A J. Neurol. 2013, 136 Pt 9, 2697–2706. [Google Scholar] [CrossRef]
- Petrovitch, H.; Ross, G.W.; Steinhorn, S.C.; Abbott, R.D.; Markesbery, W.; Davis, D.; Nelson, J.; Hardman, J.; Masaki, K.; Vogt, M.R.; et al. AD lesions and infarcts in demented and non-demented Japanese-American men. Ann. Neurol. 2005, 57, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Yang, J.J.; Kwon, H.; Kim, C.; Lee, J.M.; Chun, P.; Kim, Y.J.; Jung, N.Y.; Chin, J.; Kim, S.; et al. Relative impact of amyloid-beta, lacunes, and downstream imaging markers on cognitive trajectories. Brain 2016, 139 Pt 9, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Park, J.Y.; Jang, Y.K.; Kim, H.J.; Lee, J.S.; Na, D.L.; Noh, Y.; Lockhart, S.N.; Seong, J.K.; Seo, S.W. Distinct amyloid distribution patterns in amyloid positive subcortical vascular cognitive impairment. Sci. Rep. 2018, 8, 16178. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, G.; Kim, H.J.; Fox, Z.; Jager, H.R.; Wilson, D.; Charidimou, A.; Na, H.K.; Na, D.L.; Seo, S.W.; Werring, D.J. MRI-visible perivascular space location is associated with Alzheimer’s disease independently of amyloid burden. Brain A J. Neurol. 2017, 140, 1107–1116. [Google Scholar] [CrossRef]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; Bergstrom, M.; Savitcheva, I.; Huang, G.F.; Estrada, S.; et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 2004, 55, 306–319. [Google Scholar] [CrossRef]
- Johnson, K.A.; Sperling, R.A.; Gidicsin, C.M.; Carmasin, J.S.; Maye, J.E.; Coleman, R.E.; Reiman, E.M.; Sabbagh, M.N.; Sadowsky, C.H.; Fleisher, A.S.; et al. Florbetapir (F18-AV-45) PET to assess amyloid burden in Alzheimer’s disease dementia, mild cognitive impairment, and normal aging. Alzheimers Dement. 2013, 9 (Suppl. 5), S72–S83. [Google Scholar] [CrossRef]
- Hatashita, S.; Yamasaki, H.; Suzuki, Y.; Tanaka, K.; Wakebe, D.; Hayakawa, H. [18F]Flutemetamol amyloid-beta PET imaging compared with [11C]PIB across the spectrum of Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 290–300. [Google Scholar] [CrossRef]
- Barthel, H.; Sabri, O. Florbetaben to trace amyloid-beta in the Alzheimer brain by means of PET. J. Alzheimers Dis. 2011, 26 (Suppl. 3), 117–121. [Google Scholar] [CrossRef]
- Schwarz, A.J.; Yu, P.; Miller, B.B.; Shcherbinin, S.; Dickson, J.; Navitsky, M.; Joshi, A.D.; Devous, M.D., Sr.; Mintun, M.S. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 2016, 139 Pt 5, 1539–1550. [Google Scholar] [CrossRef]
- Pascoal, T.A.; Therriault, J.; Benedet, A.L.; Savard, M.; Lussier, F.Z.; Chamoun, M.; Tissot, C.; Qureshi, M.N.I.; Kang, M.S.; Mathotaarachchi, S.; et al. 18F-MK-6240 PET for early and late detection of neurofibrillary tangles. Brain 2020, 143, 2818–2830. [Google Scholar] [CrossRef]
- Kroth, H.; Oden, F.; Molette, J.; Schieferstein, H.; Capotosti, F.; Mueller, A.; Berndt, M.; Schmitt-Willich, H.; Darmency, V.; Gabellieri, E.; et al. Discovery and preclinical characterization of [(18)F]PI-2620, a next-generation tau PET tracer for the assessment of tau pathology in Alzheimer’s disease and other tauopathies. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2178–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwabara, H.; Comley, R.A.; Borroni, E.; Honer, M.; Kitmiller, K.; Roberts, J.; Gapasin, L.; Mathur, A.; Klein, G.; Wong, D.F. Evaluation of (18)F-RO-948 PET for Quantitative Assessment of Tau Accumulation in the Human Brain. J. Nucl. Med. 2018, 59, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Aizenstein, H.J.; Nebes, R.D.; Saxton, J.A.; Price, J.C.; Mathis, C.A.; Tsopelas, N.D.; Ziolko, S.K.; James, J.A.; Snitz, B.E.; Houck, P.R.; et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch. Neurol. 2008, 65, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Wolk, D.A.; Price, J.C.; Saxton, J.A.; Snitz, B.E.; James, J.A.; Lopez, O.L.; Aizenstein, H.J.; Cohen, A.D.; Weissfeld, L.A.; Mathis, C.A.; et al. Amyloid imaging in mild cognitive impairment subtypes. Ann. Neurol. 2009, 65, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; Leuzy, A.; Cho, H.; Sudre, C.H.; Strandberg, O.; Smith, R.; Palmqvist, S.; Mattsson-Carlgren, N.; Olsson, T.; Jogi, J.; et al. The impact of demographic, clinical, genetic, and imaging variables on tau PET status. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2245–2258. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Okamura, N.; Harada, R.; Ishiki, A.; Kikuchi, A.; Nakamura, T.; Kudo, Y. The development and validation of tau PET tracers: Current status and future directions. Clin. Transl. Imaging 2018, 6, 305–316. [Google Scholar] [CrossRef]
- Saint-Aubert, L.; Lemoine, L.; Chiotis, K.; Leuzy, A.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging: Present and future directions. Mol. Neurodegener. 2017, 12, 19. [Google Scholar] [CrossRef]
- Chong, J.S.X.; Jang, H.; Kim, H.J.; Ng, K.K.; Na, D.L.; Lee, J.H.; Seo, S.W.; Zhou, J. Amyloid and cerebrovascular burden divergently influence brain functional network changes over time. Neurology 2019, 93, e1514–e1525. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, S.H.; Kim, G.H.; Seo, S.W.; Park, H.K.; Oh, S.J.; Kim, J.S.; Cheong, H.K.; Na, D.L. Identification of pure subcortical vascular dementia using 11C-Pittsburgh compound B. Neurology 2011, 77, 18–25. [Google Scholar] [CrossRef]
- Ye, B.S.; Seo, S.W.; Kim, J.H.; Kim, G.H.; Cho, H.; Noh, Y.; Kim, H.J.; Yoon, C.W.; Woo, S.Y.; Kim, S.H.; et al. Effects of amyloid and vascular markers on cognitive decline in subcortical vascular dementia. Neurology 2015, 85, 1687–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.; Kim, H.J.; Park, S.; Park, Y.H.; Choe, Y.; Cho, H.; Lyoo, C.H.; Yoon, U.; Lee, J.S.; Kim, Y.; et al. Application of an amyloid and tau classification system in subcortical vascular cognitive impairment patients. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Noh, Y.; Seo, S.W.; Jeon, S.; Lee, J.M.; Kim, J.H.; Kim, G.H.; Cho, H.; Yoon, C.W.; Kim, H.J.; Ye, B.S.; et al. White matter hyperintensities are associated with amyloid burden in APOE4 non-carriers. J. Alzheimers Dis. 2014, 40, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Ghosh, P.M.; Madison, C.; Laforce, R., Jr.; Corbetta-Rastelli, C.; Weiner, M.W.; Greicius, M.D.; Seeley, W.W.; Gorno-Tempini, M.L.; Rosen, H.J.; et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer’s disease. Brain A J. Neurol. 2013, 136 Pt 3, 844–858. [Google Scholar] [CrossRef]
- Jang, H.; Seo, S.W. Comparison of Amyloid and Tau Patterns and Neuropsychological Findings in Cerebral Amyloid Angiopathy and Hypertensive Small Vessel Disease. In Proceedings of the Conference of Korean Dementia Association, Seoul, Korea, 4 November 2017; Korea Dementia Association: Seoul, Korea, 2017. [Google Scholar]
- Grothe, M.J.; Barthel, H.; Sepulcre, J.; Dyrba, M.; Sabri, O.; Teipel, S.J. In vivo staging of regional amyloid deposition. Neurology 2017, 89, 2031–2038. [Google Scholar] [CrossRef]
- van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 2020, 21, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, A.; Greenberg, S.M. Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 2011, 70, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Neuroanatomy and pathology of sporadic Alzheimer’s disease. Adv. Anat. Embryol. Cell Biol. 2015, 215, 1–162. [Google Scholar] [PubMed]
- Mattsson, N.; Palmqvist, S.; Stomrud, E.; Vogel, J.; Hansson, O. Staging β-Amyloid Pathology with Amyloid Positron Emission Tomography. JAMA Neurol. 2019, 76, 1319–1329. [Google Scholar] [CrossRef]
- Collij, L.E.; Heeman, F.; Salvadó, G.; Ingala, S.; Altomare, D.; de Wilde, A.; Konijnenberg, E.; van Buchem, M.; Yaqub, M.; Markiewicz, P.; et al. Multitracer model for staging cortical amyloid deposition using PET imaging. Neurology 2020, 95, e1538–e1553. [Google Scholar] [CrossRef]
- Palmqvist, S.; Schöll, M.; Strandberg, O.; Mattsson, N.; Stomrud, E.; Zetterberg, H.; Blennow, K.; Landau, S.; Jagust, W.; Hansson, O. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat. Commun. 2017, 8, 1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, B.; Ao, Q.; Wang, Z.; Liu, W.; Niu, Y.; Shen, Q.; Zuo, H.; Zhang, X.; Gong, Y. Phosphorylation of tau protein over time in rats subjected to transient brain ischemia. Neural. Regen. Res. 2013, 8, 3173–3182. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.E.; Robbins, A.B.; Hu, M.; Cao, X.; Betensky, R.A.; Clark, T.; Das, S.; Hyman, B.T. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1289–E1298. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, S.; Cho, H.; Jang, Y.K.; San Lee, J.; Jang, H.; Kim, Y.; Kim, K.W.; Ryu, Y.H.; Choi, J.Y.; et al. Assessment of Extent and Role of Tau in Subcortical Vascular Cognitive Impairment Using 18F-AV1451 Positron Emission Tomography Imaging. JAMA Neurol. 2018, 75, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Choi, J.Y.; Hwang, M.S.; Kim, Y.J.; Lee, H.M.; Lee, H.S.; Lee, J.H.; Ryu, Y.H.; Lee, M.S.; Lyoo, C.H. In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann. Neurol. 2016, 80, 247–258. [Google Scholar] [CrossRef]
- Gottesman, R.F.; Schneider, A.L.; Zhou, Y.; Coresh, J.; Green, E.; Gupta, N.; Knopman, D.S.; Mintz, A.; Rahmim, A.; Sharrett, A.R.; et al. Association Between Midlife Vascular Risk Factors and Estimated Brain Amyloid Deposition. JAMA 2017, 317, 1443–1450. [Google Scholar] [CrossRef]
- Hughes, T.M.; Wagenknecht, L.E.; Craft, S.; Mintz, A.; Heiss, G.; Palta, P.; Wong, D.; Zhou, Y.; Knopman, D.; Mosley, T.H.; et al. Arterial stiffness and dementia pathology: Atherosclerosis Risk in Communities (ARIC)-PET Study. Neurology 2018, 90, e1248–e1256. [Google Scholar] [CrossRef]
- Rabin, J.S.; Schultz, A.P.; Hedden, T.; Viswanathan, A.; Marshall, G.A.; Kilpatrick, E.; Klein, H.; Buckley, R.F.; Yang, H.S.; Properzi, M.; et al. Interactive Associations of Vascular Risk and beta-Amyloid Burden with Cognitive Decline in Clinically Normal Elderly Individuals: Findings From the Harvard Aging Brain Study. JAMA Neurol. 2018, 75, 1124–1131. [Google Scholar] [CrossRef]
- Arfanakis, K.; Evia, A.M.; Leurgans, S.E.; Cardoso, L.F.C.; Kulkarni, A.; Alqam, N.; Lopes, L.F.; Vieira, D.; Bennett, D.A.; Schneider, J.A. Neuropathologic Correlates of White Matter Hyperintensities in a Community-Based Cohort of Older Adults. J. Alzheimers Dis. 2020, 73, 333–345. [Google Scholar] [CrossRef]
- Köbe, T.; Gonneaud, J.; Pichet Binette, A.; Meyer, P.F.; McSweeney, M.; Rosa-Neto, P.; Breitner, J.C.S.; Poirier, J.; Villeneuve, S. Association of Vascular Risk Factors with β-Amyloid Peptide and Tau Burdens in Cognitively Unimpaired Individuals and Its Interaction with Vascular Medication Use. JAMA Netw. Open. 2020, 3, e1920780. [Google Scholar] [CrossRef]
- Lockhart, S.N.; Schaich, C.L.; Craft, S.; Sachs, B.C.; Rapp, S.R.; Jung, Y.; Whitlow, C.T.; Solingapuram Sai, K.K.; Cleveland, M.; Williams, B.J.; et al. Associations among vascular risk factors, neuroimaging biomarkers, and cognition: Preliminary analyses from the Multi-Ethnic Study of Atherosclerosis (MESA). Alzheimers Dement. 2022, 18, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Boulouis, G.; Gurol, M.E.; Ayata, C.; Bacskai, B.J.; Frosch, M.P.; Viswanathan, A.; Greenberg, S.M. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain A J. Neurol. 2017, 140, 1829–1850. [Google Scholar] [CrossRef] [PubMed]
- Grimmer, T.; Faust, M.; Auer, F.; Alexopoulos, P.; Forstl, H.; Henriksen, G.; Perneczky, R.; Sorg, C.; Yousefi, B.H.; Drzezga, A.; et al. White matter hyperintensities predict amyloid increase in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 2766–2773. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef]
- Terwel, D.; Muyllaert, D.; Dewachter, I.; Borghgraef, P.; Croes, S.; Devijver, H.; Van Leuven, F. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am. J. Pathol. 2008, 172, 786–798. [Google Scholar] [CrossRef]
- Hernandez, P.; Lee, G.; Sjoberg, M.; Maccioni, R.B. Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Abeta (25-35): Involvement of lipid rafts. J. Alzheimers Dis. 2009, 16, 149–156. [Google Scholar] [CrossRef]
- Gamblin, T.C.; Chen, F.; Zambrano, A.; Abraha, A.; Lagalwar, S.; Guillozet, A.L.; Lu, M.; Fu, Y.; Garcia-Sierra, F.; LaPointe, N.; et al. Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 10032–10037. [Google Scholar] [CrossRef]
- Park, S.-Y.; Ferreira, A. The Generation of a 17 kDa Neurotoxic Fragment: An Alternative Mechanism by which Tau Mediates β-Amyloid-Induced Neurodegeneration. J. Neurosci. 2005, 25, 5365–5375. [Google Scholar] [CrossRef]
- Ismail, R.; Parbo, P.; Madsen, L.S.; Hansen, A.K.; Hansen, K.V.; Schaldemose, J.L.; Kjeldsen, P.L.; Stokholm, M.G.; Gottrup, H.; Eskildsen, S.F.; et al. The relationships between neuroinflammation, beta-amyloid and tau deposition in Alzheimer’s disease: A longitudinal PET study. J. Neuroinflamm. 2020, 17, 151. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain A J. Neurol. 2015, 138 Pt 6, 1738–1755. [Google Scholar] [CrossRef]
- Korte, N.; Nortley, R.; Attwell, D. Cerebral blood flow decrease as an early pathological mechanism in Alzheimer’s disease. Acta Neuropathol. 2020, 140, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.L.; Wang, C.; Jiang, T.; Tan, L.; Xing, A.; Yu, J.T. The Role of Cdk5 in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 4328–4342. [Google Scholar] [CrossRef] [PubMed]
- Toro-Fernández, L.F.; Zuluaga-Monares, J.C.; Saldarriaga-Cartagena, A.M.; Cardona-Gómez, G.P.; Posada-Duque, R. Targeting CDK5 in Astrocytes Promotes Calcium Homeostasis Under Excitotoxic Conditions. Front. Cell. Neurosci. 2021, 15, 643717. [Google Scholar] [CrossRef] [PubMed]
- Mottet, D.; Dumont, V.; Deccache, Y.; Demazy, C.; Ninane, N.; Raes, M.; Michiels, C. Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J. Biol. Chem. 2003, 278, 31277–31285. [Google Scholar] [CrossRef]
- Curtis, D.; Bandyopadhyay, S. Mini-review: Role of the PI3K/Akt pathway and tyrosine phosphatases in Alzheimer’s disease susceptibility. Ann. Hum. Genet. 2021, 85, 1–6. [Google Scholar] [CrossRef]
- Grochowski, C.; Litak, J.; Kamieniak, P.; Maciejewski, R. Oxidative stress in cerebral small vessel disease. Role of reactive species. Free Radic. Res. 2018, 52, 1–13. [Google Scholar] [CrossRef]
- Hort, J.; Vališ, M.; Kuča, K.; Angelucci, F. Vascular Cognitive Impairment: Information from Animal Models on the Pathogenic Mechanisms of Cognitive Deficits. Int. J. Mol. Sci. 2019, 20, 2405. [Google Scholar] [CrossRef]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Ros-Bernal, F. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Lee, H.; Park, S.; Choe, Y.; Park, Y.H.; Cheon, B.K.; Hahn, A.; Ossenkoppele, R.; Kim, H.J.; Kim, S.; et al. Association between APOE epsilon2 and Abeta burden in patients with Alzheimer- and vascular-type cognitive impairment. Neurology 2020, 95, e2354–e2365. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Seo, S.W.; Kim, C.; Kim, S.H.; Kim, G.H.; Kim, S.T.; Jeon, S.; Lee, J.M.; Oh, S.J.; Kim, J.S.; et al. Effects of cerebrovascular disease and amyloid beta burden on cognition in subjects with subcortical vascular cognitive impairment. Neurobiol. Aging 2014, 35, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Im, K.; Kwon, H.; Lee, J.M.; Kim, C.; Kim, Y.J.; Jung, N.Y.; Cho, H.; Ye, B.S.; Noh, Y.; et al. Clinical effect of white matter network disruption related to amyloid and small vessel disease. Neurology 2015, 85, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.K.; Kwon, H.; Kim, Y.J.; Jung, N.Y.; Lee, J.S.; Lee, J.; Chin, J.; Im, K.; Jeon, S.; Lee, J.M.; et al. Early- vs late-onset subcortical vascular cognitive impairment. Neurology 2016, 86, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ye, B.S.; Yoon, C.W.; Cho, H.; Noh, Y.; Kim, G.H.; Choi, Y.S.; Kim, J.H.; Jeon, S.; Lee, J.M.; et al. Effects of APOE epsilon4 on brain amyloid, lacunar infarcts, and white matter lesions: A study among patients with subcortical vascular cognitive impairment. Neurobiol. Aging 2013, 34, 2482–2487. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kwon, H.K.; Lee, J.M.; Cho, H.; Kim, H.J.; Park, H.K.; Jung, N.Y.; San Lee, J.; Lee, J.; Jang, Y.K.; et al. Gray and white matter changes linking cerebral small vessel disease to gait disturbances. Neurology 2016, 86, 1199–1207. [Google Scholar] [CrossRef]
- Jang, H.; Kim, H.J.; Choe, Y.S.; Kim, S.J.; Park, S.; Kim, Y.; Kim, K.W.; Lyoo, C.H.; Cho, H.; Ryu, Y.H.; et al. The Impact of Amyloid-beta or Tau on Cognitive Change in the Presence of Severe Cerebrovascular Disease. J. Alzheimers Dis. 2020, 78, 573–585. [Google Scholar] [CrossRef]
- Byeon, H. A Prediction Model for Mild Cognitive Impairment Using Random Forests. Int. J. Adv. Comput. Sci. Appl. (IJACSA) 2015, 6, 1–5. [Google Scholar] [CrossRef]
- Ye, B.S.; Seo, S.W.; Kim, G.H.; Noh, Y.; Cho, H.; Yoon, C.W.; Kim, H.J.; Chin, J.; Jeon, S.; Lee, J.M.; et al. Amyloid burden, cerebrovascular disease, brain atrophy, and cognition in cognitively impaired patients. Alzheimers Dement. 2015, 11, 494–503.e3. [Google Scholar] [CrossRef]
- Weller, R.O.; Massey, A.; Newman, T.A.; Hutchings, M.; Kuo, Y.M.; Roher, A.E. Cerebral amyloid angiopathy: Amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am. J. Pathol. 1998, 153, 725–733. [Google Scholar] [CrossRef]
- Yamada, M. Cerebral amyloid angiopathy: Emerging concepts. J. Stroke 2015, 17, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—one peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Vernooij, M.W.; Cordonnier, C.; Viswanathan, A.; Al-Shahi Salman, R.; Warach, S.; Launer, L.J.; Van Buchem, M.A.; Breteler, M.M.; Microbleed Study, G. Cerebral microbleeds: A guide to detection and interpretation. Lancet Neurol. 2009, 8, 165–174. [Google Scholar] [CrossRef]
- Thal, D.R.; Ghebremedhin, E.; Orantes, M.; Wiestler, O.D. Vascular pathology in Alzheimer disease: Correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J. Neuropathol. Exp. Neurol. 2003, 62, 1287–1301. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.H.; Jang, H.; Park, S.B.; Choe, Y.S.; Park, Y.; Kang, S.H.; Lee, J.M.; Kim, J.S.; Kim, J.; Kim, J.P.; et al. Strictly Lobar Microbleeds Reflect Amyloid Angiopathy Regardless of Cerebral and Cerebellar Compartments. Stroke 2020, 51, 3600–3607. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Jang, Y.K.; Kim, H.J.; Werring, D.J.; Lee, J.S.; Choe, Y.S.; Park, S.; Lee, J.; Kim, K.W.; Kim, Y.; et al. Clinical significance of amyloid beta positivity in patients with probable cerebral amyloid angiopathy markers. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1287–1298. [Google Scholar] [CrossRef]
- Schmechel, D.E.; Saunders, A.M.; Strittmatter, W.J.; Crain, B.J.; Hulette, C.M.; Joo, S.H.; Pericak-Vance, M.A.; Goldgaber, D.; Roses, A.D. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 9649–9653. [Google Scholar] [CrossRef]
- Kok, E.; Haikonen, S.; Luoto, T.; Huhtala, H.; Goebeler, S.; Haapasalo, H.; Karhunen, P.J. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann. Neurol. 2009, 65, 650–657. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Vonsattel, J.-P.G.; Segal, A.Z.; Chiu, R.I.; Clatworthy, A.E.; Liao, A.; Hyman, B.T.; Rebeck, G.W. Association of apolipoprotein E ϵ2 and vasculopathy in cerebral amyloid angiopathy. Neurology 1998, 50, 961–965. [Google Scholar] [CrossRef]
- Jang, Y.K.; Kim, H.J.; Lee, J.S.; Kim, Y.J.; Kim, K.W.; Kim, Y.; Jang, H.; Lee, J.; Lee, J.M.; Kim, S.J.; et al. Distinctive Clinical Effects of Haemorrhagic Markers in Cerebral Amyloid Angiopathy. Sci. Rep. 2017, 7, 15984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagtap, A.; Gawande, S.; Sharma, S. Biomarkers in vascular dementia: A recent update. Biomark. Genom. Med. 2015, 7, 43–56. [Google Scholar] [CrossRef]
- Wahlund, L.O.; Westman, E.; van Westen, D.; Wallin, A.; Shams, S.; Cavallin, L.; Larsson, E.M. Imaging biomarkers of dementia: Recommended visual rating scales with teaching cases. Insights Imaging 2017, 8, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimers Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef]
- Hampel, H.; Cummings, J.; Blennow, K.; Gao, P.; Jack, C.R., Jr.; Vergallo, A. Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat. Rev. Neurol. 2021, 17, 580–589. [Google Scholar] [CrossRef]
- Cummings, J. The National Institute on Aging-Alzheimer’s Association Framework on Alzheimer’s disease: Application to clinical trials. Alzheimers Dement. 2019, 15, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Mattsson-Carlgren, N.; Leuzy, A.; Janelidze, S.; Palmqvist, S.; Stomrud, E.; Strandberg, O.; Smith, R.; Hansson, O. The implications of different approaches to define AT(N) in Alzheimer disease. Neurology 2020, 94, e2233–e2244. [Google Scholar] [CrossRef]
- Duron, E.; Hanon, O. Vascular risk factors, cognitive decline, and dementia. Vasc. Health Risk Manag. 2008, 4, 363–381. [Google Scholar]
- Cheng, Y.W.; Chiu, M.J.; Chen, Y.F.; Cheng, T.W.; Lai, Y.M.; Chen, T.F. The contribution of vascular risk factors in neurodegenerative disorders: From mild cognitive impairment to Alzheimer’s disease. Alzheimers Res. Ther. 2020, 12, 91. [Google Scholar] [CrossRef]
- Middleton, L.E.; Yaffe, K. Promising strategies for the prevention of dementia. Arch. Neurol. 2009, 66, 1210–1215. [Google Scholar] [CrossRef]
- Tariq, S.; Barber, P.A. Dementia risk and prevention by targeting modifiable vascular risk factors. J. Neurochem. 2018, 144, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Kuang, H.; Zhou, Z.-F.; Zhu, Y.-G.; Wan, Z.-K.; Yang, M.-W.; Hong, F.-F.; Yang, S.-L. Pharmacological Treatment of Vascular Dementia: A Molecular Mechanism Perspective. Aging Dis. 2021, 12, 308–326. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Tachibana, M.; Kanekiyo, T.; Bu, G. Role of LRP1 in the pathogenesis of Alzheimer’s disease: Evidence from clinical and preclinical studies. J. Lipid Res. 2017, 58, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Tian, D.Y.; Wang, Y.J. Peripheral clearance of brain-derived Abeta in Alzheimer’s disease: Pathophysiology and therapeutic perspectives. Transl. Neurodegener. 2020, 9, 16. [Google Scholar] [CrossRef]
- Ramanathan, A.; Nelson, A.R.; Sagare, A.P.; Zlokovic, B.V. Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: The role, regulation and restoration of LRP1. Front. Aging Neurosci. 2015, 7, 136. [Google Scholar] [CrossRef]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef]
- Schreiner, T.G.; Menéndez-González, M.; Popescu, B.O. The “Cerebrospinal Fluid Sink Therapeutic Strategy” in Alzheimer’s Disease-From Theory to Design of Applied Systems. Biomedicines 2022, 10, 1509. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Study (Country) | Length of the Study | Number of Study Participants (Age, Mean [SD]) | Vascular Risk Factors | Measurement of Brain β-Amyloid Load | Results |
|---|---|---|---|---|---|
| Gottesman et al. (2017) (USA) [67] | Evaluation of vascular risk factors since 1987–1989 with 18F-florbetapir PET scans in 2011–2013 | 322 without dementia (27% MCI) (75.8 [5.3]) | HTN, DM, BMI ≥ 30, TC ≥ 200 mg/dL, current smoking status | 18F-florbetapir PET (SUVR) | (1) Association between elevated BMI in midlife and elevated SUVR (OR: 2.06, 95% CI: 1.16–3.65) (2) OR for elevated SUVR and 1 vascular risk factor: 1.88 (95% CI: 0.95–3.72), OR for elevated SUVR and 2 or more vascular risk factors: 2.88 (95% CI: 1.46–5.69) |
| Hughes et al. (2018) (USA) [68] | Evaluation of vascular risk factors since 1987–1989 with 18F-florbetapir PET scans in 2011–2013 | 321 (27% MCI) (76 [5]) | Arterial stiffness by pulse wave velocity (PWV, carotid-femoral [cfPWV] and heart-carotid [hcPWV]) | 18F-florbetapir PET (SUVR) | (1) Association between greater central stiffness (hcPWV) and greater Aβ deposition (OR: 1.31, 95% CI: 1.01–1.7) (2) Association between cfPWV and a higher odds of Aβ-positive scans (OR: 1.4, 95% CI: 1.1–2.1). |
| Rabin et al. (2018) (USA) [69] | 7 years | 223 clinically normal older adults (73.7 [6.0]) | Framingham Heart Study general cardiovascular disease (FHS-CVD) risk score (age, sex, antihypertensive treatment, SBP, BMI, history of DM, and current cigarette smoking status) | 11C-PiB PET (DVR) | (1) Associations of a higher FHS-CVD risk score (β = −0.064; −0.094 to −0.033; p < 0.001) and higher Aβ burden (β = −0.058; −0.079 to −0.037; p < 0.001) with faster cognitive decline (2) Synergistic effect of FHS-CVD risk factors and Aβ burden (β = −0.040, 95% CI: −0.062 to −0.018; p < 0.001) |
| Arfanakis et al. (2020) (USA) [70] | 25 years | 603 (No cognitive impairment: 178, MCI: 154, dementia: 271) (age at death: 90 [7]; No cognitive impairment: 88 [7], MCI: 90 [6], dementia: 90 [7]) | HTN, DM, smoking, history of heart disease | Neuropathologic examination | Association between WMH burden and both vascular and Alzheimer’s pathologies (arteriolosclerosis (p < 10−4), gross (p < 10−4) and microscopic infarcts (p = 0.04), Aβ plaques (p = 0.028) |
| Kobe et al.(2020) (Canada) [71] | 7 years | 215 participants (PREVENT-AD cohort of cognitively unimpaired individuals) (62.3 [5.0]) | TC, HDL, LDL cholesterol levels, SBP, DBP, pulse pressure, Framingham Coronary Risk Profile (age, sex, SBP, DBP, HDL, LDL, smoking, DM) | 18F-NAV 4694 PET (SUVR) | Association of vascular risk factors with Aβ burden but not tau burden (only among individuals who were not using vascular medications) TC level (β = −0.002 [SE, 0.001]; p = 0.02), LDL cholesterol level (β = −0.002 [SE,0.001]; p = 0.006), SBP (β = −0.006 [SE, 0.002]; p = 0.02), pulse pressure (β = −0.007 [SE, 0.002]; p = 0.004), and Framingham Coronary Risk Profile score (β = −0.038 [SE, 0.011]; p = 0.001) |
| Lockhart et al. (2022) (USA) [72] | 19 years (enrollment, 2000–2002; 1st cognitive abilities screening, 2010–2012; 2nd screening, 2016–2018) | 159 participants (49.7% African-American, 50.3% White) (baseline age 55.8 [6.7]) | FSRP, CAIDE, ASCVD (All vascular risk factor scores include age, sex, SBP); FSRP, ASCVD (DM, antihypertensive treatment, smoking); CAIDE, ASCVD (TC) | 11C-PiB PET (SUVR) | Association of higher baseline Framingham stroke risk profile (FSRP) (p = 0.014) and Cardiovascular Risk Factors, Aging, and Incidence of Dementia (CAIDE) scores (p = 0.004) with global brain Aβ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.E.; Kim, H.J.; Jang, H.; Weiner, M.W.; DeCarli, C.; Na, D.L.; Seo, S.W. Interaction between Alzheimer’s Disease and Cerebral Small Vessel Disease: A Review Focused on Neuroimaging Markers. Int. J. Mol. Sci. 2022, 23, 10490. https://doi.org/10.3390/ijms231810490
Kim SE, Kim HJ, Jang H, Weiner MW, DeCarli C, Na DL, Seo SW. Interaction between Alzheimer’s Disease and Cerebral Small Vessel Disease: A Review Focused on Neuroimaging Markers. International Journal of Molecular Sciences. 2022; 23(18):10490. https://doi.org/10.3390/ijms231810490
Chicago/Turabian StyleKim, Si Eun, Hee Jin Kim, Hyemin Jang, Michael W. Weiner, Charles DeCarli, Duk L. Na, and Sang Won Seo. 2022. "Interaction between Alzheimer’s Disease and Cerebral Small Vessel Disease: A Review Focused on Neuroimaging Markers" International Journal of Molecular Sciences 23, no. 18: 10490. https://doi.org/10.3390/ijms231810490
APA StyleKim, S. E., Kim, H. J., Jang, H., Weiner, M. W., DeCarli, C., Na, D. L., & Seo, S. W. (2022). Interaction between Alzheimer’s Disease and Cerebral Small Vessel Disease: A Review Focused on Neuroimaging Markers. International Journal of Molecular Sciences, 23(18), 10490. https://doi.org/10.3390/ijms231810490