
Stearoyl-CoA Desaturase Regulates Angiogenesis and Energy Metabolism in Ischemic Cardiomyocytes

 , , and
, , and 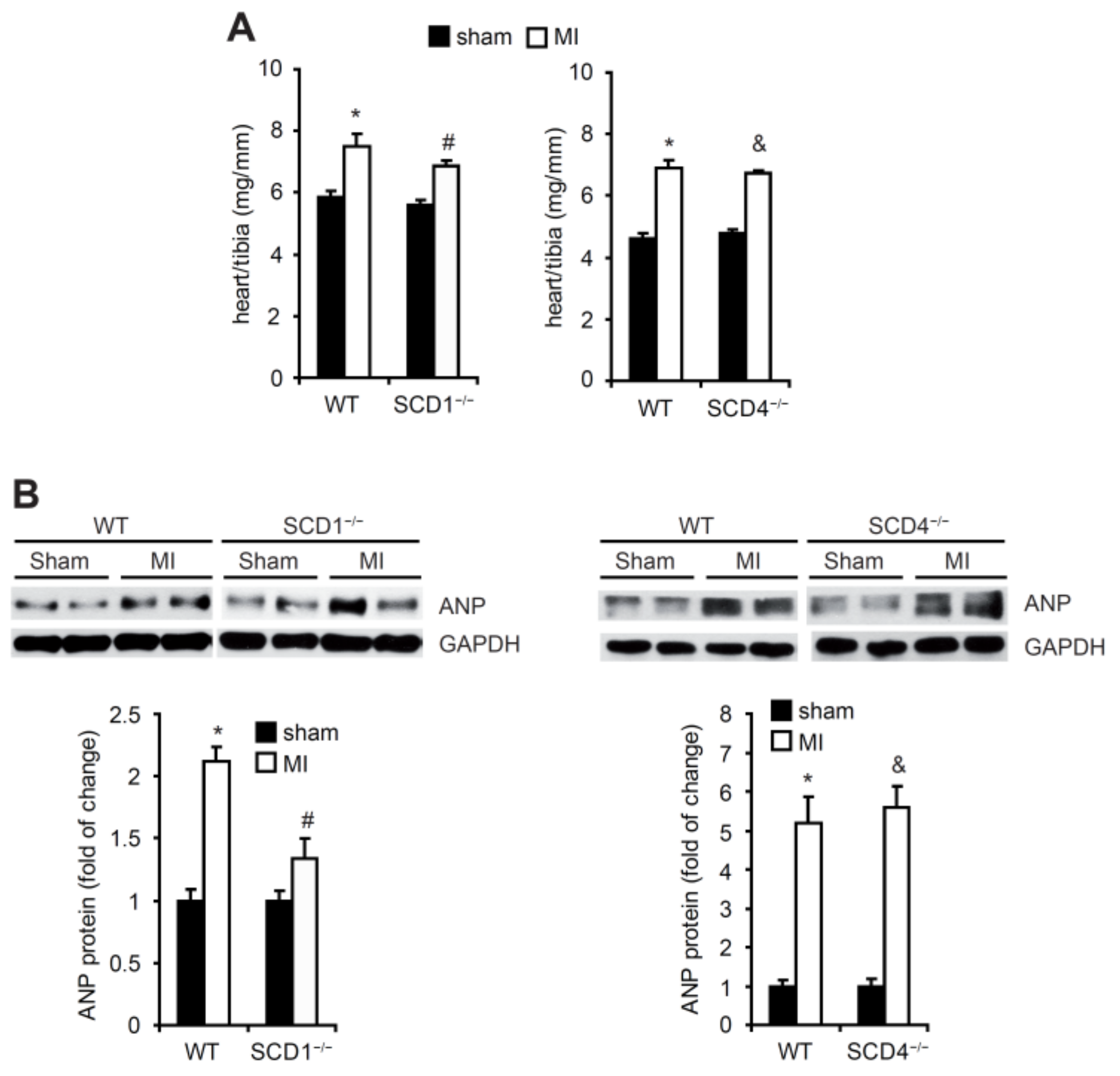{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
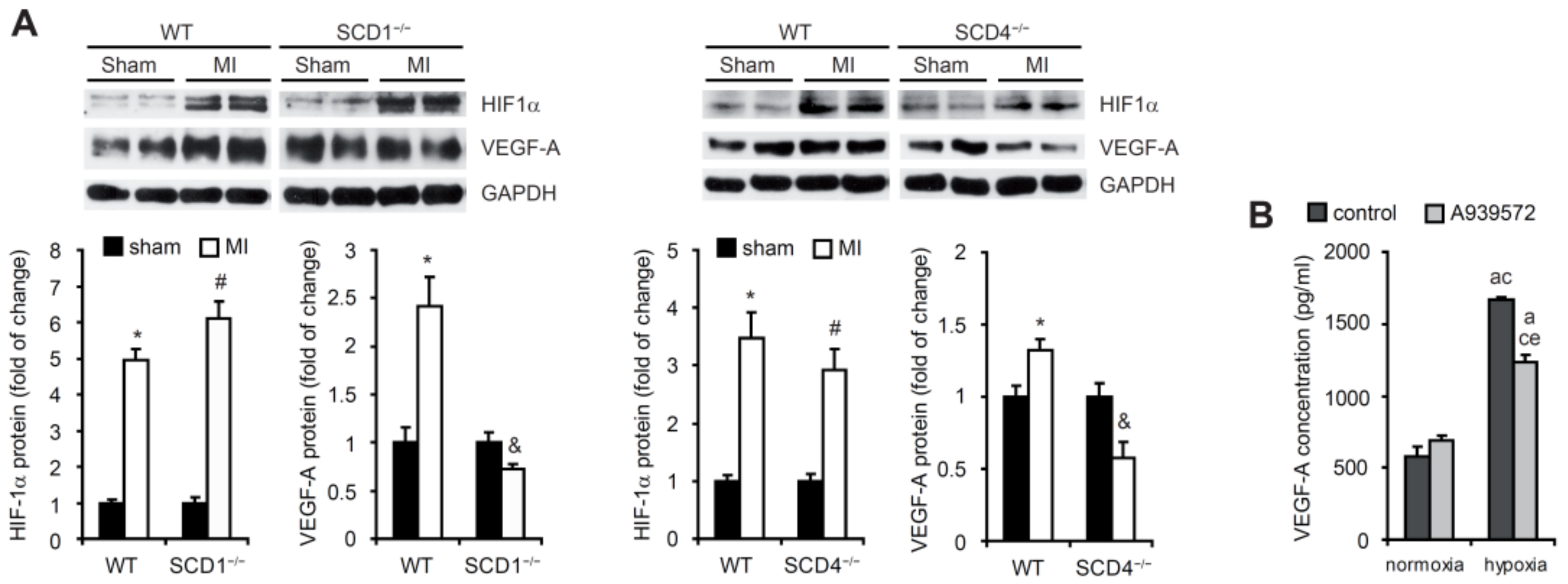
2.1. SCD Regulates VEGF-A Expression in Cardiomyocytes under Hypoxic Conditions
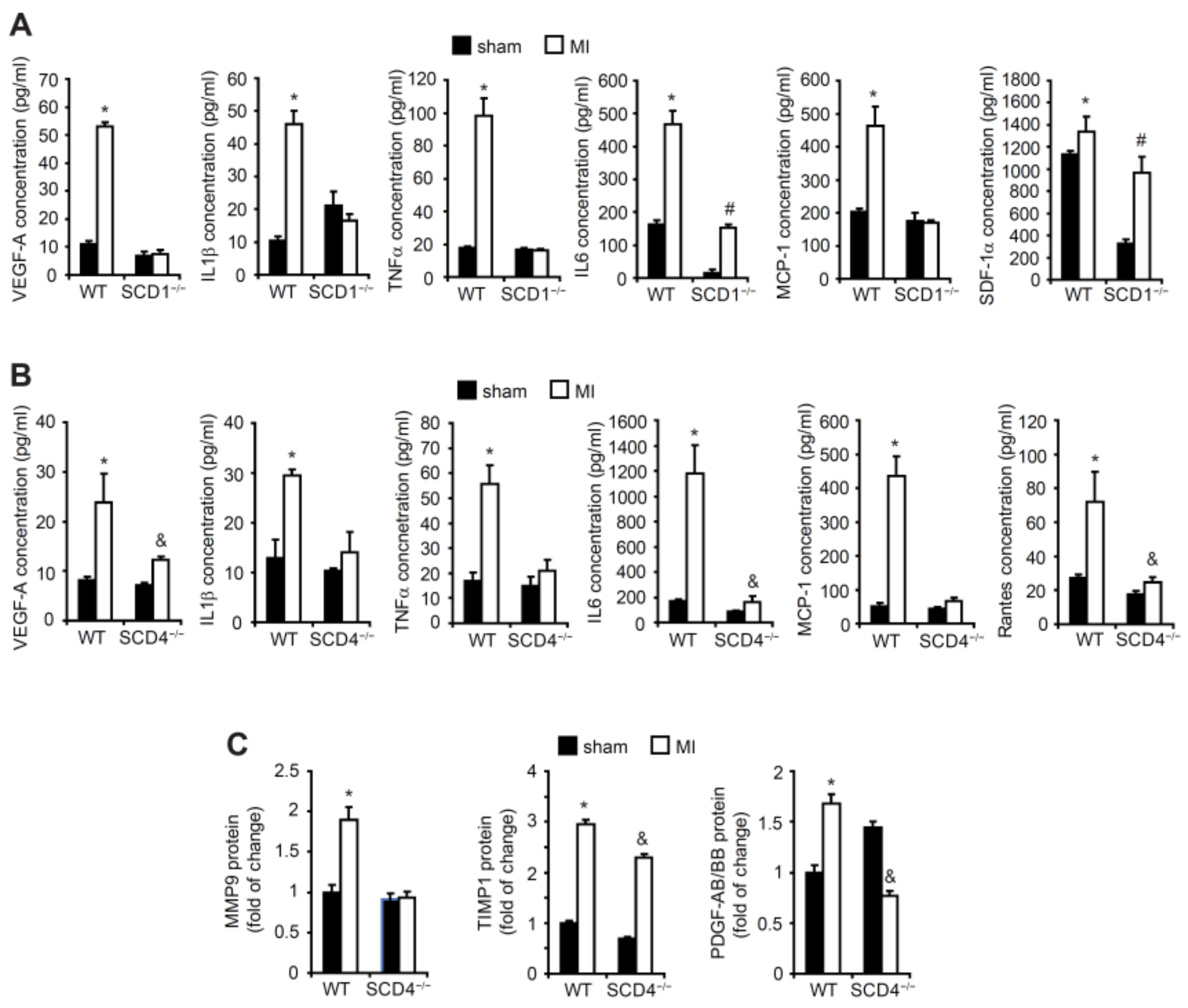
2.2. SCD Regulates the Secretion of Proangiogenic Factors to Plasma in Mice Post-MI
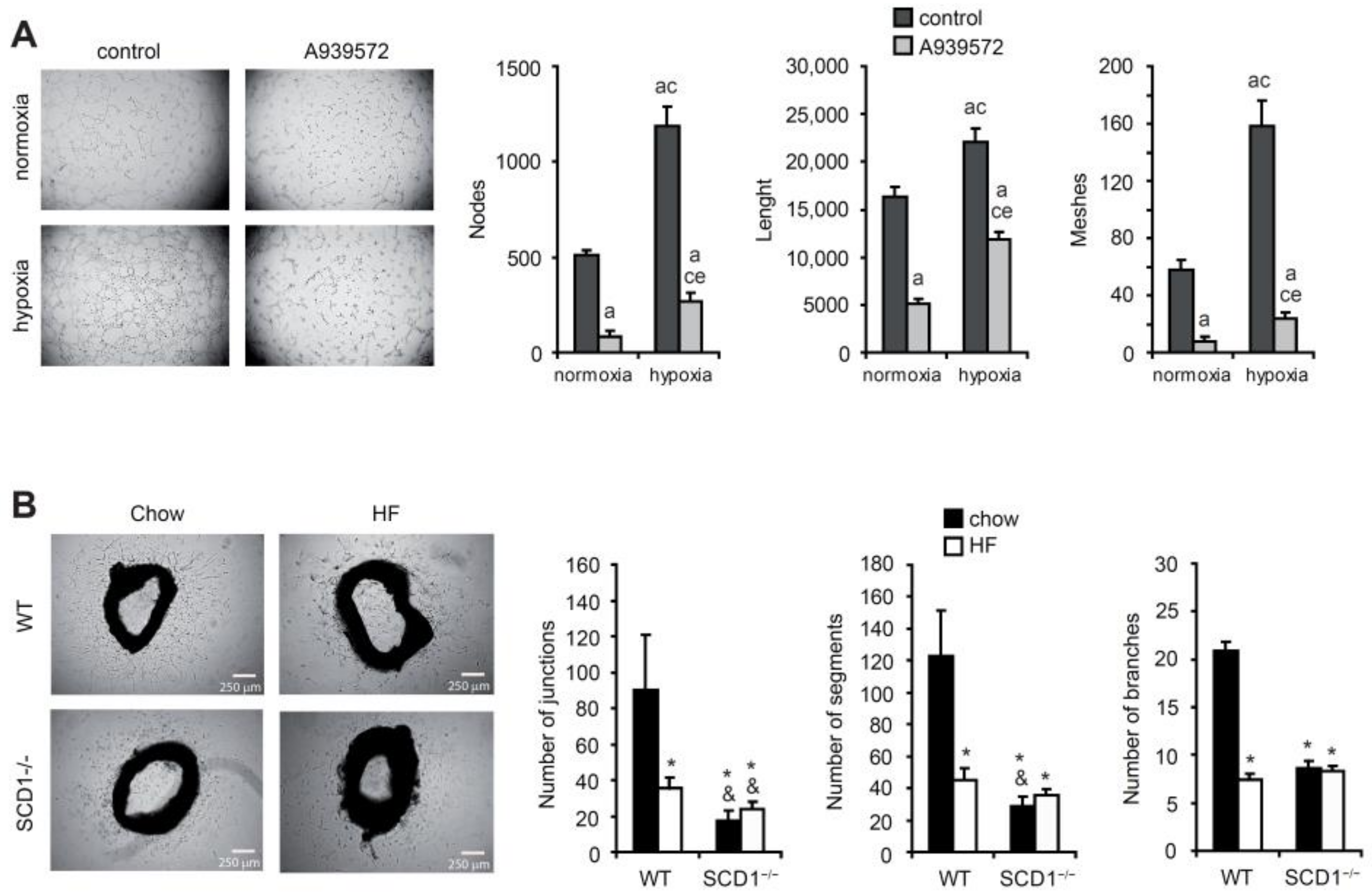
2.3. SCD Promotes New Blood Vessel Formation
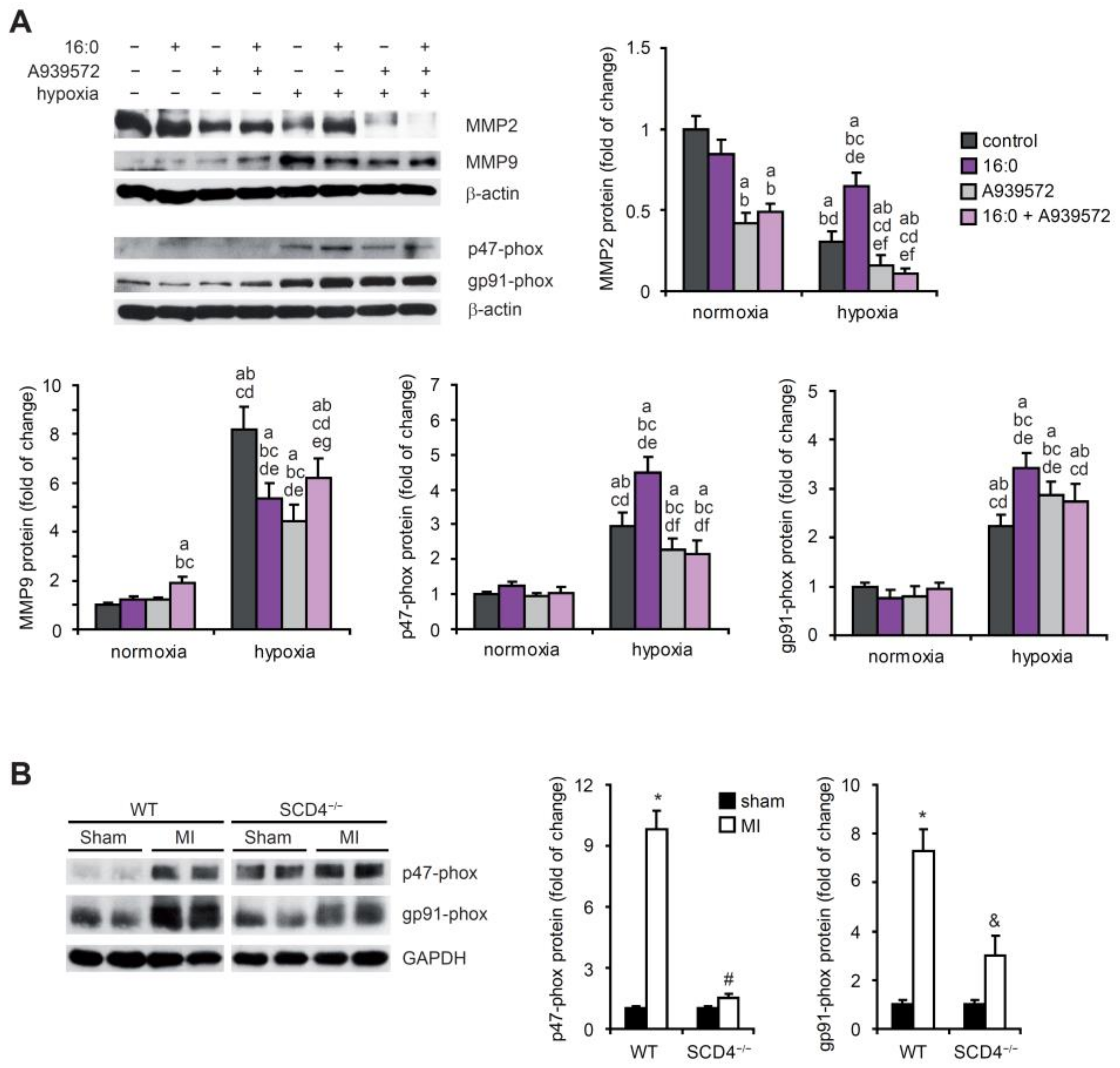
2.4. SCD Affects NADPH Oxidase Subunits and MMP Expression under Hypoxic Conditions
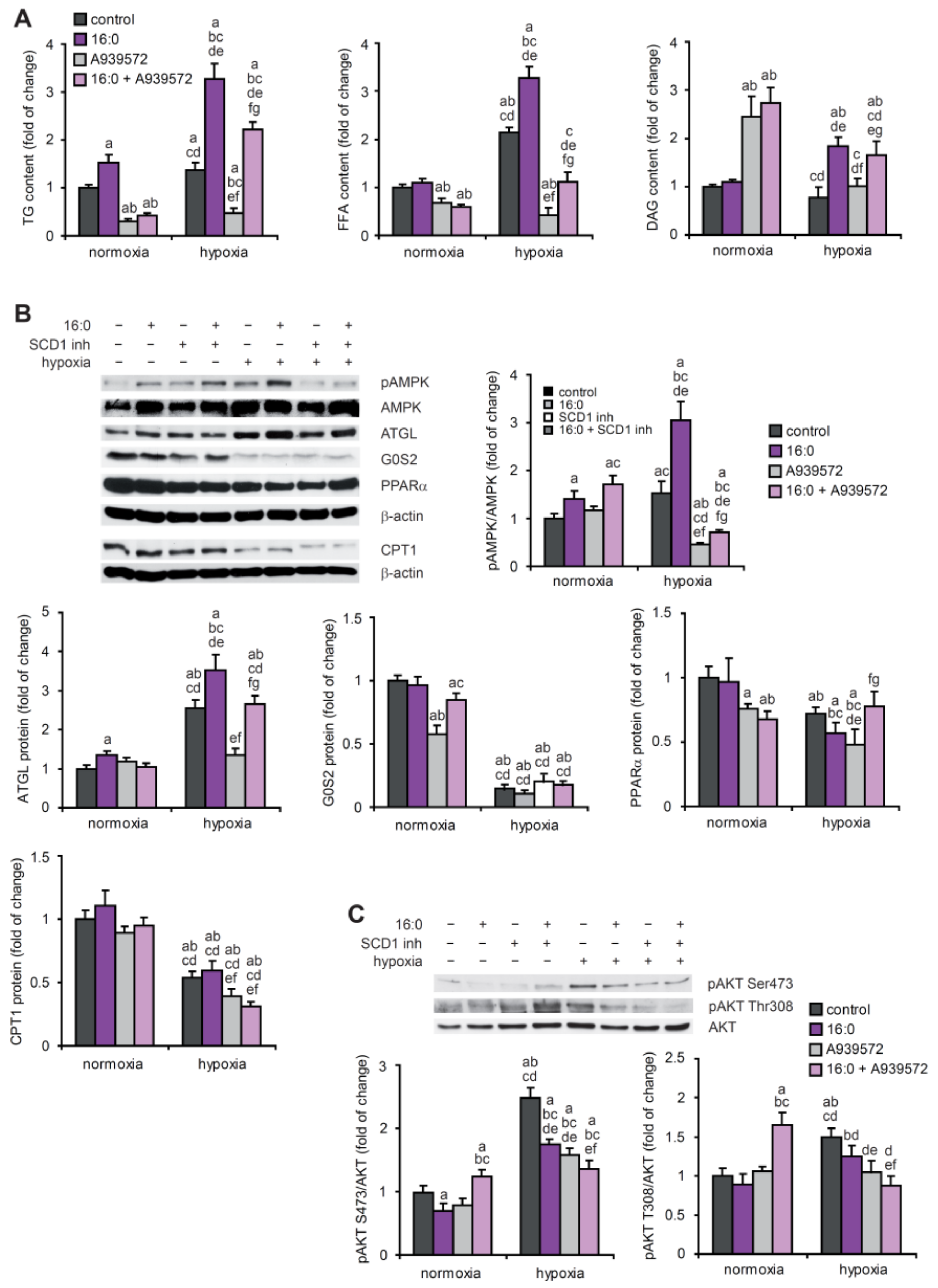
2.5. SCD1 Regulates Lipid Metabolism of Hypoxic Cardiomyocytes
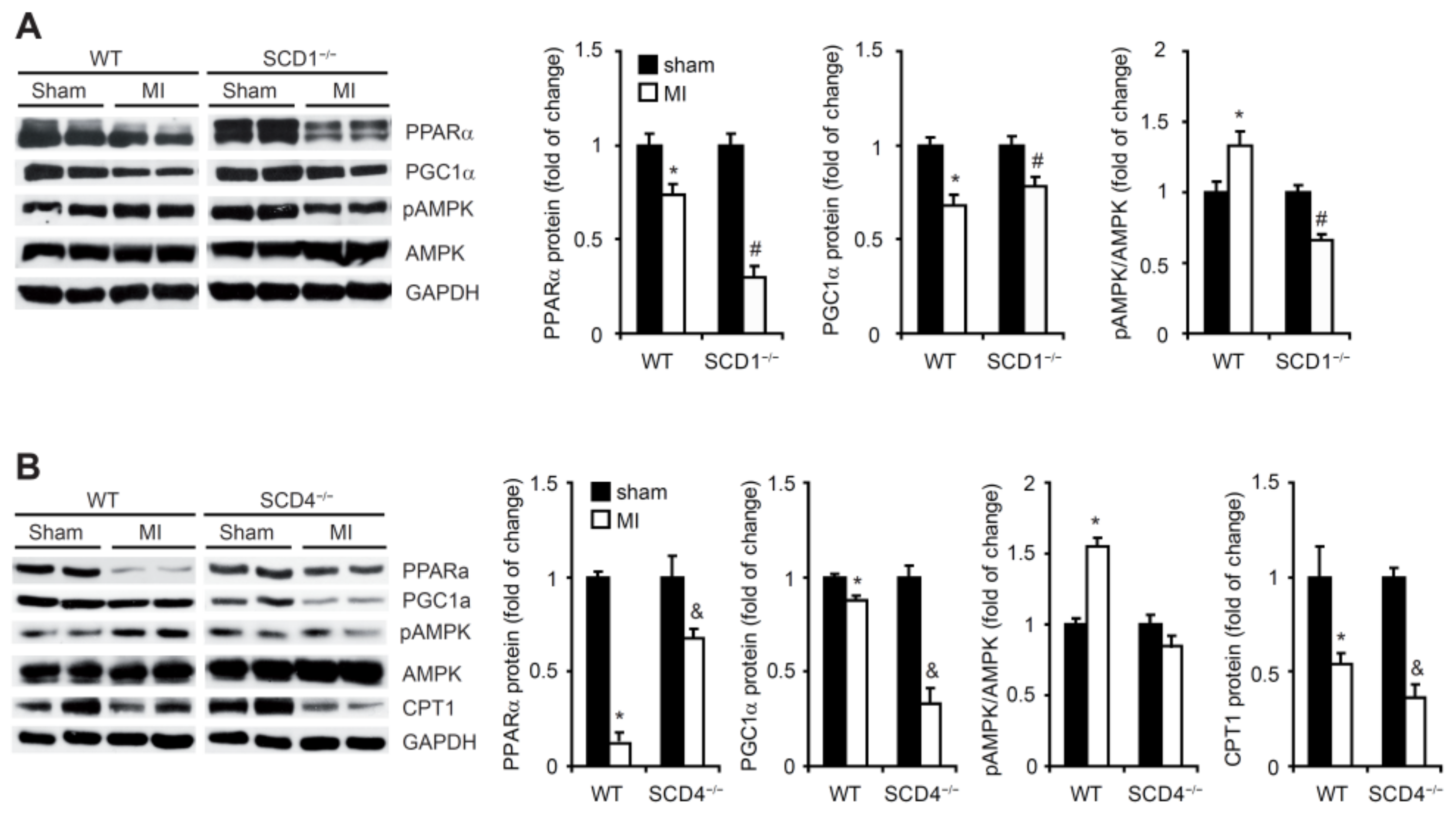
2.6. SCD1 and SCD4 Deletion Affects Metabolism in the Post-MI Heart
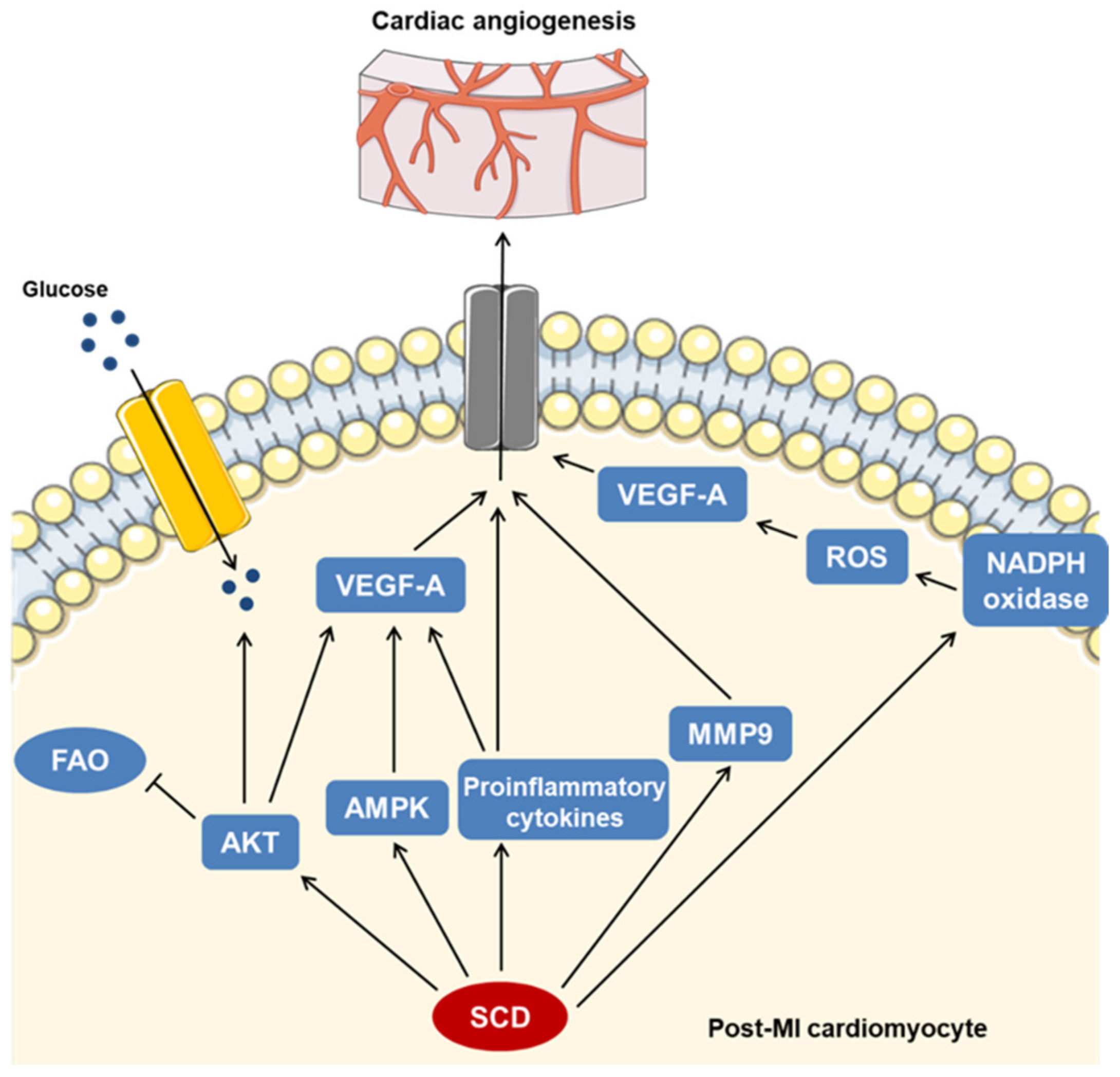
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Induction of Myocardial Infarction
4.4. Cellular Hypoxia Induction in HL-1 Cardiomyocytes
4.5. Enzyme-Linked Immunosorbent Assay
4.6. Determination of Angiogenic Profile
4.7. In Vitro Tube Formation Assay
4.8. Ex Vivo Mouse Aortic Ring Assays for Angiogenesis
4.9. Western Blot Analysis
4.10. Lipid Analysis
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dodd, M.S.; da Luz Sousa Fialho, M.; Montes Aparicio, C.M.; Kerr, M.; Timm, K.N.; Griffin, J.L.; Luiken, J.J.; Glatz, J.F.; Tyler, D.J.; Heather, L.C. Fatty acids prevent hypoxia-inducible factor-1α signaling through decreased succinate in diabetes. JACC Basic Transl. Sci. 2018, 3, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhao, W.; Chen, Y.; Liu, L.; Ahokas, R.A.; Sun, Y. Differential expression of vascular endothelial growth factor isoforms and receptor subtypes in the infarcted heart. Int. J. Cardiol. 2013, 167, 2638–2645. [Google Scholar] [CrossRef] [PubMed]
- Pluijmert, N.J.; Atsma, D.E.; Quax, P.H.A. Post-ischemic myocardial inflammatory response: A complex and dynamic process susceptible to immunomodulatory therapies. Front. Cardiovasc. Med. 2021, 8, 647785. [Google Scholar] [CrossRef] [PubMed]
- Deten, A.; Volz, H.C.; Briest, W.; Zimmer, H.G. Cardiac cytokine expression is upregulated in the acute phase after myocardial infarction. Experimental studies in rats. Cardiovasc. Res. 2002, 55, 329–340. [Google Scholar] [CrossRef]
- Ikonomidis, J.S.; Hendrick, J.W.; Parkhurst, A.M.; Herron, A.R.; Escobar, P.G.; Dowdy, K.B.; Stroud, R.E.; Hapke, E.; Zile, M.R.; Spinale, F.G. Accelerated LV remodeling after myocardial infarction in TIMP-1-deficient mice: Effects of exogenous MMP inhibition. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H149–H158. [Google Scholar] [CrossRef]
- Maulik, N.; Das, D.K. Redox signaling in vascular angiogenesis. Free Radic. Biol. Med. 2002, 33, 1047–1060. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Tang, Y.; Fukai, T.; Dikalov, S.I.; Ma, Y.; Fujimoto, M.; Quinn, M.T.; Pagano, P.J.; Johnson, C.; Alexander, R.W. Novel role of gp91(phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ. Res. 2002, 91, 1160–1167. [Google Scholar] [CrossRef]
- Staels, B.; Fruchart, J.C. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes 2005, 54, 2460–2470. [Google Scholar] [CrossRef]
- Barlaka, E.; Ledvényiová, V.; Galatou, E.; Ferko, M.; Čarnická, S.; Ravingerová, T.; Lazou, A. Delayed cardioprotective effects of WY-14643 are associated with inhibition of MMP-2 and modulation of Bcl-2 family proteins through PPAR-α activation in rat hearts subjected to global ischaemia-reperfusion. Can. J. Physiol. Pharmacol. 2013, 91, 608–816. [Google Scholar] [CrossRef]
- Duerr, G.D.; Heinemann, J.C.; Arnoldi, V.; Feisst, A.; Kley, J.; Ghanem, A.; Welz, A.; Dewald, O. Cardiomyocyte specific peroxisome proliferator-activated receptor-α overexpression leads to irreversible damage in ischemic murine heart. Life Sci. 2014, 102, 88–97. [Google Scholar] [CrossRef]
- Fraisl, P.; Mazzone, M.; Schmidt, T.; Carmeliet, P. Regulation of angiogenesis by oxygen and metabolism. Dev. Cell 2009, 16, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Foo, S.Y.; Ma, Y.; Ruas, J.L.; Bommi-Reddy, A.; Girnun, G.; Cooper, M.; Laznik, D.; Chinsomboon, J.; Rangwala, S.M.; et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature 2008, 451, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef]
- Dobrzyn, P.; Sampath, H.; Dobrzyn, A.; Miyazaki, M.; Ntambi, J.M. Loss of stearoyl-CoA desaturase 1 inhibits fatty acid oxidation and increases glucose utilization in the heart. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E357–E364. [Google Scholar] [CrossRef]
- Dobrzyn, P.; Dobrzyn, A.; Miyazaki, M.; Ntambi, J.M. Loss of stearoyl-CoA desaturase 1 rescues cardiac function in obese leptin-deficient mice. J. Lipid Res. 2010, 51, 2202–2210. [Google Scholar] [CrossRef]
- Bednarski, T.; Olichwier, A.; Opasinska, A.; Pyrkowska, A.; Gan, A.M.; Ntambi, J.M.; Dobrzyn, P. Stearoyl-CoA desaturase 1 deficiency reduces lipid accumulation in the heart by activating lipolysis independently of peroxisome proliferator-activated receptor α. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Jacobson, M.J.; Man, W.C.; Cohen, P.; Asilmaz, E.; Friedman, J.M.; Ntambi, J.M. Identification and characterization of murine SCD4, a novel heart-specific stearoyl-CoA desaturase isoform regulated by leptin and dietary factors. J. Biol. Chem. 2003, 278, 33904–33911. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, Y.; Fang, Q.; Zhong, P.; Li, W.; Wang, L.; Fu, W.; Zhang, Y.; Xu, Z.; Li, X.; et al. Saturated palmitic acid induces myocardial inflammatory injuries through direct binding to TLR4 accessory protein MD2. Nat. Commun. 2017, 8, 13997. [Google Scholar] [CrossRef]
- Yuan, L.; Mao, Y.; Luo, W.; Wu, W.; Hao, X.; Wang, X.L.; Shen, Y.H. Palmitic acid dysregulates the Hippo-YAP pathway and inhibits angiogenesis by inducing mitochondrial damage and activating the cytosolic DNA sensor cGAS-STING-IRF3 signaling mechanism. J. Biol. Chem. 2017, 292, 15002–15015. [Google Scholar] [CrossRef]
- Zhao, W.; Zhao, T.; Huang, V.; Chen, Y.; Ahokas, R.A.; Sun, Y. Platelet-derived growth factor involvement in myocardial remodeling following infarction. J. Mol. Cell. Cardiol. 2011, 51, 830–838. [Google Scholar] [CrossRef]
- Hsieh, P.C.; Davis, M.E.; Gannon, J.; MacGillivray, C.; Lee, R.T. Controlled delivery of PDGF-BB for myocardial protection using injectable self-assembling peptide nanofibers. J. Clin. Investig. 2006, 116, 237–248. [Google Scholar] [CrossRef] [PubMed]
- van Hinsbergh, V.W.; Koolwijk, P. Endothelial sprouting and angiogenesis: Matrix metalloproteinases in the lead. Cardiovasc. Res. 2008, 78, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, V.E. Impact of nutrition on cardiovascular function. Curr. Probl. Cardiol. 2020, 45, 100391. [Google Scholar] [CrossRef] [PubMed]
- Cole, M.A.; Abd Jamil, A.H.; Heather, L.C.; Murray, A.J.; Sutton, E.R.; Slingo, M.; Sebag-Montefiore, L.; Tan, S.C.; Aksentijević, D.; Gildea, O.S.; et al. On the pivotal role of PPARα in adaptation of the heart to hypoxia and why fat in the diet increases hypoxic injury. FASEB J. 2016, 30, 2684–2697. [Google Scholar] [CrossRef]
- Duncan, J.G. Peroxisome proliferator activated receptor-alpha (PPARα) and PPAR gamma coactivator-1alpha (PGC-1α) regulation of cardiac metabolism in diabetes. Pediatr. Cardiol. 2011, 32, 323–328. [Google Scholar] [CrossRef]
- Song, S.; Attia, R.R.; Connaughton, S.; Niesen, M.I.; Ness, G.C.; Elam, M.B.; Hori, R.T.; Cook, G.A.; Park, E.A. Peroxisome proliferator activated receptor alpha (PPARalpha) and PPAR gamma coactivator (PGC-1alpha) induce carnitine palmitoyltransferase IA (CPT-1A) via independent gene elements. Mol. Cell. Endocrinol. 2010, 325, 54–63. [Google Scholar] [CrossRef]
- Braile, M.; Marcella, S.; Cristinziano, L.; Galdiero, M.R.; Modestino, L.; Ferrara, A.L.; Varricchi, G.; Marone, G.; Loffredo, S. VEGF-A in cardiomyocytes and heart diseases. Int. J. Mol. Sci. 2020, 21, 5294. [Google Scholar] [CrossRef]
- Yu, J.M.; Zhang, X.B.; Jiang, W.; Wang, H.D.; Zhang, Y.N. Astragalosides promote angiogenesis via vascular endothelial growth factor and basic fibroblast growth factor in a rat model of myocardial infarction. Mol. Med. Rep. 2015, 12, 6718–6726. [Google Scholar] [CrossRef]
- Seko, Y.; Seko, Y.; Takahashi, N.; Shibuya, M.; Yazaki, Y. Pulsatile stretch stimulates vascular endothelial growth factor (VEGF) secretion by cultured rat cardiac myocytes. Biochem. Biophys. Res. Commun. 1999, 254, 462–465. [Google Scholar] [CrossRef]
- Doronzo, G.; Viretto, M.; Barale, C.; Russo, I.; Mattiello, L.; Anfossi, G.; Trovati, M. Oleic acid increases synthesis and secretion of VEGF in rat vascular smooth muscle cells: Role of oxidative stress and impairment in obesity. Int. J. Mol. Sci. 2013, 14, 18861–18880. [Google Scholar] [CrossRef]
- Smith, A.N.; Muffley, L.A.; Bell, A.N.; Numhom, S.; Hocking, A.M. Unsaturated fatty acids induce mesenchymal stem cells to increase secretion of angiogenic mediators. J. Cell. Physiol. 2012, 227, 3225–3233. [Google Scholar] [CrossRef]
- Hong, K.H.; Ryu, J.; Han, K.H. Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood 2005, 105, 1405–1407. [Google Scholar] [CrossRef] [PubMed]
- Maloney, J.P.; Gao, L. Proinflammatory cytokines increase vascular endothelial growth factor expression in alveolar epithelial cells. Med. Inflamm. 2015, 2015, 387842. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, J.; Zhang, X.; Wang, C.; Huang, Y.; Dai, K.; Zhang, X. TNF-α-induced LRG1 promotes angiogenesis and mesenchymal stem cell migration in the subchondral bone during osteoarthritis. Cell Death Dis. 2017, 8, e2715. [Google Scholar] [CrossRef] [PubMed]
- Laschke, M.W.; Elitzsch, A.; Vollmar, B.; Vajkoczy, P.; Menger, M.D. Combined inhibition of vascular endothelial growth factor (VEGF), fibroblast growth factor and platelet-derived growth factor, but not inhibition of VEGF alone, effectively suppresses angiogenesis and vessel maturation in endometriotic lesions. Hum. Reprod. 2006, 21, 262–268. [Google Scholar] [CrossRef]
- Mongiat, M.; Andreuzzi, E.; Tarticchio, G.; Paulitti, A. Extracellular matrix, a hard player in angiogenesis. Int. J. Mol. Sci. 2016, 17, 1822. [Google Scholar] [CrossRef]
- Zhang, P.; Wu, C.; Huang, X.H.; Shen, C.L.; Li, L.; Zhang, W.; Yao, C.Z. Aspirin suppresses TNF-α-induced MMP-9 expression via NF-κB and MAPK signaling pathways in RAW264.7 cells. Exp. Ther. Med. 2017, 14, 5597–5604. [Google Scholar] [CrossRef][Green Version]
- Wali, J.A.; Jarzebska, N.; Raubenheimer, D.; Simpson, S.J.; Rodionov, R.N.; O’Sullivan, J.F. Cardio-metabolic effects of high-fat diets and their underlying mechanisms—A narrative review. Nutrients 2020, 12, 1505. [Google Scholar] [CrossRef]
- Yu, S.; Kim, R.; Jiang, K.; Ogrodnik, M.; Zhu, X.Y.; Ferguson, C.M.; Tchkonia, T.; Lerman, A.; Kirkland, J.L.; Lerman, L.O. Quercetin reverses cardiac systolic dysfunction in mice fed with a high-fat diet: Role of angiogenesis. Oxid. Med. Cell. Longev. 2021, 2021, 8875729. [Google Scholar] [CrossRef]
- Broniarek, I.; Koziel, A.; Jarmuszkiewicz, W. The effect of chronic exposure to high palmitic acid concentrations on the aerobic metabolism of human endothelial EA.hy926 cells. Pflug. Arch. 2016, 468, 1541–1554. [Google Scholar] [CrossRef]
- Xia, C.; Meng, Q.; Liu, L.Z.; Rojanasakul, Y.; Wang, X.R.; Jiang, B.H. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007, 67, 10823–10830. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.I.; Barca, E.; Subramanyam, P.; Komrowski, M.; Pajvani, U.; Colecraft, H.M.; Hirano, M.; Morrow, J.P. Inhibition of NAPDH oxidase 2 (NOX2) prevents oxidative stress and mitochondrial abnormalities caused by saturated fat in cardiomyocytes. PLoS ONE 2016, 11, e0145750. [Google Scholar]
- Malla, R.R.; Raghu, H.; Rao, J.S. Regulation of NADPH oxidase (Nox2) by lipid rafts in breast carcinoma cells. Int. J. Oncol. 2010, 37, 1483–1493. [Google Scholar]
- Tan, S.H.; Shui, G.; Zhou, J.; Shi, Y.; Huang, J.; Xia, D.; Wenk, M.R.; Shen, H.M. Critical role of SCD1 in autophagy regulation via lipogenesis and lipid rafts-coupled AKT-FOXO1 signaling pathway. Autophagy 2014, 10, 226–242. [Google Scholar] [CrossRef] [PubMed]
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid use and misuse by the heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef]
- Yang, X.; Lu, X.; Lombès, M.; Bae Rha, G.; Chi, Y.I.; Guerin, T.M.; Smart, E.J.; Liu, J. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010, 11, 194–205. [Google Scholar] [CrossRef]
- Kim, S.J.; Tang, T.; Abbott, M.; Viscarra, J.A.; Wang, Y.; Sul, H.S. AMPK phosphorylates desnutrin/ATGL and hormone-sensitive lipase to regulate lipolysis and fatty acid oxidation within adipose tissue. Mol. Cell. Biol. 2016, 36, 1961–1976. [Google Scholar] [CrossRef]
- Li, Y.; Sun, R.; Zou, J.; Ying, Y.; Luo, Z. Dual roles of the AMP-activated protein kinase pathway in angiogenesis. Cells 2019, 8, 752. [Google Scholar] [CrossRef]
- Gutsaeva, D.R.; Sue Carraway, M.; Suliman, H.B.; Demchenko, I.T.; Shitara, H.; Yonekawa, H.; Piantadosi, C.A. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J. Neurosci. 2008, 28, 2015–2024. [Google Scholar] [CrossRef]
- de Lima Luna, C.A.; Forti, F.L. Modulation of SCD1 activity in hepatocyte cell lines: Evaluation of genomic stability and proliferation. Mol. Cell. Biochem. 2021, 476, 3393–3405. [Google Scholar] [CrossRef]
- Tracz-Gaszewska, Z.; Dobrzyn, P. Stearoyl-CoA desaturase 1 as a therapeutic target for the treatment of cancer. Cancers 2019, 11, 948. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.A.; Matsui, T.; Li, L.; Rosenzweig, A. Transcriptional effects of chronic Akt activation in the heart. J. Biol. Chem. 2002, 277, 22528–22533. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Man, W.C.; Ntambi, J.M. Targeted disruption of stearoyl-CoA desaturase1 gene in mice causes atrophy of sebaceous and meibomian glands and depletion of wax esters in the eyelid. J. Nutr. 2001, 131, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Bond, L.M.; Ntambi, J.M. UCP1 deficiency increases adipose tissue monounsaturated fatty acid synthesis and trafficking to the liver. J. Lipid Res. 2018, 59, 224–236. [Google Scholar] [CrossRef]
- Claycomb, W.C.; Lanson, N.A.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef]
- Paton, C.M.; Ntambi, J.M. Loss of stearoyl-CoA desaturase activity leads to free cholesterol synthesis through increased Xbp-1 splicing. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E1066–E1075. [Google Scholar] [CrossRef][Green Version]
- Liu, J.; Cinar, R.; Xiong, K.; Godlewski, G.; Jourdan, T.; Lin, Y.; Ntambi, J.M.; Kunos, G. Monounsaturated fatty acids generated via stearoyl CoA desaturase-1 are endogenous inhibitors of fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2013, 110, 18832–18837. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, G.; Berndt, S.; Ferratge, S.; Rasband, W.; Cuendet, M.; Uzan, G.; Albanese, P. Angiogenesis analyzer for ImageJ—A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci. Rep. 2020, 10, 11568. [Google Scholar] [CrossRef]
- Baker, M.; Robinson, S.D.; Lechertier, T.; Barber, P.R.; Tavora, B.; D’Amico, G.; Jones, D.T.; Vojnovic, B.; Hodivala-Dilke, K. Use of the mouse aortic ring assay to study angiogenesis. Nat. Protoc. 2011, 7, 89–104. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gan, A.-M.; Tracz-Gaszewska, Z.; Ellert-Miklaszewska, A.; Navrulin, V.O.; Ntambi, J.M.; Dobrzyn, P. Stearoyl-CoA Desaturase Regulates Angiogenesis and Energy Metabolism in Ischemic Cardiomyocytes. Int. J. Mol. Sci. 2022, 23, 10459. https://doi.org/10.3390/ijms231810459
Gan A-M, Tracz-Gaszewska Z, Ellert-Miklaszewska A, Navrulin VO, Ntambi JM, Dobrzyn P. Stearoyl-CoA Desaturase Regulates Angiogenesis and Energy Metabolism in Ischemic Cardiomyocytes. International Journal of Molecular Sciences. 2022; 23(18):10459. https://doi.org/10.3390/ijms231810459
Chicago/Turabian StyleGan, Ana-Maria, Zuzanna Tracz-Gaszewska, Aleksandra Ellert-Miklaszewska, Viktor O. Navrulin, James M. Ntambi, and Pawel Dobrzyn. 2022. "Stearoyl-CoA Desaturase Regulates Angiogenesis and Energy Metabolism in Ischemic Cardiomyocytes" International Journal of Molecular Sciences 23, no. 18: 10459. https://doi.org/10.3390/ijms231810459
APA StyleGan, A.-M., Tracz-Gaszewska, Z., Ellert-Miklaszewska, A., Navrulin, V. O., Ntambi, J. M., & Dobrzyn, P. (2022). Stearoyl-CoA Desaturase Regulates Angiogenesis and Energy Metabolism in Ischemic Cardiomyocytes. International Journal of Molecular Sciences, 23(18), 10459. https://doi.org/10.3390/ijms231810459