
State-of-the-Art Molecular Dynamics Simulation Studies of RNA-Dependent RNA Polymerase of SARS-CoV-2


Abstract
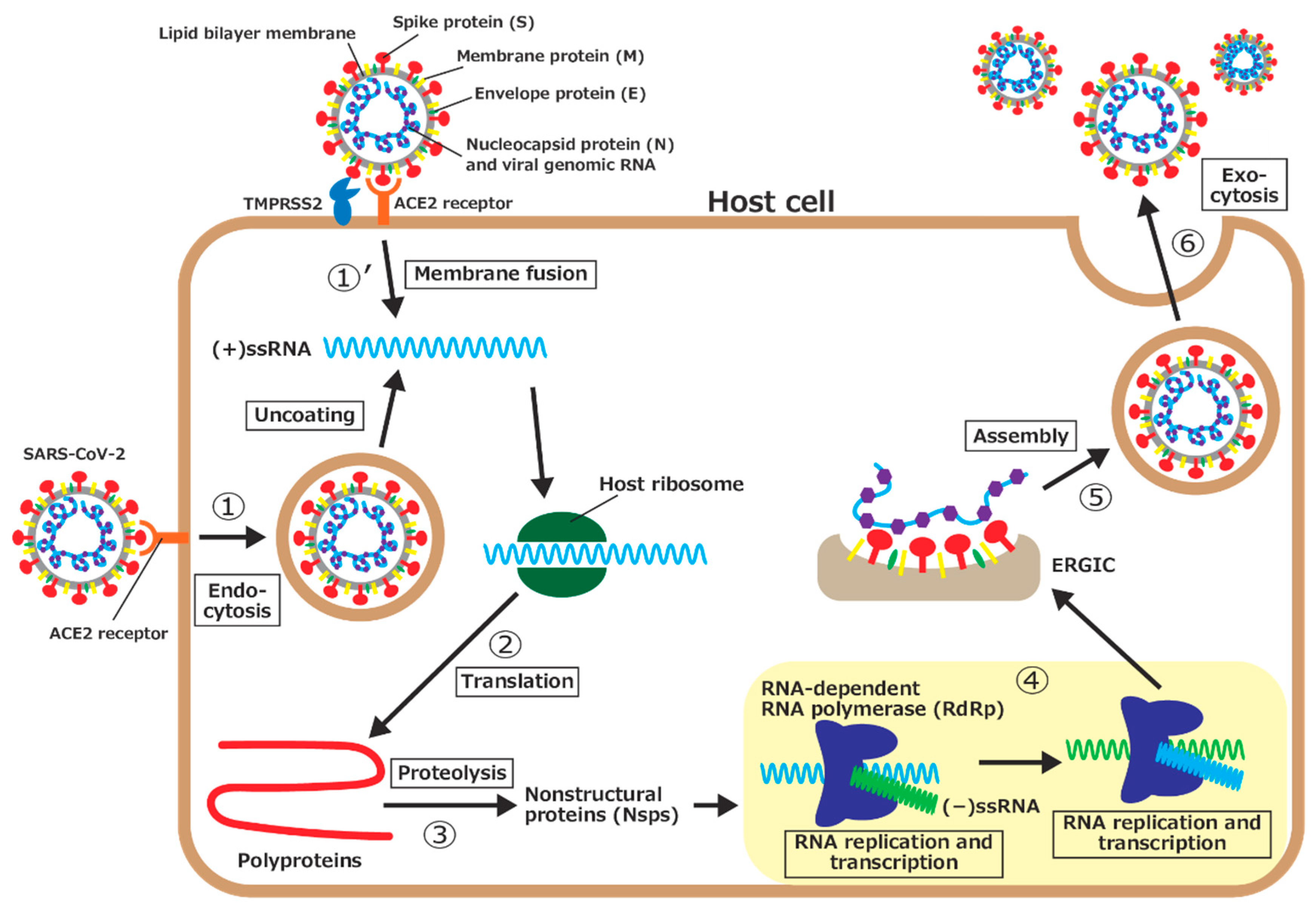
:1. Introduction
- 1.
- The spike protein on the surface of SARS-CoV-2 is bound to angiotensin-converting enzyme II (ACE2), a receptor on the host cell’s surface, and the virus enters the cell through endocytosis. The virus then uncoats, and the viral genomic RNA is released into the cytoplasm.
- 1’.
- Another way for the viral genomic RNA to enter the host cell is by membrane fusion. After the spike protein is bound to ACE2, part of the spike protein is cleaved by a type II transmembrane serine protease (TMPRSS2) on the host cell’s surface. The viral envelope is fused with the host cell’s membrane, and the viral genomic RNA is then released into the cytoplasm.
- 2.
- Because the genomic RNA of SARS-CoV-2 is (+)ssRNA, which functions as mRNAs, it is translated by ribosomes of the host cell. Two large proteins, called polyprotein 1a (pp1a) and polyprotein 1ab (pp1ab), are synthesized.
- 3.
- The polyproteins are then hydrolyzed, that is, proteolyzed, by virus-derived proteases, part of the polyprotein, to synthesize a series of nsps, which is necessary for the viral replication.
- 4.
- The RNA-dependent RNA polymerase (RdRp), one of the nsps, first synthesizes a negative-sense single-stranded RNA ((−)ssRNA) from (+)ssRNA. RdRp then uses (−)ssRNA as a template to synthesize the genomic RNAs for progeny viruses and several short subgenomic RNAs.
- 5.
- The synthesized nucleocapsid proteins are bound to the genomic RNA. In addition, various structural proteins are synthesized by translation of the subgenomic RNAs and inserted into the endoplasmic reticulum membrane. The genomic RNA and nucleocapsid protein complex is assembled with structural proteins (spike, membrane, and envelope proteins) at the endoplasmic reticulum–Golgi intermediate compartment (ERGIC) to form progeny viruses.
- 6.
- Progeny viruses are then released from the host cell through exocytosis.
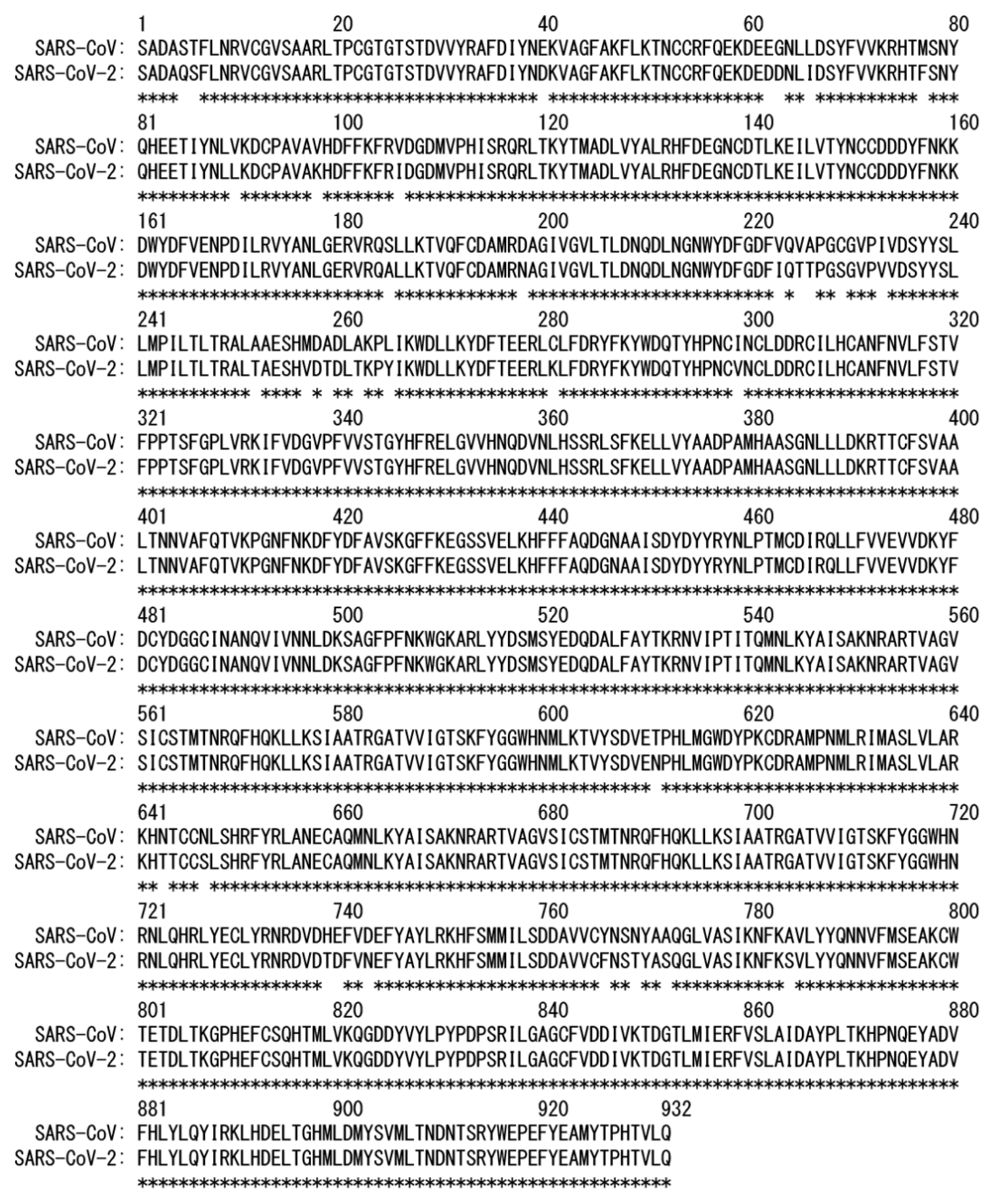
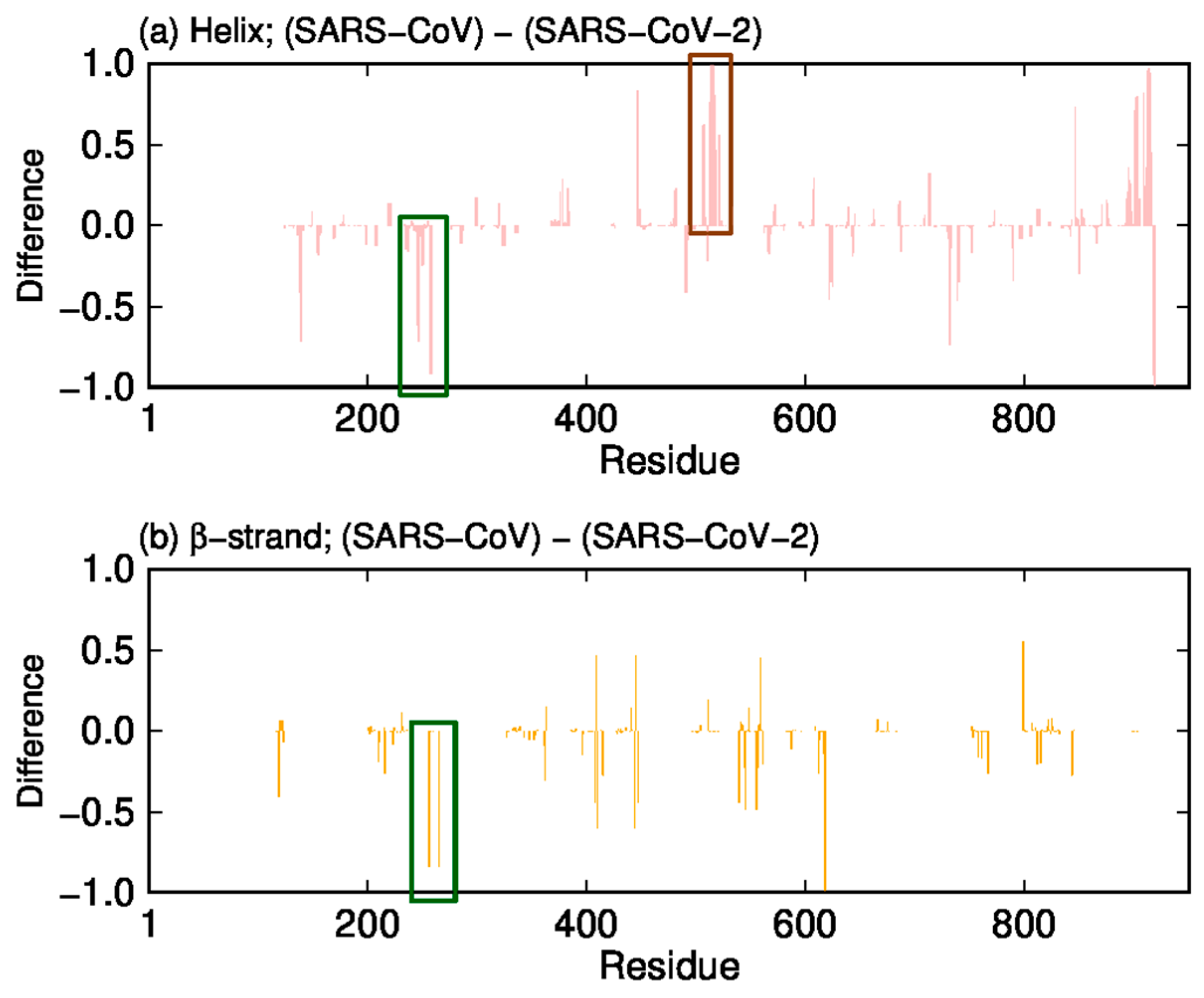
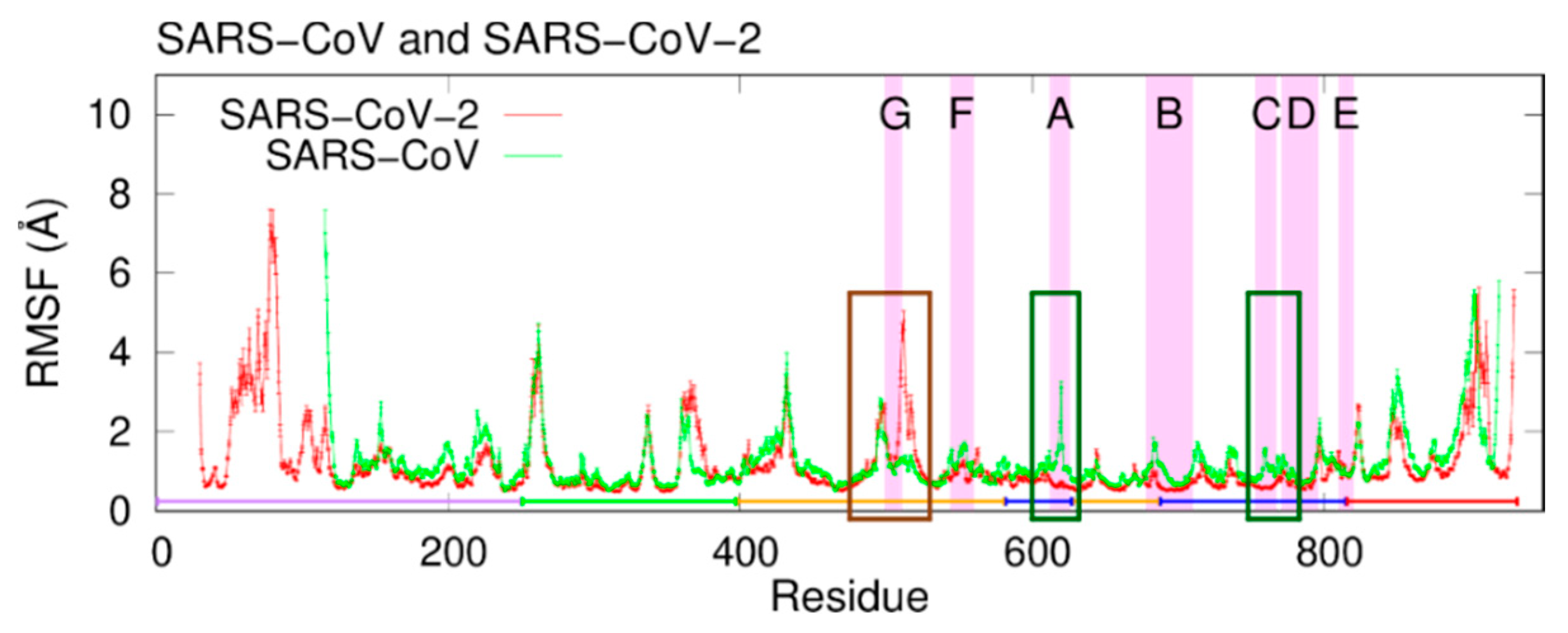
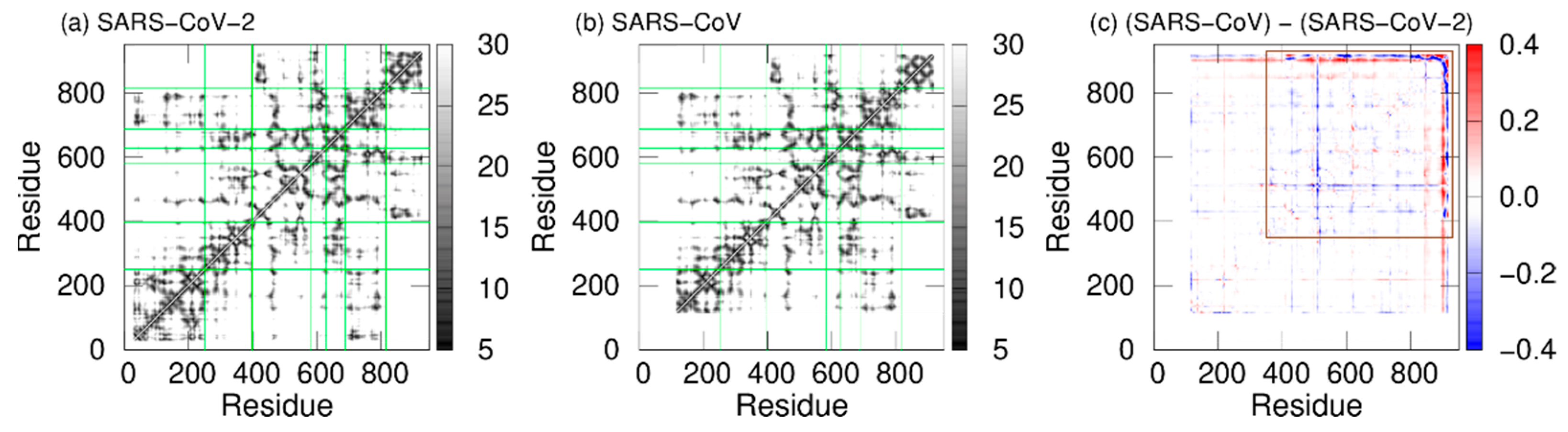
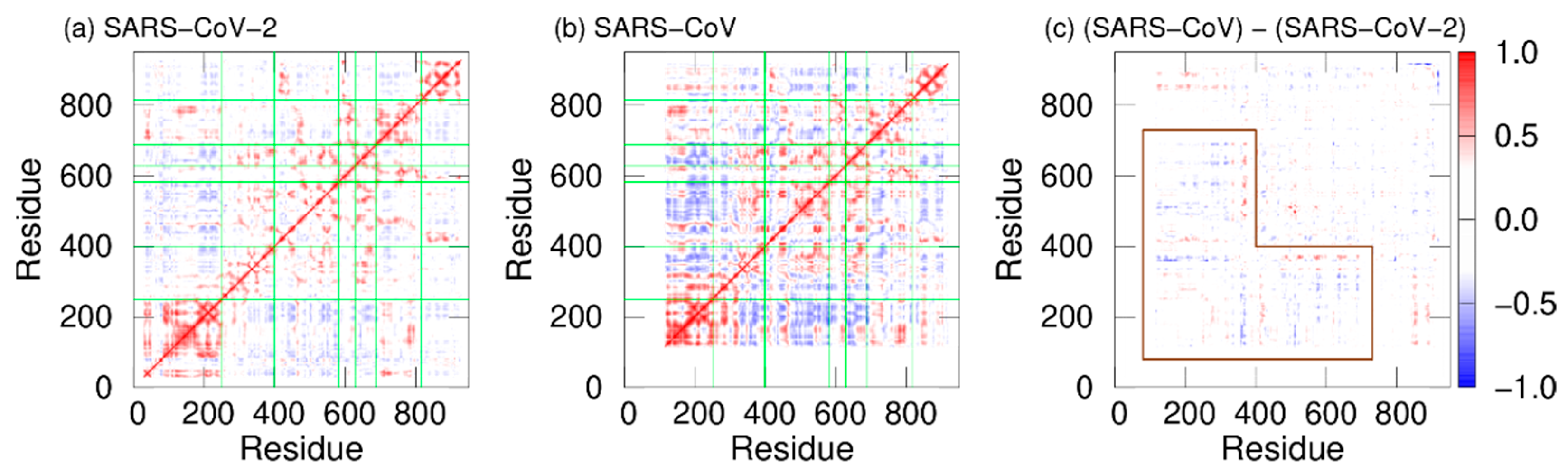
2. Difference in Dynamic Properties of SARS-CoV and SARS-CoV-2 RNA-Dependent RNA Polymerases
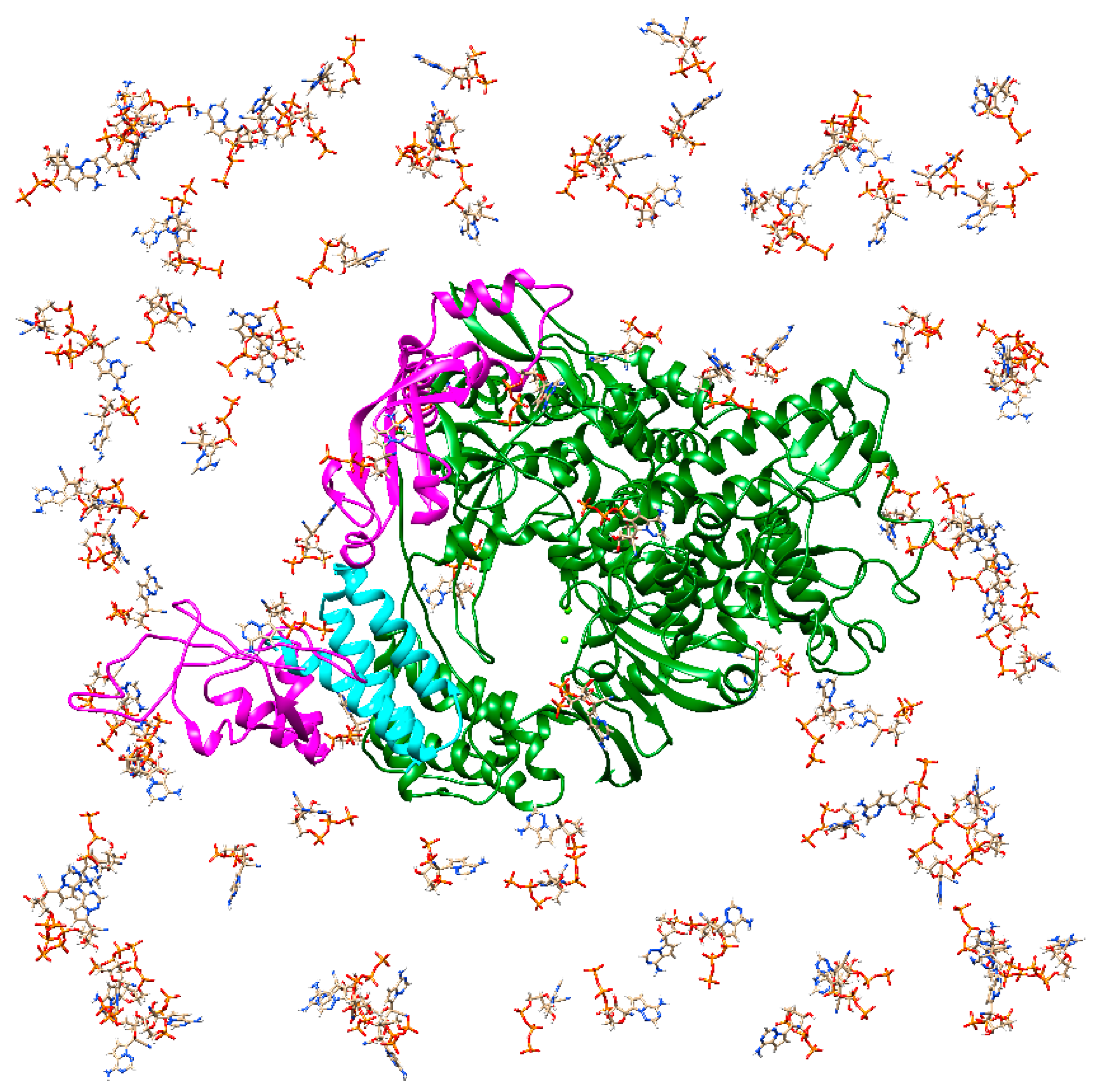
3. “Bucket Brigade” in RdRp Ligand Recognition
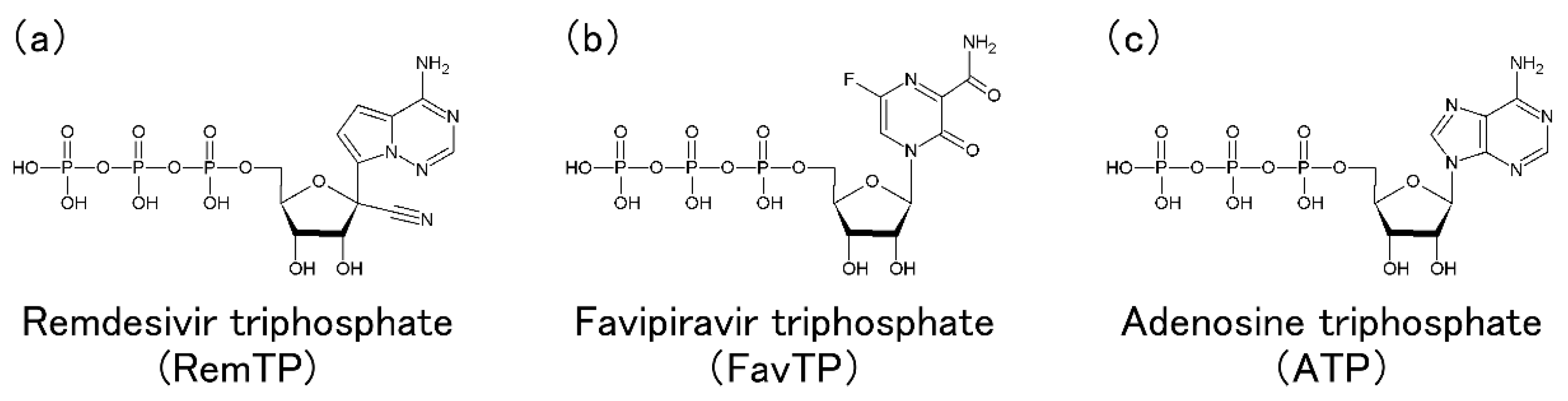
4. Molecular Simulation Studies on Inhibition Mechanisms of Nucleotide Analogs against the SARS-CoV-2 RdRp Function
4.1. SARS-CoV-2 RdRp with Remdesivir in the Triphosphate Form
4.2. SARS-CoV-2 RdRp with Other Nucleotide Analogs
4.3. Overview and Perspective of Molecular Simulations on SARS-CoV-2 RdRp with Nucleotide Analogs
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.C.; Shih, T.P.; Ko, W.C.; Tang, H.J.; Hsueh, P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- MacLaren, G.; Fisher, D.; Brodie, D. Preparing for the Most Critically Ill Patients with COVID-19: The Potential Role of Extracorporeal Membrane Oxygenation. JAMA 2020, 323, 1245–1246. [Google Scholar] [CrossRef]
- Cook, D.J.; Marshall, J.C.; Fowler, R.A. Critical Illness in Patients with COVID-19: Mounting an Effective Clinical and Research Response. JAMA 2020, 323, 1559–1560. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Sohag, A.A.M.; Hannan, M.A.; Rahman, S.; Hossain, M.; Hasan, M.; Khan, M.K.; Khatun, A.; Dash, R.; Uddin, M.J. Revisiting potential druggable targets against SARS-CoV-2 and repurposing therapeutics under preclinical study and clinical trials: A comprehensive review. Drug Dev. Res. 2020, 81, 919–941. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 323, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.-D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Anirudhan, V.; Du, R.; Cui, Q.; Rong, L. RNA-dependent RNA polymerase of SARS-CoV-2 as a therapeutic target. J. Med. Virol. 2021, 93, 300–310. [Google Scholar] [CrossRef]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. USA 2014, 111, E3900–E3909. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef]
- Zhang, W.F.; Stephen, P.; Theriault, J.F.; Wang, R.; Lin, S.X. Novel Coronavirus Polymerase and Nucleotidyl-Transferase Structures: Potential to Target New Outbreaks. J. Phys. Chem. Lett. 2020, 11, 4430–4435. [Google Scholar] [CrossRef]
- Itoh, S.G.; Tanimoto, S.; Okumura, H. Dynamic properties of SARS-CoV and SARS-CoV-2 RNA-dependent RNA polymerases studied by molecular dynamics simulations. Chem. Phys. Lett. 2021, 778, 138819. [Google Scholar] [CrossRef]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Malone, B.; Llewellyn, E.; Grasso, M.; Shelton, P.M.M.; Olinares, P.D.B.; Maruthi, K.; Eng, E.T.; Vatandaslar, H.; Chait, B.T.; et al. Structural Basis for Helicase-Polymerase Coupling in the SARS-CoV-2 Replication-Transcription Complex. Cell 2020, 182, 1560–1573. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Peng, R.; Yuan, B.; Zhao, J.; Wang, M.; Wang, X.; Wang, Q.; Sun, Y.; Fan, Z.; Qi, J.; et al. Structural and Biochemical Characterization of the nsp12-nsp7-nsp8 Core Polymerase Complex from SARS-CoV-2. Cell Rep. 2020, 31, 107774. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, S.; Itoh, S.G.; Okumura, H. “Bucket brigade” using lysine residues in RNA-dependent RNA polymerase of SARS-CoV-2. Biophys. J. 2021, 120, 3615–3627. [Google Scholar] [CrossRef] [PubMed]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef]
- Furuta, Y.; Takahashi, K.; Kuno-Maekawa, M.; Sangawa, H.; Uehara, S.; Kozaki, K.; Nomura, N.; Egawa, H.; Shiraki, K. Mechanism of action of T-705 against influenza virus. Antimicrob. Agents Chemother. 2005, 49, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Peng, R.; Yuan, B.; Wang, M.; Zhao, J.; Fu, L.; Qi, J.; Shi, Y. Structural Basis of SARS-CoV-2 Polymerase Inhibition by Favipiravir. Innovation 2021, 2, 100080. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, J.; Wang, H.; Gao, Y.; Liu, Q.; Mu, A.; Ji, W.; Yan, L.; Zhu, Y.; Zhu, C.; et al. Structural Basis for RNA Replication by the SARS-CoV-2 Polymerase. Cell 2020, 182, 417–428. [Google Scholar] [CrossRef]
- Kokic, G.; Hillen, H.S.; Tegunov, D.; Dienemann, C.; Seitz, F.; Schmitzova, J.; Farnung, L.; Siewert, A.; Hobartner, C.; Cramer, P. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021, 12, 279. [Google Scholar] [CrossRef]
- Naydenova, K.; Muir, K.W.; Wu, L.F.; Zhang, Z.; Coscia, F.; Peet, M.J.; Castro-Hartmann, P.; Qian, P.; Sader, K.; Dent, K.; et al. Structure of the SARS-CoV-2 RNA-dependent RNA polymerase in the presence of favipiravir-RTP. Proc. Natl. Acad. Sci. USA 2021, 118, e2021946118. [Google Scholar] [CrossRef]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Bixler, S.L.; Bocan, T.M.; Wells, J.; Wetzel, K.S.; Van Tongeren, S.A.; Garza, N.L.; Donnelly, G.; Cazares, L.H.; Soloveva, V.; Welch, L.; et al. Intracellular conversion and in vivo dose response of favipiravir (T-705) in rodents infected with Ebola virus. Antivir. Res. 2018, 151, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cao, L.; Li, G.; Cong, F.; Li, Y.; Sun, J.; Luo, Y.; Chen, G.; Li, G.; Wang, P.; et al. Remdesivir Metabolite GS-441524 Effectively Inhibits SARS-CoV-2 Infection in Mouse Models. J. Med. Chem. 2022, 65, 2785–2793. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 449–463. [Google Scholar] [CrossRef]
- Narayanan, N.; Nair, D.T. Vitamin B12 may inhibit RNA-dependent-RNA polymerase activity of nsp12 from the SARS-CoV-2 virus. IUBMB Life 2020, 72, 2112–2120. [Google Scholar] [CrossRef]
- Ebrahimi, K.S.; Ansari, M.; Moghaddam, M.S.H.; Ebrahimi, Z.; Salehi, Z.; Shahlaei, M.; Moradi, S. In silico investigation on the inhibitory effect of fungal secondary metabolites on RNA dependent RNA polymerase of SARS-CoV-II: A docking and molecular dynamic simulation study. Comput. Biol. Med. 2021, 135, 10. [Google Scholar]
- Kumar, S.; Sharma, P.P.; Upadhyay, C.; Kempaiah, P.; Rathi, B.; Poonam. Multi-targeting approach for nsp3, nsp9, nsp12 and nsp15 proteins of SARS-CoV-2 by Diosmin as illustrated by molecular docking and molecular dynamics simulation methodologies. Methods 2021, 195, 44–56. [Google Scholar] [CrossRef]
- Begum, F.; Srivastava, A.K.; Ray, U. Repurposing nonnucleoside antivirals against SARS-CoV2 NSP12 (RNA dependent RNA polymerase): In silico-molecular insight. Biochem. Biophys. Res. Commun. 2021, 571, 26–31. [Google Scholar] [CrossRef]
- Ghosh, D.; Ghosh Dastidar, D.; Roy, K.; Ghosh, A.; Mukhopadhyay, D.; Sikdar, N.; Biswas, N.K.; Chakrabarti, G.; Das, A. Computational prediction of the molecular mechanism of statin group of drugs against SARS-CoV-2 pathogenesis. Sci. Rep. 2022, 12, 6241. [Google Scholar] [CrossRef]
- Samy, M.N.; Gomaa, A.A.-R.; Attia, E.Z.; Ibrahim, M.A.A.; Desoukey, S.Y.; Kamel, M.S. Flavonoids of Zinnia elegans: Chemical profile and in vitro antioxidant and in silico anti-COVID-19 activities. S. Afr. J. Bot. 2022, 147, 576–585. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Yoshida, H. Construction of higher order symplectic integrators. Phys. Lett. A 1990, 150, 262–268. [Google Scholar] [CrossRef]
- Miller, T.F.; Eleftheriou, M.; Pattnaik, P.; Ndirango, A.; Newns, D.; Martyna, G.J. Symplectic quaternion scheme for biophysical molecular dynamics. J. Chem. Phys. 2002, 116, 8649–8659. [Google Scholar] [CrossRef]
- Okumura, H.; Itoh, S.G.; Okamoto, Y. Explicit symplectic integrators of molecular dynamics algorithms for rigid-body molecules in the canonical, isobaric-isothermal, and related ensembles. J. Chem. Phys. 2007, 126, 084103. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Andersen, H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef]
- Okumura, H. Temperature and pressure denaturation of chignolin: Folding and unfolding simulation by multibaric-multithermal molecular dynamics method. Proteins 2012, 80, 2397–2416. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.L.; Chen, C.J.; Okumura, H.; Hu, C.K. Transformation between α-helix and β-sheet structures of one and two polyglutamine peptides in explicit water molecules by replica-exchange molecular dynamics simulations. J. Comput. Chem. 2014, 35, 1430–1437. [Google Scholar] [CrossRef]
- Okumura, H.; Itoh, S.G. Amyloid fibril disruption by ultrasonic cavitation: Nonequilibrium molecular dynamics simulations. J. Am. Chem. Soc. 2014, 136, 10549–10552. [Google Scholar] [CrossRef]
- Okumura, H.; Itoh, S.G. Structural and fluctuational difference between two ends of Aβ amyloid fibril: MD simulations predict only one end has open conformations. Sci. Rep. 2016, 6, 38422. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- McCammon, J.A. Protein dynamics. Rep. Prog. Phys. 1984, 47, 1. [Google Scholar] [CrossRef]
- Lehmann, K.C.; Gulyaeva, A.; Zevenhoven-Dobbe, J.C.; Janssen, G.M.; Ruben, M.; Overkleeft, H.S.; van Veelen, P.A.; Samborskiy, D.V.; Kravchenko, A.A.; Leontovich, A.M.; et al. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res. 2015, 43, 8416–8434. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Giambasu, G.; et al. AMBER 2019; University of California: San Francisco, CA, USA, 2019. [Google Scholar]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Gotte, M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, R. Structural Basis of the Potential Binding Mechanism of Remdesivir to SARS-CoV-2 RNA-Dependent RNA Polymerase. J. Phys. Chem. B 2020, 124, 6955–6962. [Google Scholar] [CrossRef]
- Srivastava, M.; Mittal, L.; Kumari, A.; Asthana, S. Molecular Dynamics Simulations Reveal the Interaction Fingerprint of Remdesivir Triphosphate Pivotal in Allosteric Regulation of SARS-CoV-2 RdRp. Front. Mol. Biosci. 2021, 8, 639614. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.E.; Long, C.; La Rocco, D.; Keerthi, A.M.; Xu, D.; Yu, J. Probing remdesivir nucleotide analogue insertion to SARS-CoV-2 RNA dependent RNA polymerase in viral replication. Mol. Syst. Des. Eng. 2021, 6, 888–902. [Google Scholar] [CrossRef]
- Luo, X.; Xu, T.; Gao, X.; Zhang, L. Alternative role of motif B in template dependent polymerase inhibition. Chin. J. Chem. Phys. 2022, 35, 407–412. [Google Scholar] [CrossRef]
- Tchesnokov, E.P.; Gordon, C.J.; Woolner, E.; Kocinkova, D.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Gotte, M. Template-dependent inhibition of coronavirus RNA-dependent RNA polymerase by remdesivir reveals a second mechanism of action. J. Biol. Chem. 2020, 295, 16156–16165. [Google Scholar] [CrossRef] [PubMed]
- Olotu, F.A.; Omolabi, K.F.; Soliman, M.E.S. Piece of the puzzle: Remdesivir disassembles the multimeric SARS-CoV-2 RNA-dependent RNA polymerase complex. Cell Biochem. Biophys. 2021, 79, 175–187. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, D.; Wang, X.; Yuan, C.; Li, Y.; Jia, X.; Gao, X.; Yen, H.L.; Cheung, P.P.; Huang, X. 1′-Ribose cyano substitution allows Remdesivir to effectively inhibit nucleotide addition and proofreading during SARS-CoV-2 viral RNA replication. Phys. Chem. Chem. Phys. 2021, 23, 5852–5863. [Google Scholar] [CrossRef]
- Naseem-Khan, S.; Berger, M.B.; Leddin, E.M.; Maghsoud, Y.; Cisneros, G.A. Impact of Remdesivir Incorporation along the Primer Strand on SARS-CoV-2 RNA-Dependent RNA Polymerase. J. Chem. Inf. Model. 2022, 62, 2456–2465. [Google Scholar] [CrossRef]
- Bylehn, F.; Menendez, C.A.; Perez-Lemus, G.R.; Alvarado, W.; de Pablo, J.J. Modeling the Binding Mechanism of Remdesivir, Favilavir, and Ribavirin to SARS-CoV-2 RNA-Dependent RNA Polymerase. ACS Cent. Sci. 2021, 7, 164–174. [Google Scholar] [CrossRef]
- Aranda, J.; Wieczor, M.; Terrazas, M.; Brun-Heath, I.; Orozco, M. Mechanism of reaction of RNA-dependent RNA polymerase from SARS-CoV-2. Chem Catal. 2022, 2, 1084–1099. [Google Scholar] [CrossRef]
- Goswami, D. Comparative assessment of RNA-dependent RNA polymerase (RdRp) inhibitors under clinical trials to control SARS-CoV2 using rigorous computational workflow. RSC Adv. 2021, 11, 29015–29028. [Google Scholar] [CrossRef]
- Defant, A.; Dosi, F.; Innocenti, N.; Mancini, I. Synthesis of Nucleoside-like Molecules from a Pyrolysis Product of Cellulose and Their Computational Prediction as Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitors. Int. J. Mol. Sci. 2022, 23, 518. [Google Scholar] [CrossRef] [PubMed]
- Sonousi, A.; Mahran, H.A.; Ibrahim, I.M.; Ibrahim, M.N.; Elfiky, A.A.; Elshemey, W.M. Novel adenosine derivatives against SARS-CoV-2 RNA-dependent RNA polymerase: An in silico perspective. Pharmacol. Rep. 2021, 73, 1754–1764. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A.; Mahran, H.A.; Ibrahim, I.M.; Ibrahim, M.N.; Elshemey, W.M. Molecular dynamics simulations and MM-GBSA reveal novel guanosine derivatives against SARS-CoV-2 RNA dependent RNA polymerase. RSC Adv. 2022, 12, 2741–2750. [Google Scholar] [CrossRef] [PubMed]
- Arba, M.; Paradis, N.; Wahyudi, S.T.; Brunt, D.J.; Hausman, K.R.; Lakernick, P.M.; Singh, M.; Wu, C. Unraveling the binding mechanism of the active form of Remdesivir to RdRp of SARS-CoV-2 and designing new potential analogues: Insights from molecular dynamics simulations. Chem. Phys. Lett. 2022, 799, 139638. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Goonetilleke, E.C.; Unarta, I.C.; Huang, X. Incorporation efficiency and inhibition mechanism of 2′-substituted nucleotide analogs against SARS-CoV-2 RNA-dependent RNA polymerase. Phys. Chem. Chem. Phys. 2021, 23, 20117–20128. [Google Scholar] [CrossRef] [PubMed]
- Jockusch, S.; Tao, C.; Li, X.; Anderson, T.K.; Chien, M.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. A library of nucleotide analogues terminate RNA synthesis catalyzed by polymerases of coronaviruses that cause SARS and COVID-19. Antivir. Res. 2020, 180, 104857. [Google Scholar] [CrossRef]
- Chien, M.; Anderson, T.K.; Jockusch, S.; Tao, C.; Li, X.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. Nucleotide Analogues as Inhibitors of SARS-CoV-2 Polymerase, a Key Drug Target for COVID-19. J. Proteome Res. 2020, 19, 4690–4697. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Zhang, X.; Zheng, W.; Sun, J.; Hua, L.; Xu, L.; Chu, X.-J.; Ding, S.; Xiong, W. Development of a Simple In Vitro Assay to Identify and Evaluate Nucleotide Analogs against SARS-CoV-2 RNA-Dependent RNA Polymerase. Antimicrob. Agents Chemother. 2021, 65, e01508–e01520. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, D.; Gao, X.; Wang, X.; Zhang, L. 2′- and 3′-Ribose Modifications of Nucleotide Analogues Establish the Structural Basis to Inhibit the Viral Replication of SARS-CoV-2. J. Phys. Chem. Lett. 2022, 13, 4111–4118. [Google Scholar] [CrossRef]
- Venkataraman, S.; Prasad, B.V.L.S.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef]
- Mitsutake, A.; Sugita, Y.; Okamoto, Y. Generalized-Ensemble Algorithms for Molecular Simulations of Biopolymers. Pept. Sci. 2001, 60, 96–123. [Google Scholar] [CrossRef]
- Itoh, S.G.; Okumura, H.; Okamoto, Y. Generalized-ensemble algorithms for molecular dynamics simulations. Mol. Simul. 2007, 33, 47–56. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Itoh, S.G.; Okumura, H.; Okamoto, Y. Replica-exchange method in van der Waals radius space: Overcoming steric restrictions for biomolecules. J. Chem. Phys. 2010, 132, 134105. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.G.; Okumura, H. Coulomb replica-exchange method: Handling electrostatic attractive and repulsive forces for biomolecules. J. Comput. Chem. 2013, 34, 622–639. [Google Scholar] [CrossRef]
- Itoh, S.G.; Okumura, H. Replica-Permutation Method with the Suwa-Todo Algorithm beyond the Replica-Exchange Method. J. Chem. Theory Comput. 2013, 9, 570–581. [Google Scholar] [CrossRef]
- Itoh, S.G.; Okumura, H. Hamiltonian replica-permutation method and its applications to an alanine dipeptide and amyloid-β(29–42) peptides. J. Comput. Chem. 2013, 34, 2493–2497. [Google Scholar] [CrossRef]
- Yamauchi, M.; Okumura, H. Development of isothermal-isobaric replica-permutation method for molecular dynamics and Monte Carlo simulations and its application to reveal temperature and pressure dependence of folded, misfolded, and unfolded states of chignolin. J. Chem. Phys. 2017, 147, 184107. [Google Scholar] [CrossRef]
- Yamauchi, M.; Okumura, H. Replica sub-permutation method for molecular dynamics and monte carlo simulations. J. Comput. Chem. 2019, 40, 2694–2711. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]

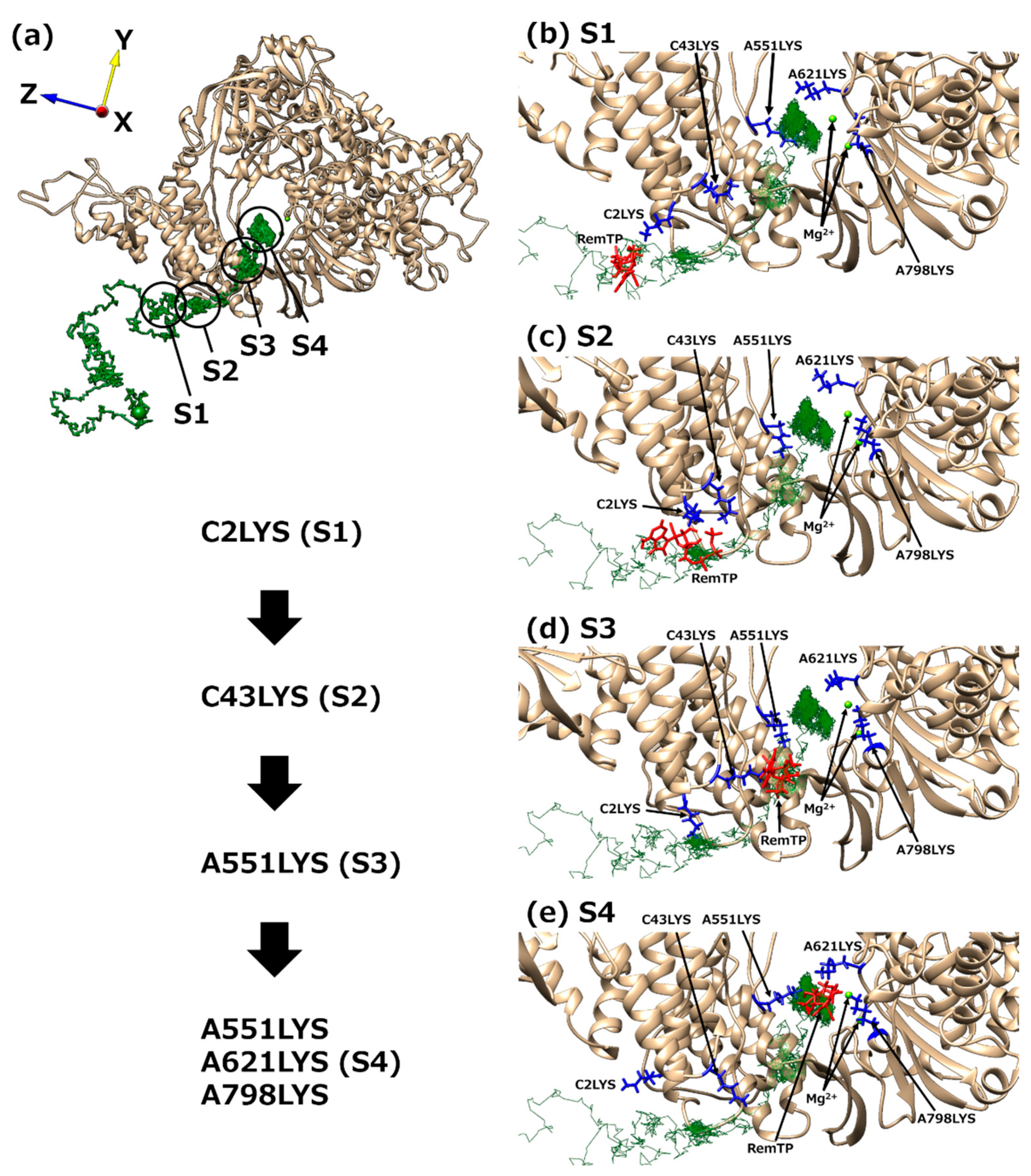
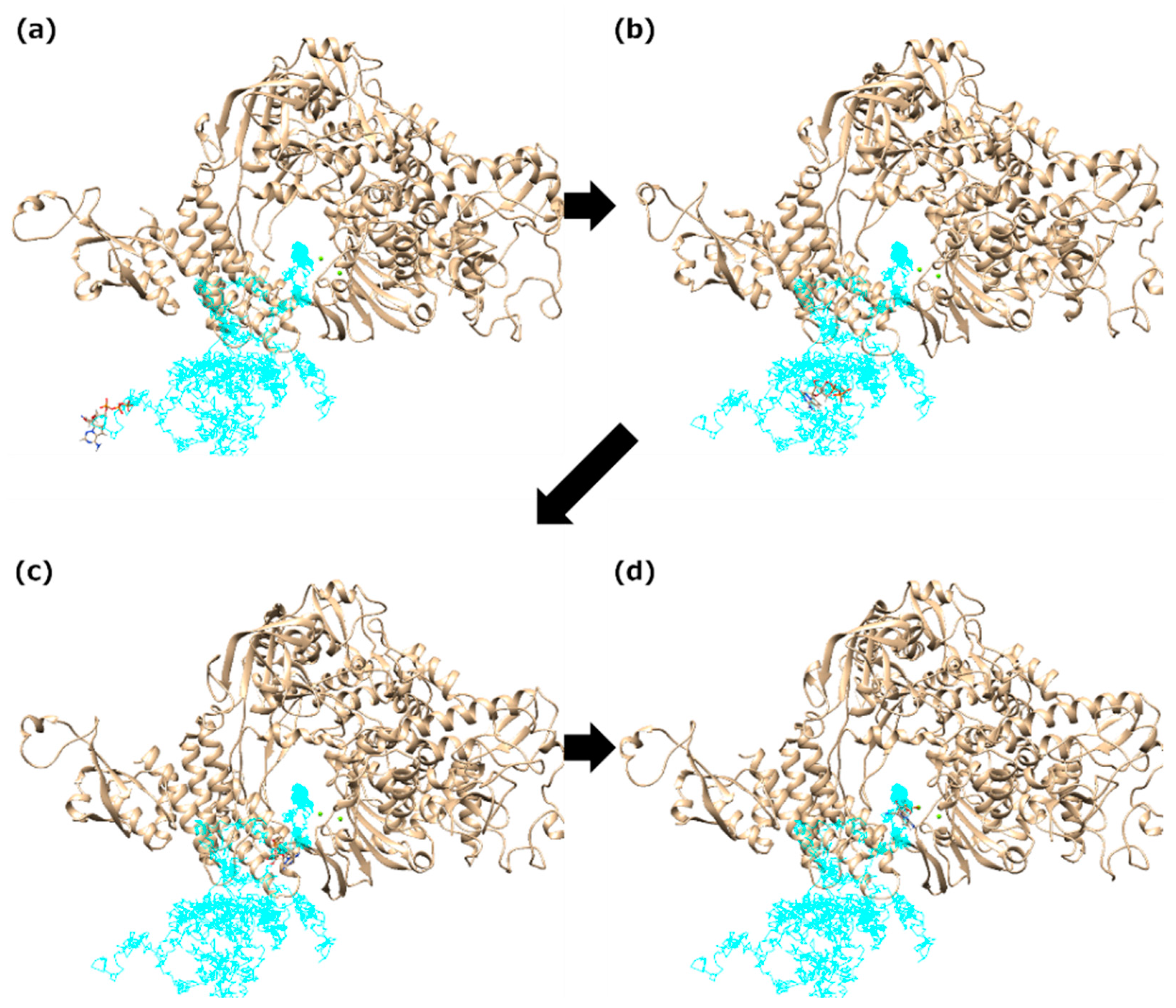

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Ligand Recognition/Total | Ligand Recognition Probability |
|---|---|---|
| RemTP | 12/50 | 0.24 ± 0.07 |
| FavTP | 9/50 | 0.18 ± 0.06 |
| ATP | 7/50 | 0.14 ± 0.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanimoto, S.; Itoh, S.G.; Okumura, H. State-of-the-Art Molecular Dynamics Simulation Studies of RNA-Dependent RNA Polymerase of SARS-CoV-2. Int. J. Mol. Sci. 2022, 23, 10358. https://doi.org/10.3390/ijms231810358
Tanimoto S, Itoh SG, Okumura H. State-of-the-Art Molecular Dynamics Simulation Studies of RNA-Dependent RNA Polymerase of SARS-CoV-2. International Journal of Molecular Sciences. 2022; 23(18):10358. https://doi.org/10.3390/ijms231810358
Chicago/Turabian StyleTanimoto, Shoichi, Satoru G. Itoh, and Hisashi Okumura. 2022. "State-of-the-Art Molecular Dynamics Simulation Studies of RNA-Dependent RNA Polymerase of SARS-CoV-2" International Journal of Molecular Sciences 23, no. 18: 10358. https://doi.org/10.3390/ijms231810358
APA StyleTanimoto, S., Itoh, S. G., & Okumura, H. (2022). State-of-the-Art Molecular Dynamics Simulation Studies of RNA-Dependent RNA Polymerase of SARS-CoV-2. International Journal of Molecular Sciences, 23(18), 10358. https://doi.org/10.3390/ijms231810358