
The Entero-Mammary Pathway and Perinatal Transmission of Gut Microbiota and SARS-CoV-2

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
2.1. Presence of SARS-CoV-2 Genomic RNA in the Mother–Neonate Pairs
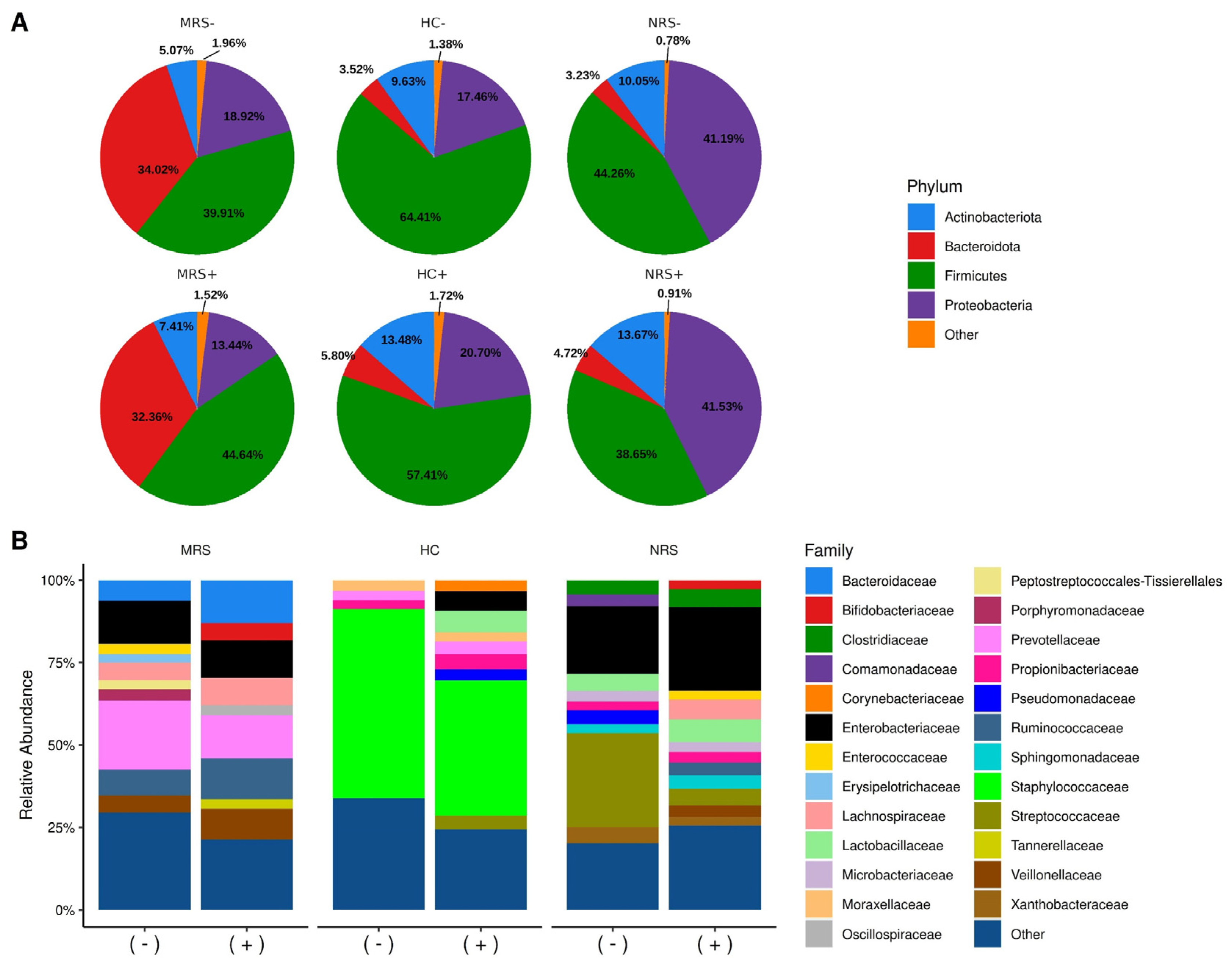
2.2. Diversity of Microbiota Taxa and SARS-CoV-2 Positivity
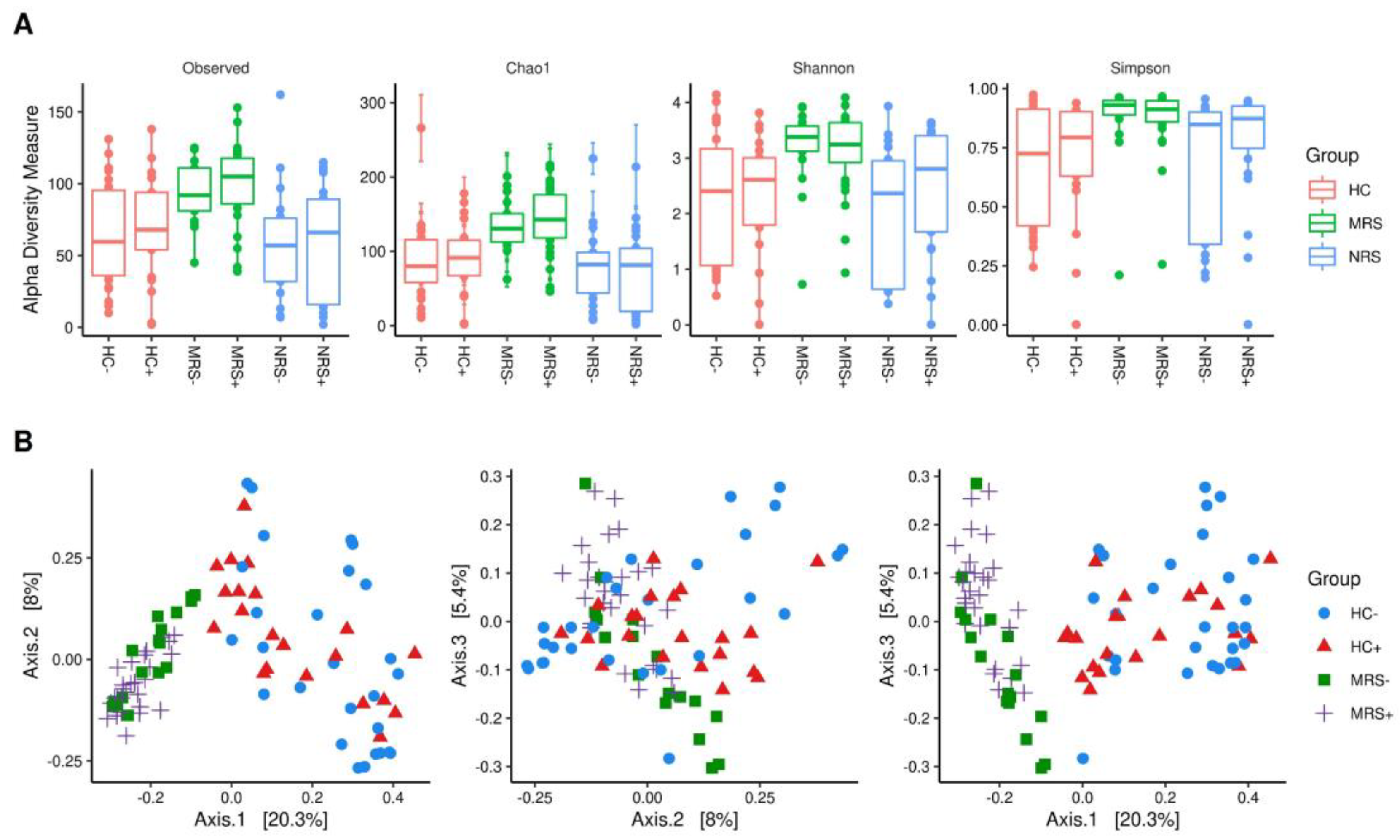
2.3. Alfa and Beta Diversity of the Bacterial Microbiota and SARS-CoV-2 Positivity
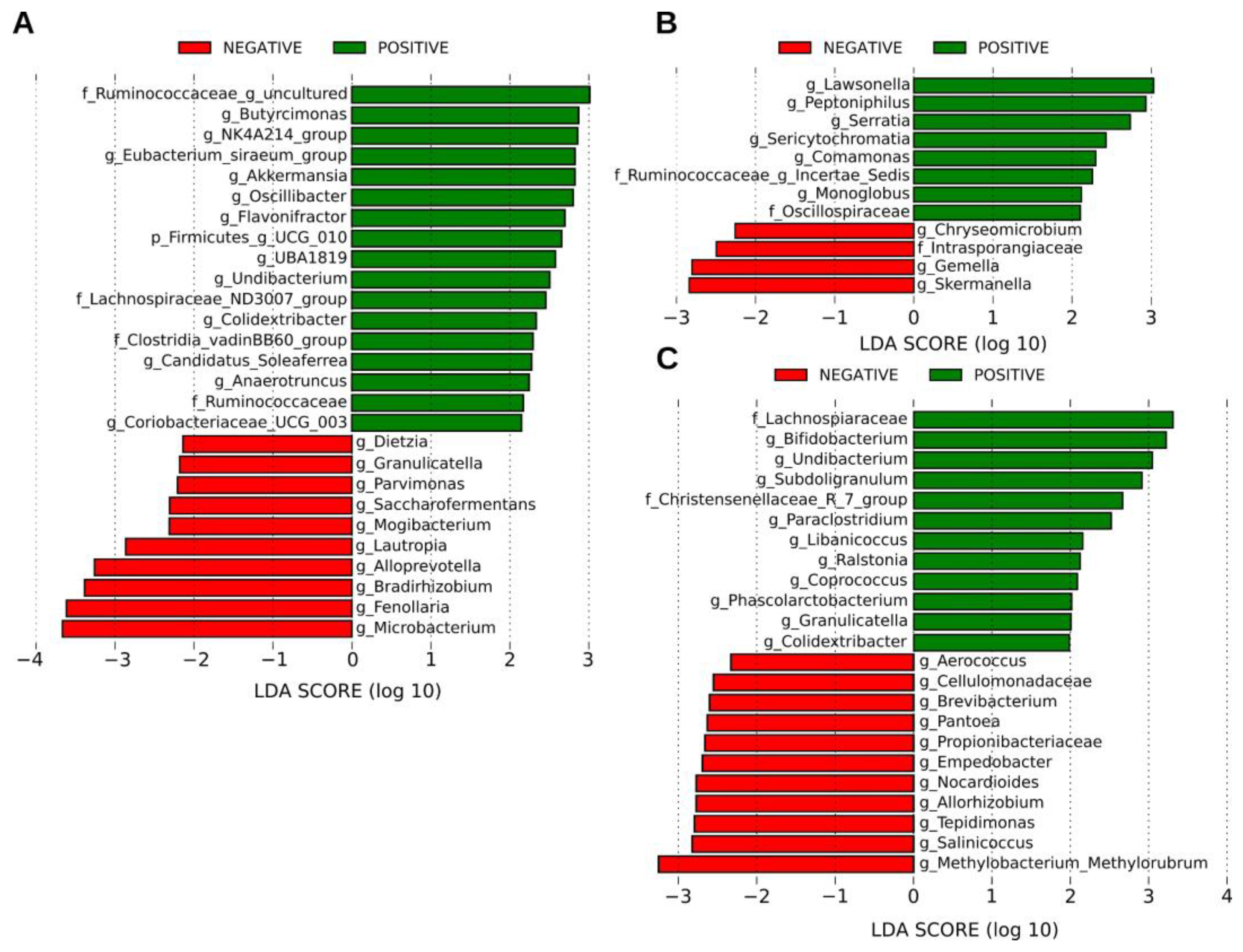
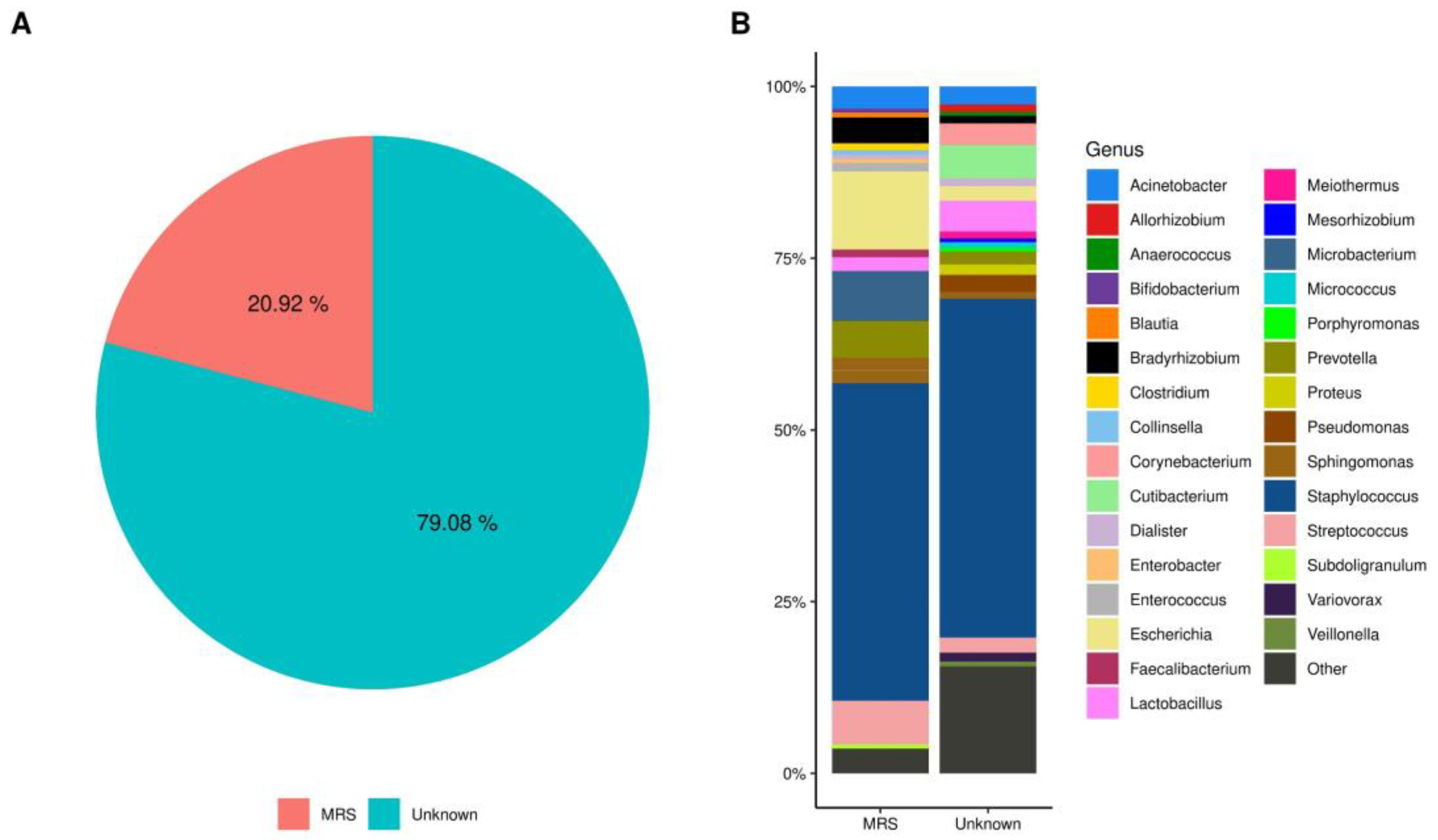
2.4. SARS-CoV-2 Positivity Is Associated with an Increased Abundance of Several Bacterial Taxa
2.5. Shared Taxa and Differential Frequency of Taxa among Samples
2.6. Origin of Bacterial Taxa Present in the Human Colostrum Samples
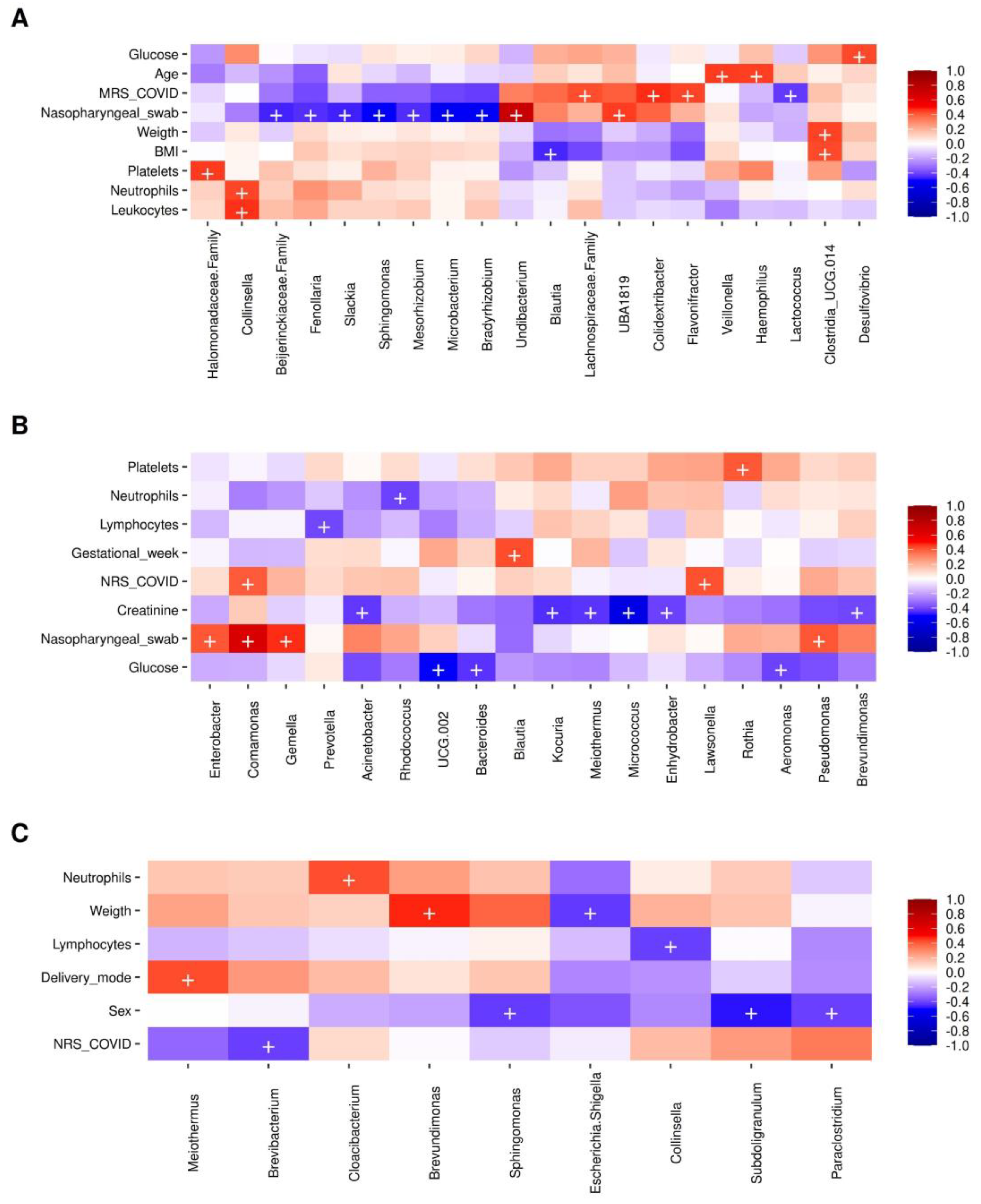
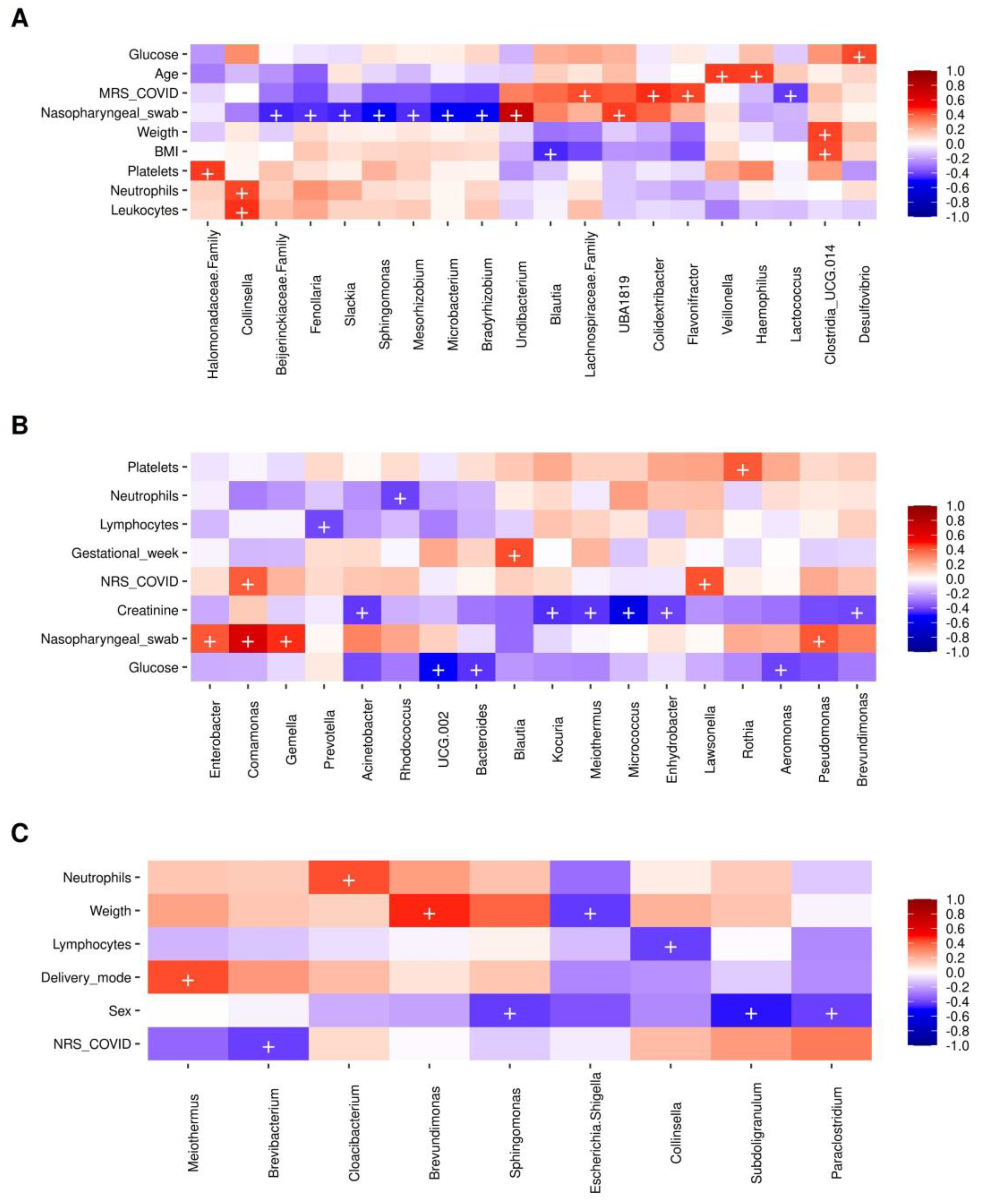
2.7. Bacterial Taxa Correlating with the Presence of SARS-CoV-2 and Metadata Collected in Mothers
3. Discussion
4. Materials and Methods
4.1. Study Type and Selection of Subjects
4.2. Sample Collection
4.3. DNA and Viral Nucleic Acid Extraction
4.4. SARS-CoV-2 Virus Genomic RNA Detection by RT-qPCR
4.5. SARS-CoV-2 Virus Genomic RNA Detection by Digital Droplet PCR (RT-ddPCR)
4.6. Preparation of the V3 16S rRNA Gene Library
4.7. High-Throughput DNA Sequencing
4.8. ASV Determination and Taxonomic Annotation
4.9. Bacterial Relative Abundance and Diversity Analyses
4.10. SourceTracker 2 and LEfSe Analyses
4.11. Statistical Analyses
4.12. Sequence Accession Numbers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramírez-Rosas, A.; Benitez-Guerrero, T.; Corona-Cervantes, K.; Vélez-Ixta, J.M.; Zavala-Torres, N.G.; Cuenca-Leija, J.; Martínez-Pichardo, S.; Landero-Montes-de-Oca, M.E.; Bastida-González, F.G.; Zárate-Segura, P.B.; et al. Study of Perinatal Transmission of SARS-CoV-2 in a Mexican Public Hospital. Int. J. Infect. Dis. 2021, 113, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Nana, M.; Nelson-Piercy, C. COVID-19 in Pregnancy. Clin. Med. 2021, 21, E446–E450. [Google Scholar] [CrossRef] [PubMed]
- Mark, E.G.; McAleese, S.; Golden, W.C.; Gilmore, M.M.; Sick-Samuels, A.; Curless, M.S.; Nogee, L.M.; Milstone, A.M.; Johnson, J. Coronavirus Disease 2019 in Pregnancy and Outcomes among Pregnant Women and Neonates: A Literature Review. Pediatric Infect. Dis. J. 2021, 40, 473–478. [Google Scholar] [CrossRef]
- Campbell, K.H.; Tornatore, J.M.; Lawrence, K.E.; Illuzzi, J.L.; Sussman, L.S.; Lipkind, H.S.; Pettker, C.M. Prevalence of SARS-CoV-2 among Patients Admitted for Childbirth in Southern Connecticut. J. Am. Med. Assoc. 2020, 323, 2520–2522. [Google Scholar] [CrossRef]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.Y.; Zhang, F.; Liu, Q.; Li, A.Y.L.; Chung, A.C.K.; Cheung, C.P.; Tso, E.Y.K.; Fung, K.S.C.; et al. Gut Microbiota Composition Reflects Disease Severity and Dysfunctional Immune Responses in Patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef]
- Xu, Y.; Li, X.; Zhu, B.; Liang, H.; Fang, C.; Gong, Y.; Guo, Q.; Sun, X.; Zhao, D.; Shen, J.; et al. Characteristics of Pediatric SARS-CoV-2 Infection and Potential Evidence for Persistent Fecal Viral Shedding. Nat. Med. 2020, 26, 502–505. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, L.; Wang, Y.; Dai, T.; Qin, Z.; Zhou, F.; Zhang, L. Alterations in Microbiota of Patients with COVID-19: Potential Mechanisms and Therapeutic Interventions. Signal Transduct. Target. Ther. 2022, 7, 143. [Google Scholar] [CrossRef]
- Hussain, I.; Cher, G.L.Y.; Abid, M.A.; Abid, M.B. Role of Gut Microbiome in COVID-19: An Insight Into Pathogenesis and Therapeutic Potential. Front. Immunol. 2021, 12, 4164. [Google Scholar] [CrossRef]
- Gómez-Torres, N.; Sánchez-García, L.; Castro, I.; Arroyo, R.; Cabañas, F.; González-Sánchez, R.; López-Azorín, M.; Moral-Pumarega, M.T.; Escuder-Vieco, D.; Cabañes-Alonso, E.; et al. Metataxonomic Analysis of Milk Samples From SARS-CoV-2-Positive and SARS-CoV-2-Negative Women. Front. Nutr. 2022, 9, 448. [Google Scholar] [CrossRef]
- Usami, K.; Niimi, K.; Matsuo, A.; Suyama, Y.; Sakai, Y.; Sato, S.; Fujihashi, K.; Kiyono, H.; Uchino, S.; Furukawa, M.; et al. The Gut Microbiota Induces Peyer’s-Patch-Dependent Secretion of Maternal IgA into Milk. Cell Rep. 2021, 36, 109655. [Google Scholar] [CrossRef]
- Mu, Q.; Swartwout, B.K.; Edwards, M.; Zhu, J.; Lee, G.; Eden, K.; Cabana-Puig, X.; McDaniel, D.K.; Mao, J.; Abdelhamid, L.; et al. Regulation of Neonatal IgA Production by the Maternal Microbiota. Proc. Natl. Acad. Sci. USA 2021, 118, e2015691118. [Google Scholar] [CrossRef]
- Gopalakrishna, K.P.; Hand, T.W. Influence of Maternal Milk on the Neonatal Intestinal Microbiome. Nutrients 2020, 12, 823. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.M.; Fernández, L.; Verhasselt, V. The Gut—Breast Axis: Programming Health for Life. Nutrients 2021, 13, 606. [Google Scholar] [CrossRef]
- Rodríguez, J.M. The Origin of Human Milk Bacteria: Is There a Bacterial Entero-Mammary Pathway during Late Pregnancy and Lactation? Adv. Nutr. 2014, 5, 779. [Google Scholar] [CrossRef]
- Corona-Cervantes, K.; García-González, I.; Villalobos-Flores, L.E.; Hernández-Quiroz, F.; Piña-Escobedo, A.; Hoyo-Vadillo, C.; Rangel-Calvillo, M.N.; García-Mena, J. Human Milk Microbiota Associated with Early Colonization of the Neonatal Gut in Mexican Newborns. PeerJ 2020, 8, e9205. [Google Scholar] [CrossRef]
- Mantziari, A.; Rautava, S. Factors Influencing the Microbial Composition of Human Milk. Semin. Perinatol. 2021, 45, 151507. [Google Scholar] [CrossRef]
- Fernández, L.; Langa, S.; Martín, V.; Maldonado, A.; Jiménez, E.; Martín, R.; Rodríguez, J.M. The Human Milk Microbiota: Origin and Potential Roles in Health and Disease. Pharmacol. Res. 2013, 69, 1–10. [Google Scholar] [CrossRef]
- Spatz, D.L.; Davanzo, R.; Müller, J.A.; Powell, R.; Rigourd, V.; Yates, A.; Geddes, D.T.; van Goudoever, J.B.; Bode, L. Promoting and Protecting Human Milk and Breastfeeding in a COVID-19 World. Front. Pediatr. 2021, 8, 633700. [Google Scholar] [CrossRef]
- Romano-Keeler, J.; Zhang, J.; Sun, J. COVID-19 and the Neonatal Microbiome: Will the Pandemic Cost Infants Their Microbes? Gut Microbes 2021, 13, 1912562. [Google Scholar] [CrossRef]
- Li, F.; Lu, H.; Li, X.; Wang, X.; Zhang, Q.; Mi, L. The Impact of COVID-19 on Intestinal Flora: A Protocol for Systematic Review and Meta Analysis. Medicine 2020, 99, e22273. [Google Scholar] [CrossRef]
- Marsland, B.J.; Trompette, A.; Gollwitzer, E.S. The Gut-Lung Axis in Respiratory Disease. Ann. Am. Thorac. Soc. 2015, 12, S150–S156. [Google Scholar] [CrossRef]
- Knight, M.; Bunch, K.; Vousden, N.; Morris, E.; Simpson, N.; Gale, C.; Obrien, P.; Quigley, M.; Brocklehurst, P.; Kurinczuk, J.J. Characteristics and Outcomes of Pregnant Women Admitted to Hospital with Confirmed SARS-CoV-2 Infection in UK: National Population Based Cohort Study. BMJ 2020, 369, m2107. [Google Scholar] [CrossRef]
- Kalamdani, P.; Kalathingal, T.; Manerkar, S.; Mondkar, J. Clinical Profile of SARS-CoV-2 Infected Neonates From a Tertiary Government Hospital in Mumbai, India. Indian Pediatr. 2020, 57, 1143. [Google Scholar] [CrossRef]
- de Bernardo, G.; Giordano, M.; Zollo, G.; Chiatto, F.; Sordino, D.; de Santis, R.; Perrone, S. The Clinical Course of SARS-CoV-2 Positive Neonates. J. Perinatol. 2020, 40, 1462. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Fett, C.; Mack, M.; ten Eyck, P.P.; Meyerholz, D.K.; Perlman, S. Sex-Based Differences in Susceptibility to SARS-CoV Infection. J. Immunol. 2017, 198, 4046. [Google Scholar] [CrossRef]
- Sridhar, S.; Nicholls, J. Pathophysiology of Infection with SARS-CoV-2—What Is Known and What Remains a Mystery. Respirology 2021, 26, 652. [Google Scholar] [CrossRef] [PubMed]
- Bwire, G.M.; Majigo, M.v.; Njiro, B.J.; Mawazo, A. Detection Profile of SARS-CoV-2 Using RT-PCR in Different Types of Clinical Specimens: A Systematic Review and Meta-Analysis. J. Med. Virol. 2021, 93, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Mazzarelli, A.; Letizia Giancola, M.; Farina, A.; Marchioni, L.; Rueca, M.; Ernesto Maria GruberID, C.; Bartolini, B.; Ascoli Bartoli, T.; Maffongelli, G.; Rosaria Capobianchi, M.; et al. 16S RRNA Gene Sequencing of Rectal Swab in Patients Affected by COVID-19. PLoS ONE 2021, 16, e0247041. [Google Scholar] [CrossRef]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020, 159, 944. [Google Scholar] [CrossRef]
- Crovetto, F.; Selma-Royo, M.; Crispi, F.; Carbonetto, B.; Pascal, R.; Larroya, M.; Casas, I.; Tortajada, M.; Escudero, N.; Muñoz-Almagro, C.; et al. Nasopharyngeal Microbiota Profiling of Pregnant Women with SARS-CoV-2 Infection. Sci. Rep. 2022, 12, 1–12. [Google Scholar] [CrossRef]
- Ricaboni, D.; Mailhe, M.; Cadoret, F.; Vitton, V.; Fournier, P.E.; Raoult, D. ‘Colidextribacter Massiliensis’ Gen. Nov., Sp. Nov., Isolated from Human Right Colon. New Microbes New Infect. 2017, 17, 27–29. [Google Scholar] [CrossRef]
- Bardanzellu, F.; Puddu, M.; Fanos, V. Breast Milk and COVID-19: From Conventional Data to “Omics” Technologies to Investigate Changes Occurring in SARS-CoV-2 Positive Mothers. Int. J. Environ. Res. Public Health 2021, 18, 5668. [Google Scholar] [CrossRef]
- Miko, E.; Csaszar, A.; Bodis, J.; Kovacs, K. The Maternal-Fetal Gut Microbiota Axis: Physiological Changes, Dietary Influence, and Modulation Possibilities. Life 2022, 12, 424. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Dhakan, D.B.; Maji, A.; Saxena, R.; Vishnu Prasoodanan, P.K.; Mahajan, S.; Pulikkan, J.; Kurian, J.; Gomez, A.M.; Scaria, J.; et al. Association of Flavonifractor Plautii, a Flavonoid-Degrading Bacterium, with the Gut Microbiome of Colorectal Cancer Patients in India. Msystems 2019, 4, e00438-19. [Google Scholar] [CrossRef]
- Miquel, S.; Martín, R.; Rossi, O.; Bermúdez-Humarán, L.G.; Chatel, J.M.; Sokol, H.; Thomas, M.; Wells, J.M.; Langella, P. Faecalibacterium Prausnitzii and Human Intestinal Health. Curr. Opin. Microbiol. 2013, 16, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Hasain, Z.; Mokhtar, N.M.; Kamaruddin, N.A.; Mohamed Ismail, N.A.; Razalli, N.H.; Gnanou, J.V.; Raja Ali, R.A. Gut Microbiota and Gestational Diabetes Mellitus: A Review of Host-Gut Microbiota Interactions and Their Therapeutic Potential. Front. Cell Infect. Microbiol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Hu, Q.; Niu, Y.; Yang, Y.; Mao, Q.; Lu, Y.; Ran, H.; Zhang, H.; Li, X.; Gu, H.; Su, Q. Polydextrose Alleviates Adipose Tissue Inflammation and Modulates the Gut Microbiota in High-Fat Diet-Fed Mice. Front. Pharmacol. 2022, 12, 4032. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Radjabzadeh, D.; Chen, L.; Kurilshikov, A.; Kavousi, M.; Ahmadizar, F.; Ikram, M.A.; Uitterlinden, A.G.; Zhernakova, A.; Fu, J.; et al. Association of Insulin Resistance and Type 2 Diabetes with Gut Microbial Diversity: A Microbiome-Wide Analysis from Population Studies. JAMA Netw. Open 2021, 4, e2118811. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, W.; Huang, X.; Feng, Y.; Lu, J.; Gao, F. Intestinal Flora Differences between Patients with Ulcerative Colitis of Different Ethnic Groups in China. Medicine 2021, 100, e26932. [Google Scholar] [CrossRef]
- Zhang, X.; Coker, O.O.; Chu, E.S.H.; Fu, K.; Lau, H.C.H.; Wang, Y.X.; Chan, A.W.H.; Wei, H.; Yang, X.; Sung, J.J.Y.; et al. Dietary Cholesterol Drives Fatty Liver-Associated Liver Cancer by Modulating Gut Microbiota and Metabolites. Gut 2021, 70, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Sencio, V.; Machelart, A.; Robil, C.; Benech, N.; Hoffmann, E.; Galbert, C.; Deryuter, L.; Heumel, S.; Hantute-Ghesquier, A.; Flourens, A.; et al. Alteration of the Gut Microbiota Following SARS-CoV-2 Infection Correlates with Disease Severity in Hamsters. Gut Microbes 2022, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Guo, J.; Song, Y.; Ariff, A.; O’Sullivan, M.; Hales, B.; Mullins, B.J.; Zhang, G. Dysfunctional Gut Microbiome Networks in Childhood IgE-Mediated Food Allergy. Int. J. Mol. Sci. 2021, 22, 2079. [Google Scholar] [CrossRef]
- Ferrocino, I.; Ponzo, V.; Gambino, R.; Zarovska, A.; Leone, F.; Monzeglio, C.; Goitre, I.; Rosato, R.; Romano, A.; Grassi, G.; et al. Changes in the Gut Microbiota Composition during Pregnancy in Patients with Gestational Diabetes Mellitus (GDM). Sci. Rep. 2018, 8, 12216. [Google Scholar] [CrossRef]
- Farsi, Y.; Tahvildari, A.; Arbabi, M.; Vazife, F.; Sechi, L.A.; Shahidi Bonjar, A.H.; Jamshidi, P.; Nasiri, M.J.; Mirsaeidi, M. Diagnostic, Prognostic, and Therapeutic Roles of Gut Microbiota in COVID-19: A Comprehensive Systematic Review. Front. Cell Infect. Microbiol. 2022, 12, 182. [Google Scholar] [CrossRef]
- Uehara, O.; Abiko, Y.; Nagasawa, T.; Morikawa, T.; Hiraki, D.; Harada, F.; Kawano, Y.; Toraya, S.; Matsuoka, H.; Paudel, D.; et al. Alterations in the Oral Microbiome of Individuals with a Healthy Oral Environment Following COVID-19 Vaccination. BMC Oral Health 2022, 22, 50. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Jiang, H.; Chen, Y.; Gu, S.; Xia, J.; Zhang, H.; Lu, Y.; Yan, R.; Li, L. The Faecal Metabolome in COVID-19 Patients Is Altered and Associated with Clinical Features and Gut Microbes. Anal. Chim. Acta 2021, 1152, 338267. [Google Scholar] [CrossRef]
- Sakamoto, M.; Tanaka, Y.; Benno, Y.; Ohkuma, M. Butyricimonas Faecihominis Sp. Nov. and Butyricimonas Paravirosa Sp. Nov., Isolated from Human Faeces, and Emended Description of the Genus Butyricimonas. Int. J. Syst. Evol. Microbiol. 2014, 64, 2992–2997. [Google Scholar] [CrossRef]
- Ren, Z.; Wang, H.; Cui, G.; Lu, H.; Wang, L.; Luo, H.; Chen, X.; Ren, H.; Sun, R.; Liu, W.; et al. Alterations in the Human Oral and Gut Microbiomes and Lipidomics in COVID-19. Gut 2021, 70, 1253–1265. [Google Scholar] [CrossRef]
- Moossavi, S.; Sepehri, S.; Robertson, B.; Bode, L.; Goruk, S.; Field, C.J.; Lix, L.M.; de Souza, R.J.; Becker, A.B.; Mandhane, P.J.; et al. Composition and Variation of the Human Milk Microbiota Are Influenced by Maternal and Early-Life Factors. Cell Host Microbe 2019, 25, 324–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermansson, H.; Kumar, H.; Collado, M.C.; Salminen, S.; Isolauri, E.; Rautava, S. Breast Milk Microbiota Is Shaped by Mode of Delivery and Intrapartum Antibiotic Exposure. Front. Nutr. 2019, 6, 4. [Google Scholar] [CrossRef]
- Pannaraj, P.S.; Li, F.; Cerini, C.; Bender, J.M.; Yang, S.; Rollie, A.; Adisetiyo, H.; Zabih, S.; Lincez, P.J.; Bittinger, K.; et al. Association Between Breast Milk Bacterial Communities and Establishment and Development of the Infant Gut Microbiome. JAMA Pediatr. 2017, 171, 647–654. [Google Scholar] [CrossRef]
- Tuominen, H.; Rautava, S.; Collado, M.C.; Syrjänen, S.; Rautava, J. HPV Infection and Bacterial Microbiota in Breast Milk and Infant Oral Mucosa. PLoS ONE 2018, 13, e0207016. [Google Scholar] [CrossRef]
- Davé, V.; Street, K.; Francis, S.; Bradman, A.; Riley, L.; Eskenazi, B.; Holland, N. Bacterial Microbiome of Breast Milk and Child Saliva from Low-Income Mexican-American Women and Children. Pediatr. Res. 2016, 79, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Lineamiento Estandarizado Para La Vigilancia Epidemiológica y Por Laboratorio de La Enfermedad Respiratoria Viral | Secretaría de Salud | Gobierno | Gob.Mx. Available online: https://www.gob.mx/salud/documentos/lineamiento-estandarizado-para-la-vigilancia-epidemiologica-y-por-laboratorio-de-la-enfermedad-respiratoria-viral (accessed on 5 July 2022).
- Secretaria de Salud Gobierno de Mexico. Lineamiento Estandarizado para la Vigilancia Epidemiológica y por Laboratorio de la Enfermedad Respiratoria Viral. 2022. Available online: https://www.gob.mx/salud/documentos/lineamiento-estandarizado-para-la-vigilancia-epidemiologica-y-por-laboratorio-de-la-enfermedad-respiratoria-viral (accessed on 5 July 2022).
- World Health Organization. Guidelines for the Collection of Clinical Specimens during Field Investigation of Outbreaks; WHO WHO/CDS/CSR/EDC/2000.4; World Health Organization: Geneva, Switzerland, 2000. [Google Scholar]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.W.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 Novel Coronavirus (2019-NCoV) by Real-Time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef]
- Murugesan, S.; Ulloa-Martínez, M.; Martínez-Rojano, H.; Galván-Rodríguez, F.M.; Miranda-Brito, C.; Romano, M.C.; Piña-Escobedo, A.; Pizano-Zárate, M.L.; Hoyo-Vadillo, C.; García-Mena, J. Study of the Diversity and Short-Chain Fatty Acids Production by the Bacterial Community in Overweight and Obese Mexican Children. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Kaehler, B.D.; Bokulich, N.A.; McDonald, D.; Knight, R.; Caporaso, J.G.; Huttley, G.A. Species Abundance Information Improves Sequence Taxonomy Classification Accuracy. Nat. Commun. 2019, 10, 4643. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- RStudio Team. RStudio: Integrated Development Environment for R; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Bisanz, J.E. Tutorial: Integrating QIIME2 and R for Data Visualization and Analysis Using Qiime2r (v0.99.6). 2018. Available online: https://github.com/jbisanz/qiime2R (accessed on 5 July 2022).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; D’, L.; Mcgowan, A.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Wickham, H.; François, R.; Henry, L.; Müller, K. Dplyr: A Grammar of Data Manipulation dplyr. 1.09. 2022. Available online: https://dplyr.tidyverse.org/ (accessed on 5 July 2022).
- Barnett, D.J.M.; Arts, I.C.W.; Penders, J. MicroViz: An R Package for Microbiome Data Visualization and Statistics. J. Open Source Softw. 2021, 6, 3201. [Google Scholar] [CrossRef]
- Kassambara, A. Ggpubr: “ggplot2” Based Publication Ready Plots ggplot2. 3.3.6. 2020. Available online: https://ggplot2.tidyverse.org/ (accessed on 5 July 2022).
- Wickham, H.; Seidel, D. Scales: Scale Functions for Visualization scales. 1.2.0. 2022. Available online: https://CRAN.R-project.org/package=scales (accessed on 5 July 2022).
- Auguie, B. GridExtra: Miscellaneous Functions for “Grid” Graphics gridExtra. 2.3. 2017. Available online: https://CRAN.R-project.org/package=gridExtra (accessed on 5 July 2022).
- Yu, G. Ggplotify: Convert Plot to “Grob” or “Ggplot” Object ggplotify. 0.1.0. 2021. Available online: https://CRAN.R-project.org/package=ggplotify (accessed on 5 July 2022).
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Vegan: Community Ecology Package vegan. 2.6-2. 2022. Available online: https://CRAN.R-project.org/package=vegan (accessed on 5 July 2022).
- Lahti, L.; Shetty, S. Microbiome R Package microbiome. 1.18.0. 2019. Available online: https://bioconductor.org/packages/release/bioc/html/microbiome.html (accessed on 5 July 2022).
- Larsson, J. Eulerr: Area-Proportional Euler and Venn Diagrams with Ellipses eulerr. 6.1.1. 2021. Available online: https://CRAN.R-project.org/package=eulerr (accessed on 5 July 2022).
- Knights, D.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Mozer, M.C.; Collman, R.G.; Bushman, F.D.; Knight, R.; Kelley, S.T. Bayesian Community-Wide Culture-Independent Microbial Source Tracking. Nat. Methods 2011, 8, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, 1–18. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Positive | Negative | p-Value | ||
|---|---|---|---|---|---|
| Number of subjects (n = 50) | 33 (66%) | 17 (34%) | nd | ||
| Age (years) | 25.48 (±5.19) | 25.47 (±6.10) | 0.993 | ||
| Age range | 18 to 38 | 16 to 36 | nd | ||
| Gestational age (weeks) | 38.69 (±1.85) | 38.95 (±1.39) | 0.571 | ||
| Weeks range | 34 to 41 | 36.6 to 41.2 | nd | ||
| Anthropometry | Height (m) | 1.57 (±0.05) | 1.55 (±0.05) | 0.499 | |
| Weight (kg) | 74.25 (±13.65) | 76.17 (±12.61) | 0.624 | ||
| Normal (BMI 18.5–24.9) | 7 (21.21%) | 0 (0.0%) | nd | ||
| Overweight (BMI 25.0–29.9) | 10 (30.30%) | 9 (52.94%) | nd | ||
| Obesity (BMI > 30.0) | 16 (48.48%) | 8 (47.06%) | nd | ||
| COVID-19 Symptoms | Symptoms | 5 (15.15%) | 1 (5.88%) | nd | |
| Asymptomatic | 28 (84.84%) | 16 (94.11%) | nd | ||
| HC samples (n = 50) | 22 (44.0%) | 28 (56.0%) | nd | ||
| SARS-CoV-2 nasopharyngeal swab * | Positive | 9 (27.3%) | 0 (0.0%) | nd | |
| Negative | 24 (72.7%) | 17 (100.0%) | nd | ||
| Blood test | Reference range | ||||
| Leukocytes (×109/L) | 4.5–10 | 9.17 (±3.31) | 8.3 (±2.02) | 0.239 | |
| Neutrophils (×109/L) | 1.8–8.0 | 5.97 (±2.69) | 5.60 (±1.89) | 0.576 | |
| Neutrophils (%) | 43.0–65.0 | 69.29 (±13.86) | 66.81 (±14.25) | 0.563 | |
| Lymphocytes (×109/L) | 1.1–3.2 | 2.26 (±2.60) | 1.7 (±0.59) | 0.247 | |
| Lymphocytes (%) | 21.0–48.0 | 23.69 (±20.61) | 20.49 (±5.86) | 0.520 | |
| Platelet count (×109/L) | 150–450 | 218.68 (±65.89) | 225.17 (±49.45) | 0.700 | |
| Fasting glucose (mg/dL) | 74–06 | 82.34 (±11.79) | 83.35 (±12.32) | 0.783 | |
| Creatinine (mg/dL) | 0.5–0.9 | 0.65 (±0.18) | 0.66 (±0.11) | 0.808 | |
| Risk factors | Alcoholism | 0 | 1 (0.17%) | nd | |
| Smoking | 0 | 1 (0.17%) | nd | ||
| Parity | Total | 63 | 40 | nd | |
| Current parities | 33 | 17 | nd | ||
| Vaginal | 23 (69.69%) | 16 (94.11%) | nd | ||
| Cesarean | 10 (30.30%) | 1 (5.88%) | nd | ||
| Gravity | Uniparous | 15 (45.45%) | 6 (35.30%) | nd | |
| Multiparous | 18 (54.54%) | 11 (64.70%) | nd | ||
| Previous parities | 30 | 23 | nd | ||
| Vaginal | 18 (72.0%) | 8 (34.8%) | nd | ||
| Cesarean | 7 (23.3%) | 8 (34.8%) | nd | ||
| Abortions | 5 (16.7%) | 7 (30.4%) | nd | ||
| Socioeconomic data | |||||
| Educational level | Primary school (6 years) | 6 (18.18%) | 3 (17.64%) | nd | |
| Secondary school (3 years) | 12 (36.36%) | 7 (41.17%) | nd | ||
| High school (3 years) | 12 (36.36%) | 6 (35.29%) | nd | ||
| University (4–5 years) | 2 (6.06%) | 1 (5.88%) | nd | ||
| None | 1 (3.03%) | 0 (0.0%) | nd | ||
| Marital status | Free union | 24 (48.97%) | 9 (52.94%) | nd | |
| Married | 5 (15.15%) | 1 (5.88%) | nd | ||
| Single | 4 (12.12%) | 7 (41.7%) | nd | ||
| Main activity | Housewife | 28 (84.84%) | 14 (82.35%) | nd | |
| General employees | 5 (15.15%) | 3 (17.64%) | nd | ||
| Variable | Positive | Negative | p-Value | |
|---|---|---|---|---|
| Number of subjects (n = 50) | 25 (50%) | 25 (50%) | nd | |
| Sex | M | 17 (68%) | 17 (68%) | nd |
| F | 8 (32%) | 8 (32%) | nd | |
| SARS-CoV-2 nasopharyngeal swab * | Positive | 5 (20.0%, M) | 0.0 (0.0%) | nd |
| Negative | 20 (80.0%, F) | 25 (100.0%) | nd | |
| Qualification status | Reference range | |||
| APGAR (1 to 10) | 6/7 | 1 (4.0%) | 0 (0.0%) | nd |
| 7/8 | 1 (4.0%) | 0 (0.0%) | nd | |
| 7/9 | 1 (4.0%) | 2 (8.0%) | nd | |
| 8/9 | 19 (76%) | 19 (76%) | nd | |
| 9/9 | 3 (12%) | 4 (16%) | nd | |
| Silverman Andersen | 0 | 20 (80%) | 19 (76%) | nd |
| 0/1 | 4 (16%) | 6 (24%) | nd | |
| 0/3 | 1 (4.0%) | 0 (0.0%) | nd | |
| Capurro (weeks) | Average | 38.67 (±1.57) | 39.00 (±1.36) | 0.430 |
| Preterm (22–36) | 2 (8.0%) | 1 (4.0%) | nd | |
| Term (37–42) | 23 (92%) | 24 (96%) | nd | |
| Somatometry | Reference range | |||
| Macrosomia (g) | >4000 | 0 (0.0%) | 0 (0.0%) | nd |
| Proper weight (g) | (2500 to 4000) | 24 (96%) | 24 (96%) | nd |
| Low weight (g) | ≤2500 | 1 (4.0%) | 1 (4.0%) | nd |
| IUGR | 1 (4.0%) | 0 (0.0%) | nd | |
| Weight (kg) | 2.88(±0.43) | 2.79(±0.41) | 0.508 | |
| Weight range (kg) | 1.66–3.68 | 2.12–3.257 | nd | |
| Size (cm) | 49.36 (±1.77) | 49.66 (±2.68) | 0.644 | |
| Size range (cm) | 45–52 | 40–53 | nd | |
| Cephalic perimeter (cm) | 33 (±1.36) | 33 (±1.32) | 0.526 | |
| Abdominal perimeter (cm) | 31 (±2.04) | 31 (±2.19) | 0.812 | |
| Perinatal asphyxiation | 1 (4.0%) | 0 (0.0%) | nd | |
| Breathing difficulty | 4 (16%) | 0 (0.0%) | nd | |
| Infection | 1 (4.0%) | 0 (0.0%) | nd |
| MRS (n = 45) | HC (n = 50) | NRS (n = 49) | |
|---|---|---|---|
| Parameter before trimming | |||
| Total forward reads | 2,451,400.00 | 1,617,526.00 | 2,188,670.00 |
| Forward reads mean | 54,475.55 | 32,350.52 | 44,666.00 |
| Min-Max forward reads | 9241.00–183,829.00 | 6557.00–231,169.00 | 2723.00–240,577.00 |
| Sequence length (median) | 193 | 198 | 198 |
| Samples with <10,000 reads | 1 | 10 | 19 |
| Parameter after trimming | |||
| QS (median) | 32 | 32 | 30 |
| Percentage of identity (97%) | |||
| Total ASV counts | 2052 | 2028 | 1560 |
| Identified ASVs | 346 | 475 | 371 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juárez-Castelán, C.J.; Vélez-Ixta, J.M.; Corona-Cervantes, K.; Piña-Escobedo, A.; Cruz-Narváez, Y.; Hinojosa-Velasco, A.; Landero-Montes-de-Oca, M.E.; Davila-Gonzalez, E.; González-del-Olmo, E.; Bastida-Gonzalez, F.; et al. The Entero-Mammary Pathway and Perinatal Transmission of Gut Microbiota and SARS-CoV-2. Int. J. Mol. Sci. 2022, 23, 10306. https://doi.org/10.3390/ijms231810306
Juárez-Castelán CJ, Vélez-Ixta JM, Corona-Cervantes K, Piña-Escobedo A, Cruz-Narváez Y, Hinojosa-Velasco A, Landero-Montes-de-Oca ME, Davila-Gonzalez E, González-del-Olmo E, Bastida-Gonzalez F, et al. The Entero-Mammary Pathway and Perinatal Transmission of Gut Microbiota and SARS-CoV-2. International Journal of Molecular Sciences. 2022; 23(18):10306. https://doi.org/10.3390/ijms231810306
Chicago/Turabian StyleJuárez-Castelán, Carmen Josefina, Juan Manuel Vélez-Ixta, Karina Corona-Cervantes, Alberto Piña-Escobedo, Yair Cruz-Narváez, Alejandro Hinojosa-Velasco, María Esther Landero-Montes-de-Oca, Eduardo Davila-Gonzalez, Eduardo González-del-Olmo, Fernando Bastida-Gonzalez, and et al. 2022. "The Entero-Mammary Pathway and Perinatal Transmission of Gut Microbiota and SARS-CoV-2" International Journal of Molecular Sciences 23, no. 18: 10306. https://doi.org/10.3390/ijms231810306
APA StyleJuárez-Castelán, C. J., Vélez-Ixta, J. M., Corona-Cervantes, K., Piña-Escobedo, A., Cruz-Narváez, Y., Hinojosa-Velasco, A., Landero-Montes-de-Oca, M. E., Davila-Gonzalez, E., González-del-Olmo, E., Bastida-Gonzalez, F., Zárate-Segura, P. B., & García-Mena, J. (2022). The Entero-Mammary Pathway and Perinatal Transmission of Gut Microbiota and SARS-CoV-2. International Journal of Molecular Sciences, 23(18), 10306. https://doi.org/10.3390/ijms231810306