

Novel Splicing Mutation in MTM1 Leading to Two Abnormal Transcripts Causes Severe Myotubular Myopathy
 , , ,
, , ,
Abstract
1. Introduction
2. Results
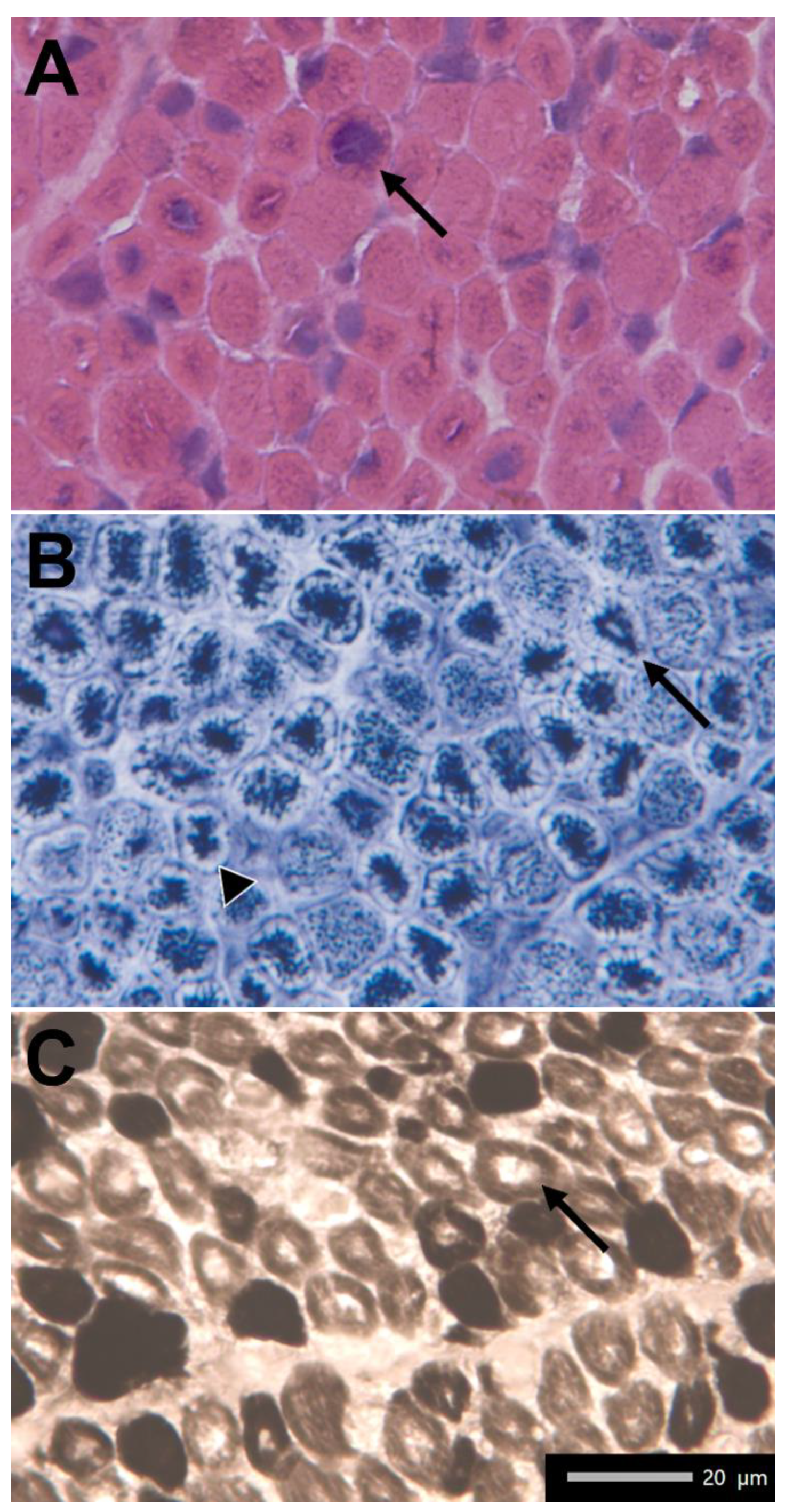
2.1. Clinical Findings
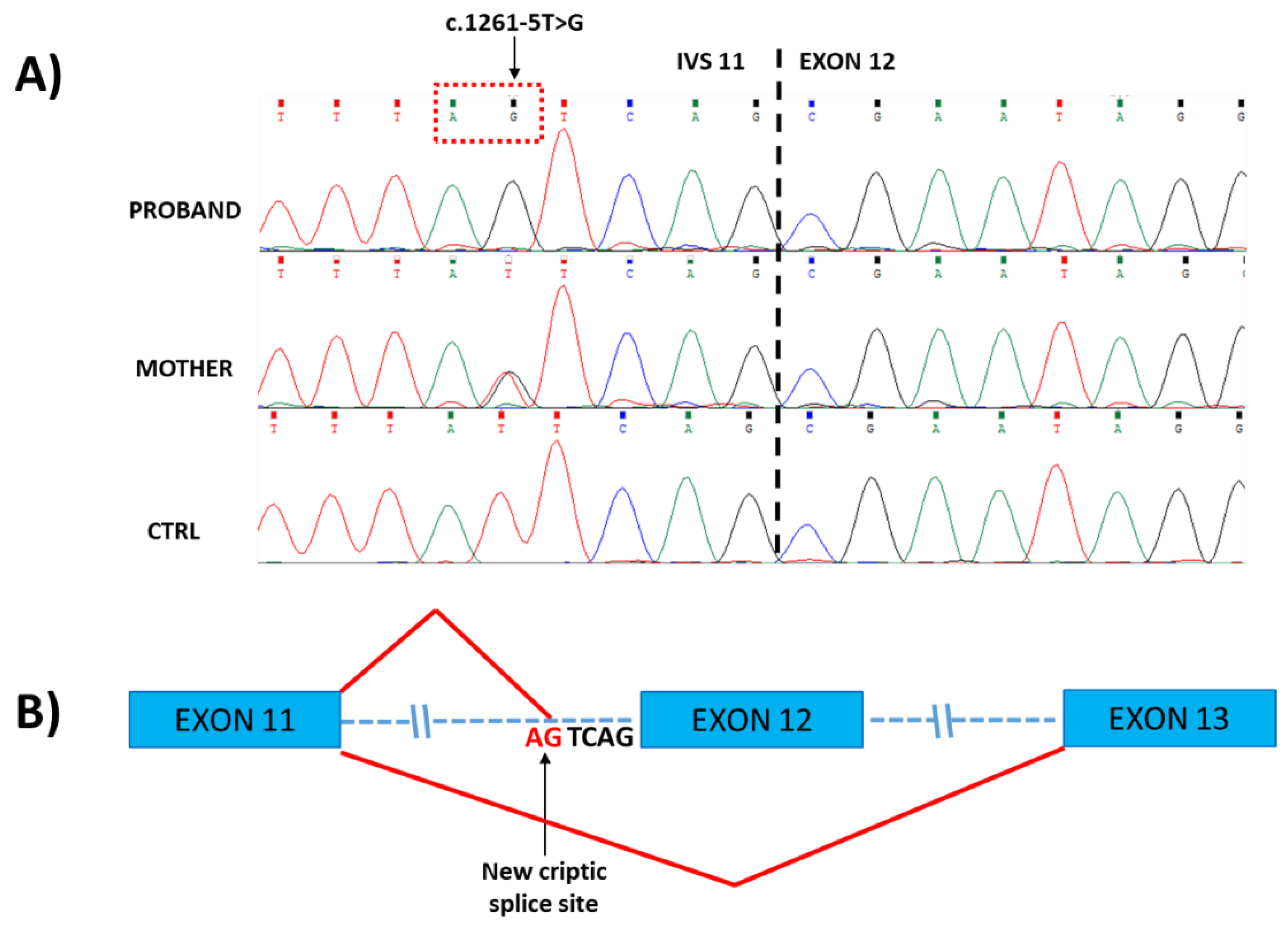
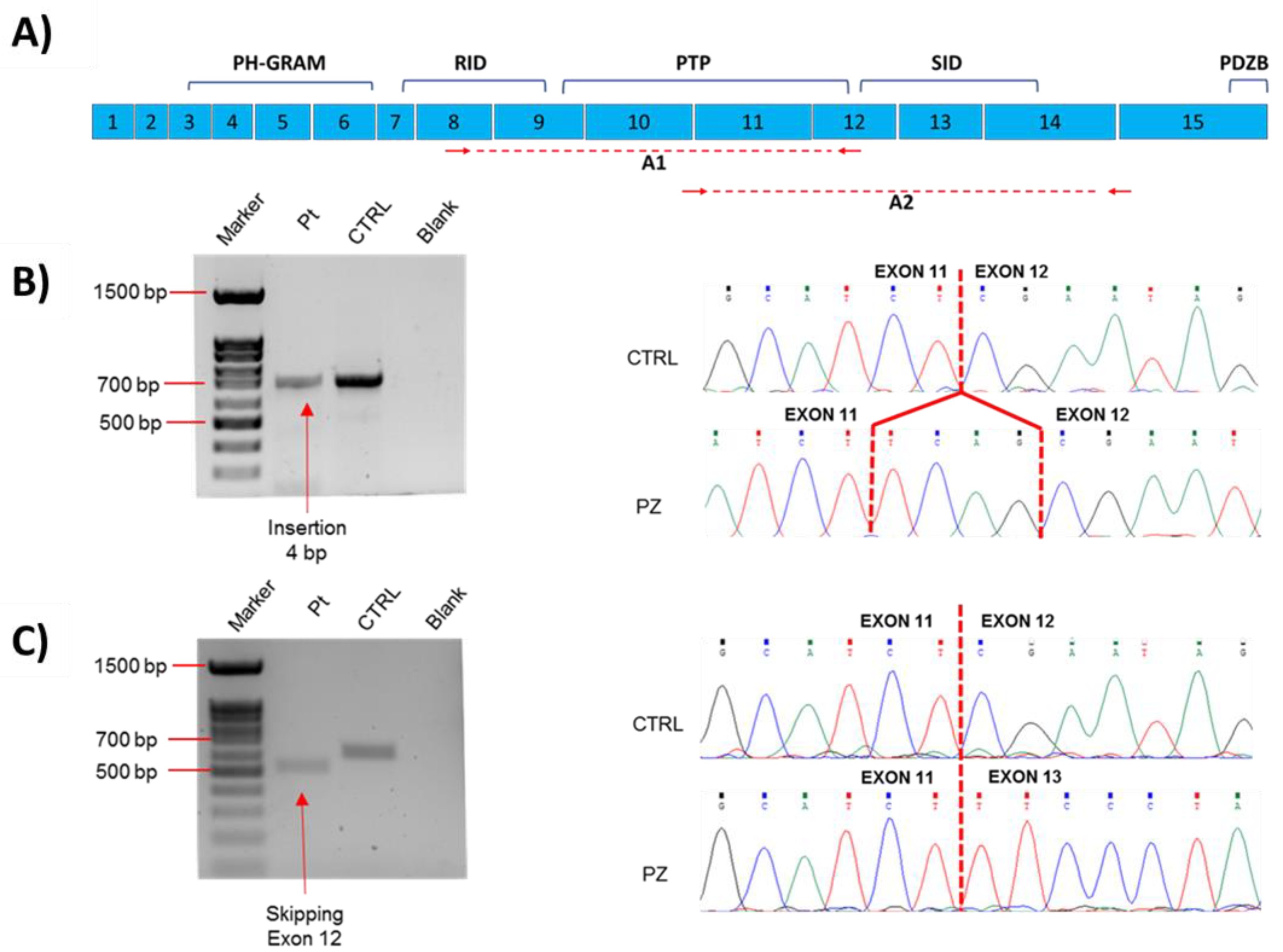
2.2. Molecular Data
3. Discussion
4. Materials and Methods
4.1. Muscle Biopsy
4.2. Genetics Analysis
4.3. Transcript Analysis
4.4. In Silico Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fattori, F.; Maggi, L.; Bruno, C.; Cassandrini, D.; Codemo, V.; Catteruccia, M.; Tasca, G.; Berardinelli, A.; Magri, F.; Pane, M.; et al. Centronuclear myopathies: Genotype–phenotype correlation and frequency of defined genetic forms in an Italian cohort. J. Neurol. 2015, 262, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Annoussamy, M.; Lilien, C.; Gidaro, T.; Gargaun, E.; Chê, V.; Schara, U.; Gangfuß, A.; D’Amico, A.; Dowling, J.J.; Darras, B.T.; et al. X-linked myotubular myopathy: A prospective international natural history study. Neurology 2019, 92, e1852–e1867. [Google Scholar] [CrossRef] [PubMed]
- Fidani, L.; Karagianni, P.; Tsakalidis, C.; Mitsiako, G.; Hatziioannidis, I.; Biancalana, V.; Nikolaidis, N. Identification of a mutation in the MTM1 gene, associated with X-linked myotubular myopathy, in a Greek family. Hippokratia 2011, 15, 278–279. [Google Scholar] [CrossRef]
- Claeys, K.G. Congenital myopathies: An update. Dev. Med. Child Neurol. 2020, 62, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Gurgel-Giannetti, J.; Zanoteli, E.; Concentino, E.L.D.C.; Neto, O.L.A.; Pesquero, J.B.; Reed, U.C.; Vainzof, M. Necklace fibers as histopathological marker in a patient with severe form of X-linked myotubular myopathy. Neuromuscul. Disord. 2012, 22, 541–545. [Google Scholar] [CrossRef]
- Amoasii, L.; Hnia, K.; Laporte, J. Myotubularin Phosphoinositide Phosphatases in Human Diseases. Curr. Top Microbiol. Immunol. 2012, 362, 209–233. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.; Feist, C.; Dietz, P.; Moore, S.; Basel, D. A Deep Intronic Variant Activates a Pseudoexon in the MTM1Gene in a Family with X-Linked Myotubular Myopathy. Mol. Syndr. 2020, 11, 264–270. [Google Scholar] [CrossRef]
- Laporte, J.; Biancalana, V.; Tanner, S.M.; Kress, W.; Schneider, V.; Wallgren-Pettersson, C.; Herger, F.; Buj-Bello, A.; Blondeau, F.; Liechti-Gallati, S.; et al. MTM1 mutations in X-linked myotubular myopathy. Hum. Mutat. 2000, 15, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.; Oliveira, M.E.; Kress, W.; Taipa, R.; Pires, M.; Hilbert, P.; Baxter, P.; Santos, M.; Buermans, H. Expanding the MTM1 mutational spectrum: Novel variants including the first multi-exonic duplication and development of a locus-specific database. Eur. J. Hum. Genet. 2012, 21, 540–549. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Laporte, J.; Hu, L.J.; Kretz, C.; Mandel, J.-L.; Kioschis, P.; Coy, J.F.; Klauck, S.M.; Poustka, A.; Dahl, N. A gene mutated in X–linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat. Genet. 1996, 13, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Goryunov, D.; Nightingale, A.; Bornfleth, L.; Leung, C.; Liem, R.K.H. Multiple disease-linked myotubularin mutations cause NFL assembly defects in cultured cells and disrupt myotubularin dimerization. J. Neurochem. 2008, 104, 1536–1552. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, M.W.; Dowling, J.J. X-linked myotubular myopathy. Neuromuscul. Disord. 2021, 31, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Robinson, F.L.; Dixon, J.E. Myotubularin phosphatases: Policing 3-phosphoinositides. Trends Cell Biol. 2006, 16, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Pierson, C.R.; Agrawal, P.B.; Blasko, J.; Beggs, A.H. Myofiber size correlates with MTM1 mutation type and outcome in X-linked myotubular myopathy. Neuromuscul. Disord. 2007, 17, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Tosch, V.; Vasli, N.; Kretz, C.; Nicot, A.-S.; Gasnier, C.; Dondaine, N.; Oriot, D.; Barth, M.; Puissant, H.; Romero, N.B.; et al. Novel molecular diagnostic approaches for X-linked centronuclear (myotubular) myopathy reveal intronic mutations. Neuromuscul. Disord. 2010, 20, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Bryen, S.J.; Oates, E.C.; Evesson, F.J.; Lu, J.K.; Waddell, L.B.; Joshi, H.; Ryan, M.M.; Cummings, B.B.; McLean, C.A.; MacArthur, D.G.; et al. Pathogenic deep intronic MTM1 variant activates a pseudo-exon encoding a nonsense codon resulting in severe X-linked myotubular myopathy. Eur. J. Hum. Genet. 2020, 29, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Vasli, N.; Laugel, V.; Böhm, J.; Lannes, B.; Biancalana, V.; Laporte, J. Myotubular myopathy caused by multiple abnormal splicing variants in the MTM1 RNA in a patient with a mild phenotype. Eur. J. Hum. Genet. 2012, 20, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Tasca, G.; Ricci, E.; Penttilä, S.; Monforte, M.; Giglio, V.; Ottaviani, P.; Camastra, G.; Silvestri, G.; Udd, B. New phenotype and pathology features in MYH7-related distal myopathy. Neuromuscul. Disord. 2012, 22, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Fattori, F.; Fiorillo, C.; Rodolico, C.; Tasca, G.; Verardo, M.; Bellacchio, E.; Pizzi, S.; Ciolfi, A.; Fagiolari, G.; Lupica, A.; et al. Expanding the histopathological spectrum of CFL2-related myopathies. Clin. Genet. 2018, 93, 1234–1239. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| AMPLICON | PRIMER SEQUENCE (5′-3′) |
|---|---|
| Amplicon A1 | Fw: GTTCCGTATCGTGCCTCAG |
| Rev: GGAGAACGGTCAGCATCGG | |
| Amplicon A2 | Fw: CATATCAAGCTCGTTTTGACAG |
| Rev: GGATTCGGCTGTTGTTGCTTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosco, L.; Leone, D.; Costa Comellas, L.; Monforte, M.; Pane, M.; Mercuri, E.; Bertini, E.; D’Amico, A.; Fattori, F. Novel Splicing Mutation in MTM1 Leading to Two Abnormal Transcripts Causes Severe Myotubular Myopathy. Int. J. Mol. Sci. 2022, 23, 10274. https://doi.org/10.3390/ijms231810274
Bosco L, Leone D, Costa Comellas L, Monforte M, Pane M, Mercuri E, Bertini E, D’Amico A, Fattori F. Novel Splicing Mutation in MTM1 Leading to Two Abnormal Transcripts Causes Severe Myotubular Myopathy. International Journal of Molecular Sciences. 2022; 23(18):10274. https://doi.org/10.3390/ijms231810274
Chicago/Turabian StyleBosco, Luca, Daniela Leone, Laura Costa Comellas, Mauro Monforte, Marika Pane, Eugenio Mercuri, Enrico Bertini, Adele D’Amico, and Fabiana Fattori. 2022. "Novel Splicing Mutation in MTM1 Leading to Two Abnormal Transcripts Causes Severe Myotubular Myopathy" International Journal of Molecular Sciences 23, no. 18: 10274. https://doi.org/10.3390/ijms231810274
APA StyleBosco, L., Leone, D., Costa Comellas, L., Monforte, M., Pane, M., Mercuri, E., Bertini, E., D’Amico, A., & Fattori, F. (2022). Novel Splicing Mutation in MTM1 Leading to Two Abnormal Transcripts Causes Severe Myotubular Myopathy. International Journal of Molecular Sciences, 23(18), 10274. https://doi.org/10.3390/ijms231810274