
Rubella Virus Triggers Type I Interferon Antiviral Response in Cultured Human Neural Cells: Involvement in the Control of Viral Gene Expression and Infectious Progeny Production
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
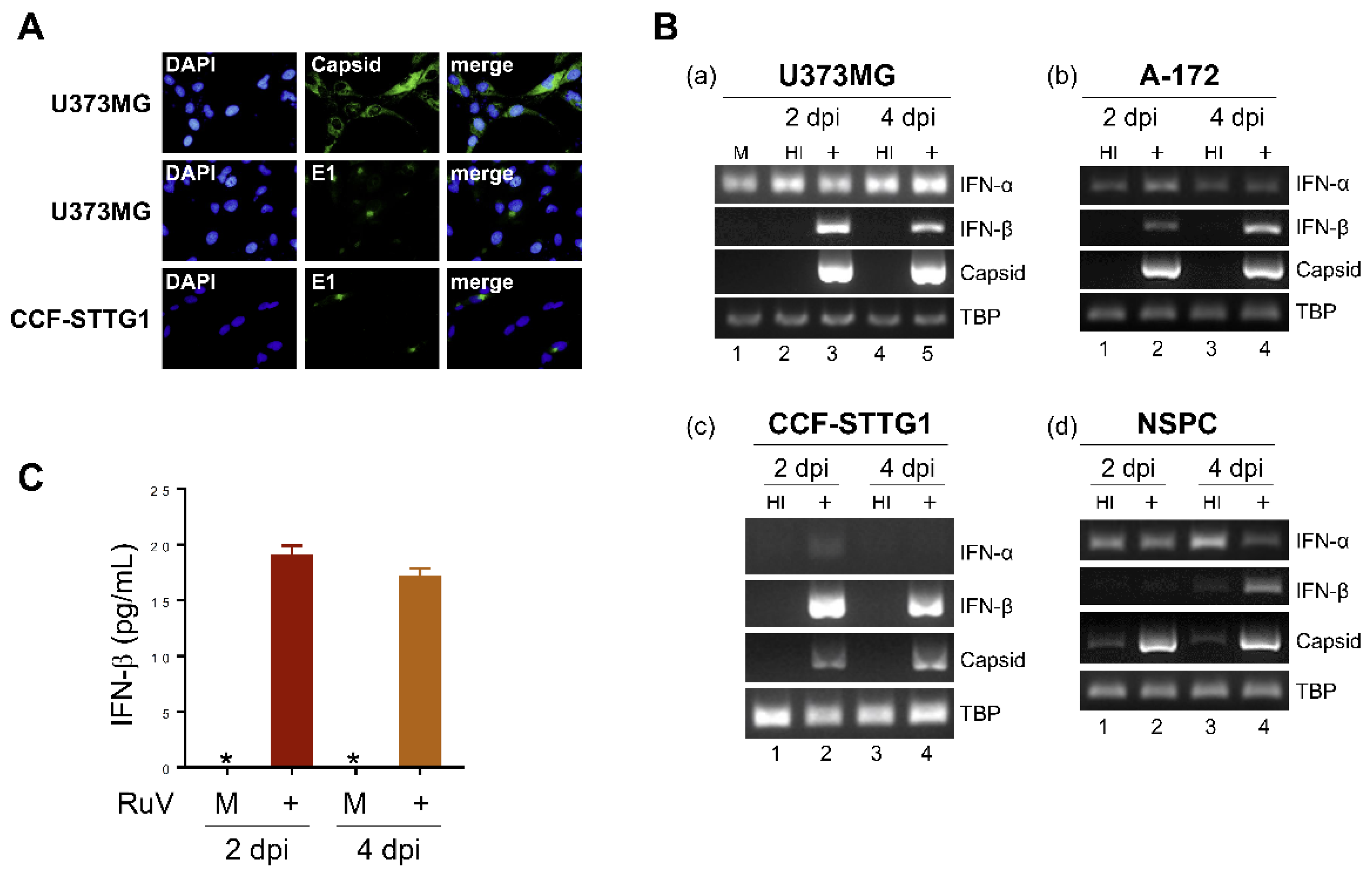
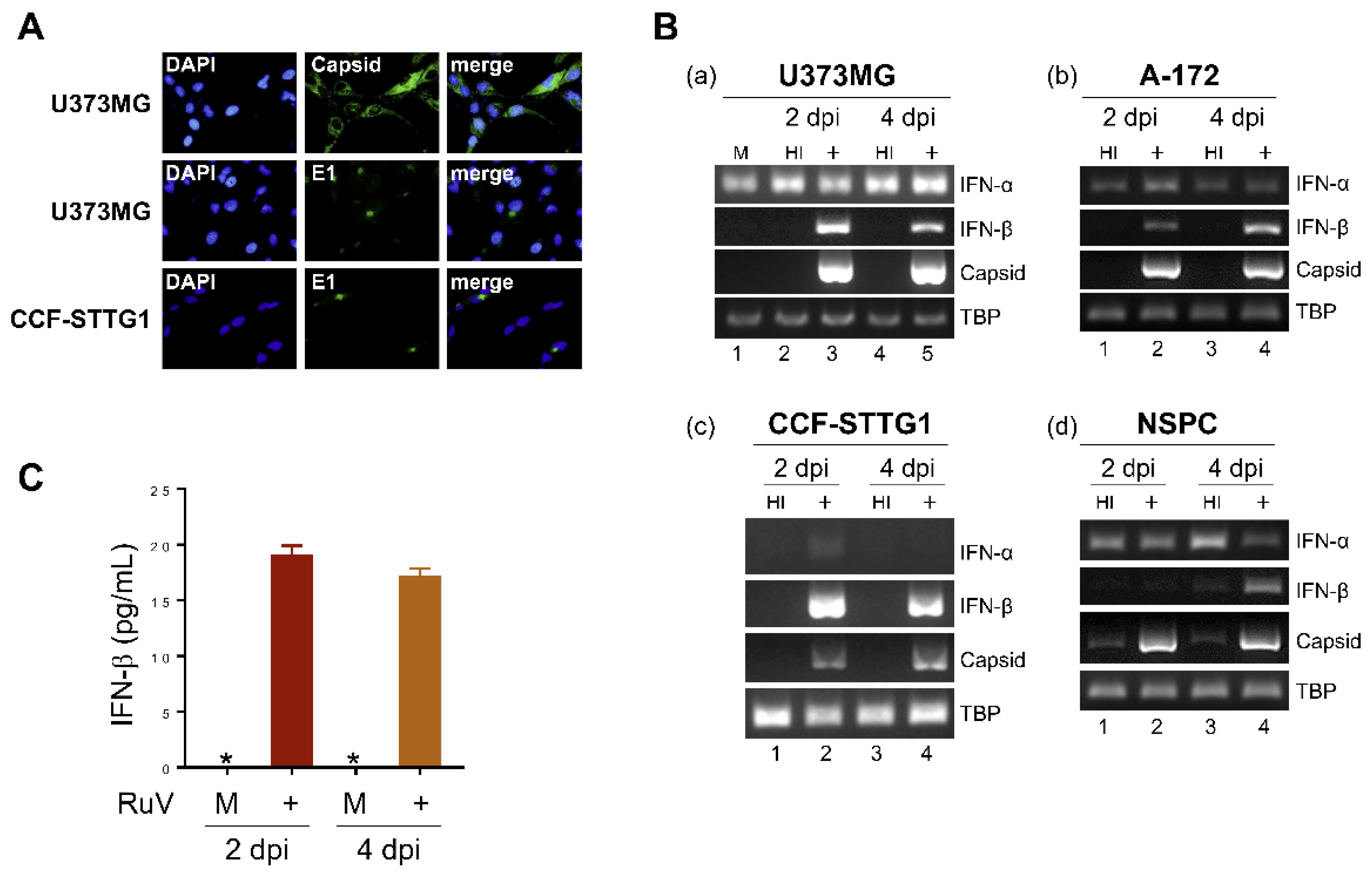
2.1. RuV Infects Cultured Human Neural Cells and Induces IFN-β Production
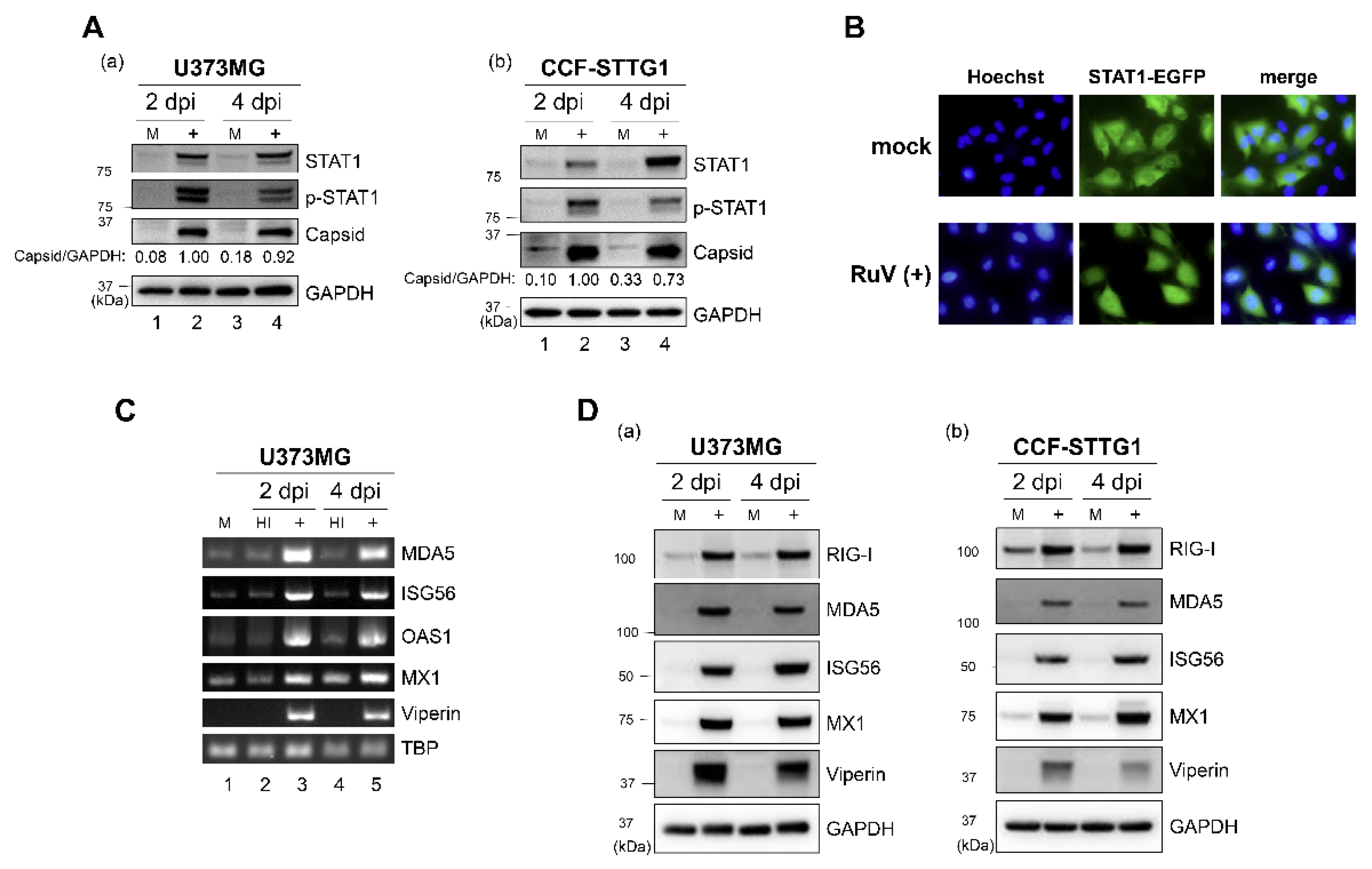
2.2. RuV Infection Induces STAT1 Activation and ISGs Expression in Human Neural Cells
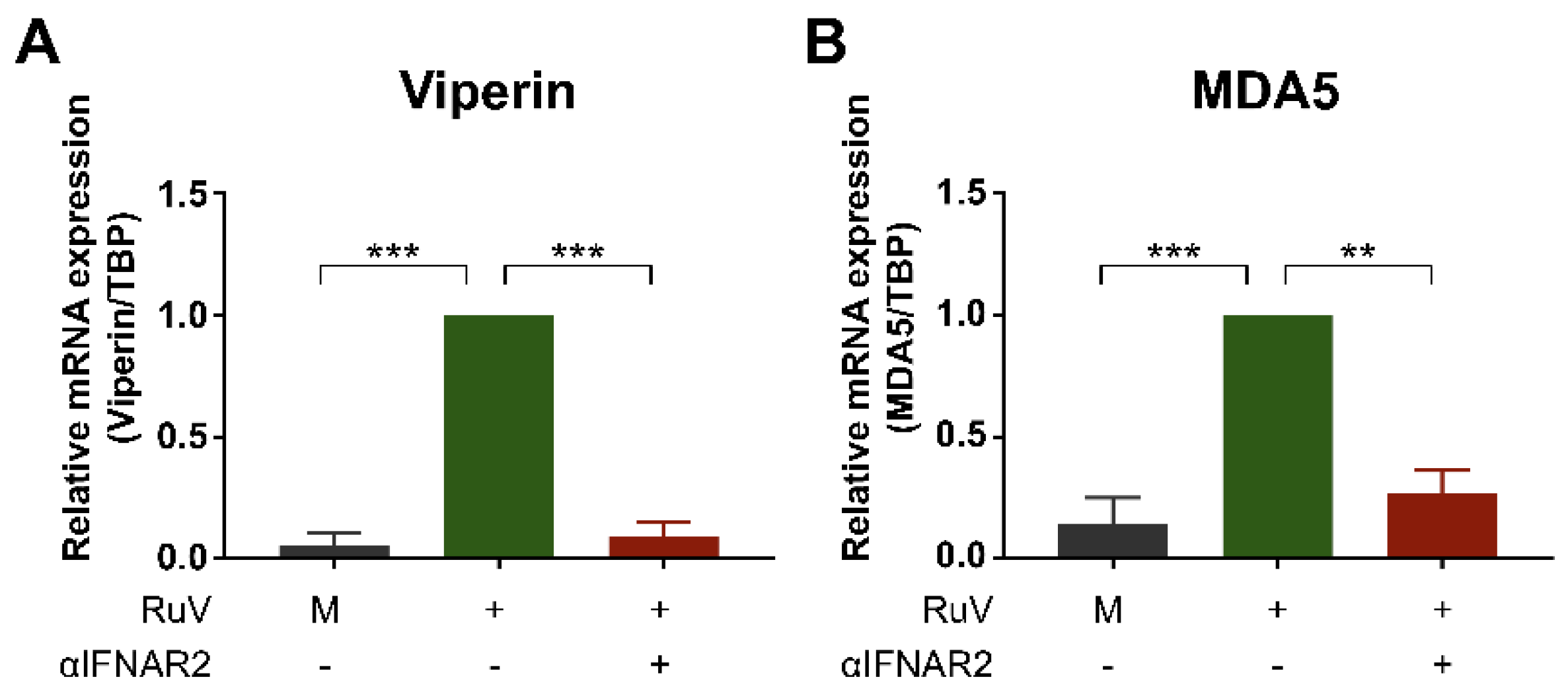
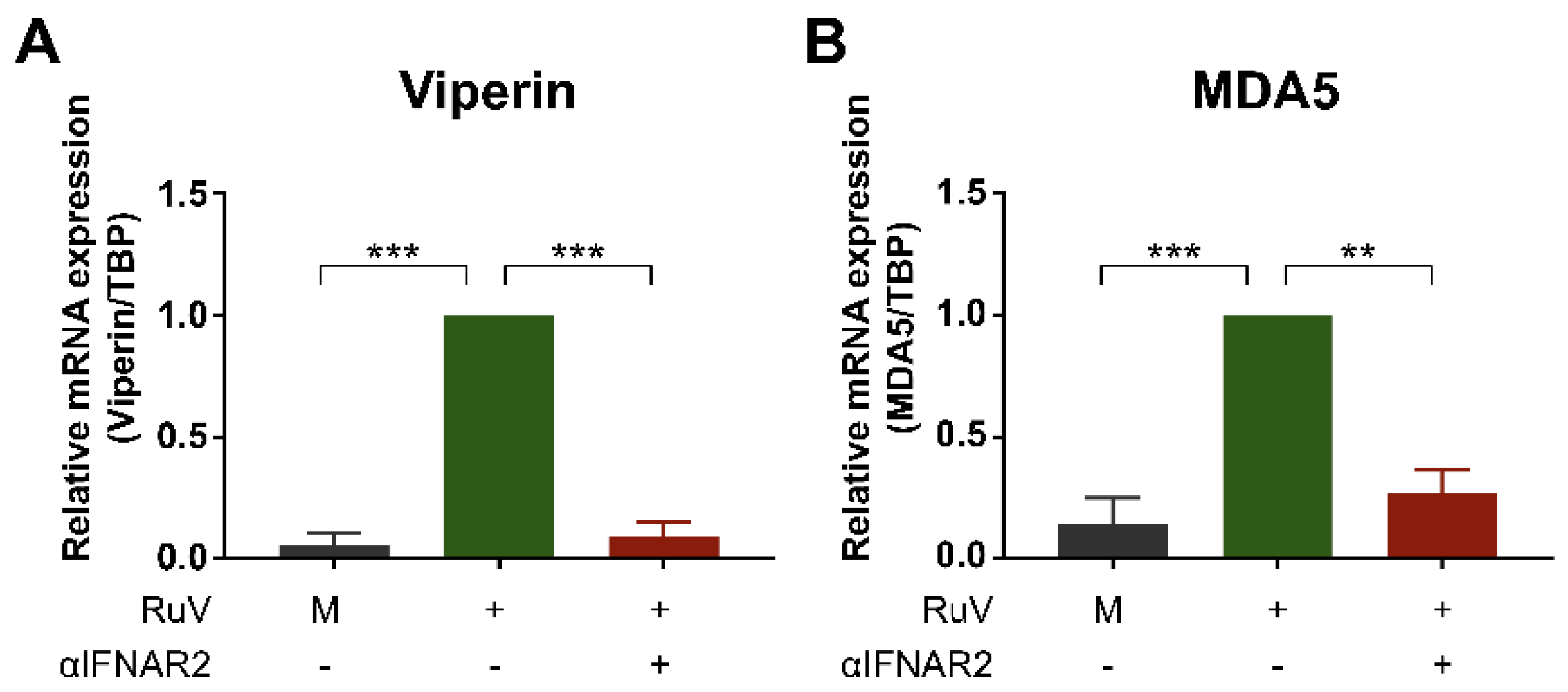
2.3. Anti-IFN-α/β Receptor Neutralizing Antibody Attenuates RuV-Induced Expression of ISGs
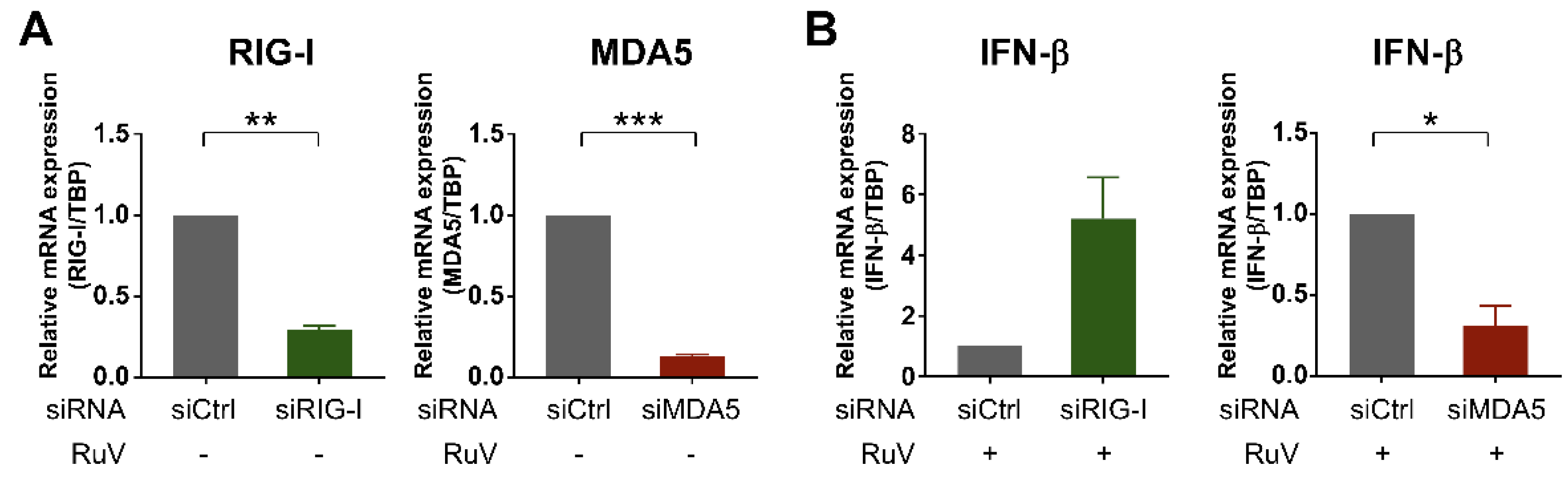
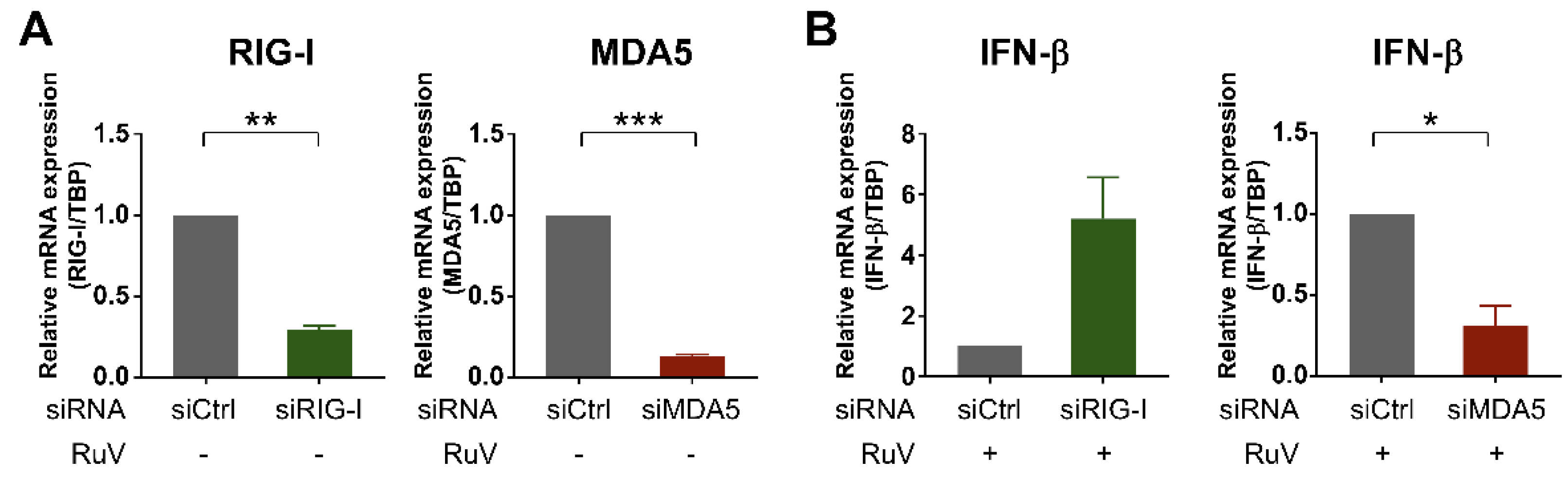
2.4. MDA5 Is Required for RuV-Triggered IFN-β Induction in U373MG Cells
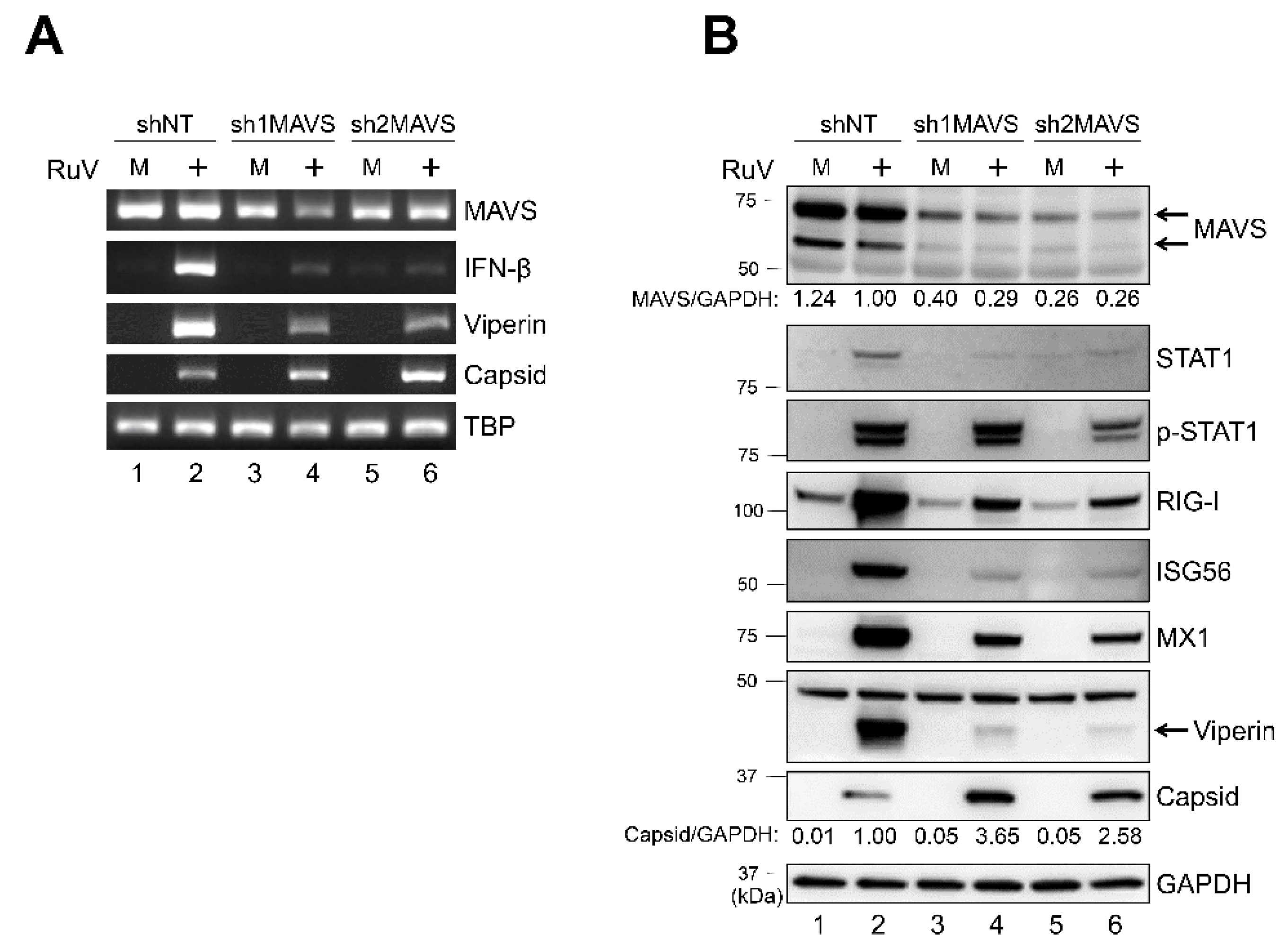
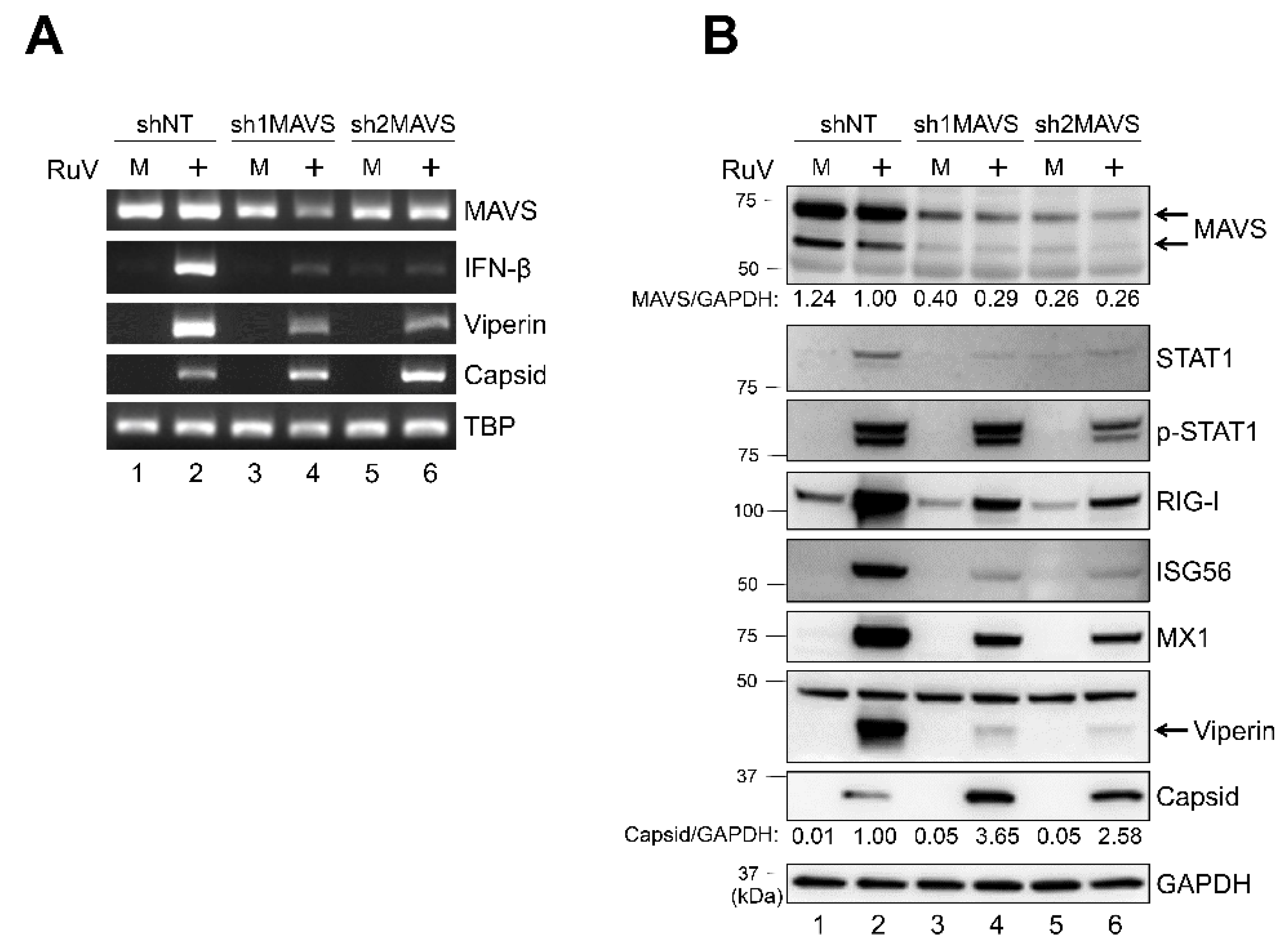
2.5. Knockdown of MAVS Attenuates Innate Immune Responses against RuV Infection
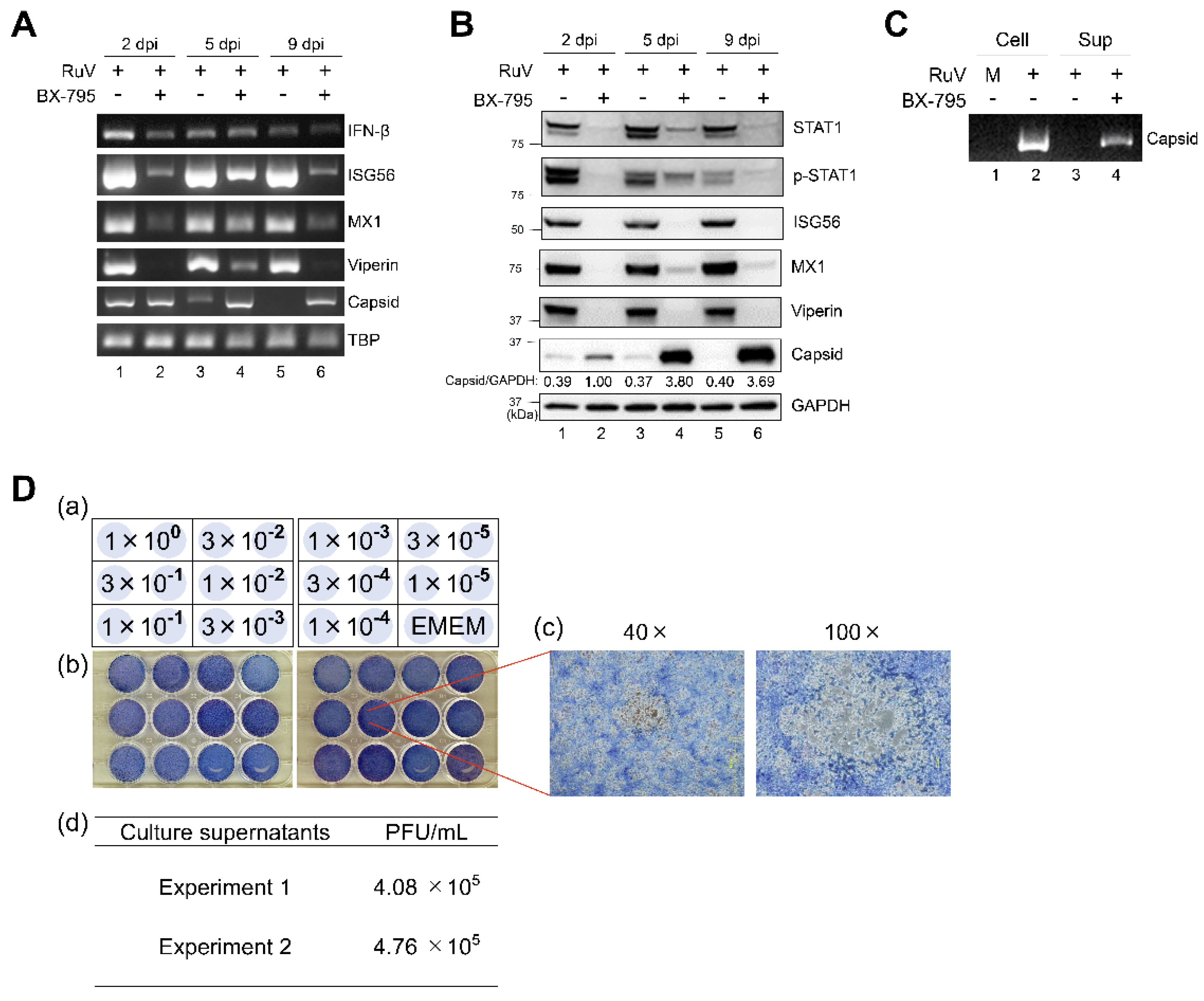
2.6. BX-795, a TBK1/IKKε Inhibitor, Abrogates the RuV-Triggered Innate Immune Response and Augments the Production of Infectious Progeny Virions
3. Discussion
4. Materials and Methods
4.1. Cells and Virus
4.2. Reagents and Plasmids
4.3. RNA Extraction and RT-PCR
4.4. Immunoblotting Assay
4.5. Indirect Immunofluorescence Assay
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. Interferon α/β Receptor Neutralization Assay
4.8. Lentiviral shRNA Transduction
4.9. BX-795 Treatment
4.10. Small Interfering RNA Transfection
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Andrejeva, J.; Childs, K.S.; Young, D.F.; Carlos, T.S.; Stock, N.; Goodbourn, S.; Randall, R.E. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. USA 2004, 101, 17264–17269. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef]
- Chan, Y.K.; Gack, M.U. RIG-I-like receptor regulation in virus infection and immunity. Curr. Opin. Virol. 2015, 12, 7–14. [Google Scholar] [CrossRef]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Nan, Y.; Nan, G.; Zhang, Y.J. Interferon induction by RNA viruses and antagonism by viral pathogens. Viruses 2014, 6, 4999–5027. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Chen, X.; Tian, J.H.; Chen, L.J.; Li, K.; Wang, W.; Eden, J.S.; Shen, J.J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Grimwood, R.M.; Holmes, E.C.; Geoghegan, J.L. A Novel Rubi-Like Virus in the Pacific Electric Ray (Tetronarce californica) Reveals the Complex Evolutionary History of the Matonaviridae. Viruses 2021, 13, 585. [Google Scholar] [CrossRef]
- Geoghegan, J.L.; Di Giallonardo, F.; Wille, M.; Ortiz-Baez, A.S.; Costa, V.A.; Ghaly, T.; Mifsud, J.C.O.; Turnbull, O.M.H.; Bellwood, D.R.; Williamson, J.E.; et al. Virome composition in marine fish revealed by meta-transcriptomics. Virus Evol. 2021, 7, veab005. [Google Scholar] [CrossRef]
- Bennett, A.J.; Paskey, A.C.; Ebinger, A.; Pfaff, F.; Priemer, G.; Hoper, D.; Breithaupt, A.; Heuser, E.; Ulrich, R.G.; Kuhn, J.H.; et al. Relatives of rubella virus in diverse mammals. Nature 2020, 586, 424–428. [Google Scholar] [CrossRef]
- Das, P.K.; Kielian, M. Molecular and Structural Insights into the Life Cycle of Rubella Virus. J. Virol. 2021, 95, e02349-20. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Bowden, D.S. Rubella virus replication and links to teratogenicity. Clin. Microbiol. Rev. 2000, 13, 571–587. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Viswanathan, R.; Sapkal, G.N. Molecular aspects of the teratogenesis of rubella virus. Biol. Res. 2019, 52, 47. [Google Scholar] [CrossRef] [PubMed]
- Atreya, C.D.; Mohan, K.V.; Kulkarni, S. Rubella virus and birth defects: Molecular insights into the viral teratogenesis at the cellular level. Birth Defects Res. Part A Clin. Mol. Teratol. 2004, 70, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Hobman, T.C. Rubella virus. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 687–711. [Google Scholar]
- Webster, W.S. Teratogen update: Congenital rubella. Teratology 1998, 58, 13–23. [Google Scholar] [CrossRef]
- Dontigny, L.; Arsenault, M.Y.; Martel, M.J.; Clinical Practice Obstetrics, C. Rubella in pregnancy. J. Obs. Gynaecol. Can. 2008, 30, 152–158. [Google Scholar] [CrossRef]
- Winter, A.K.; Moss, W.J. Rubella. Lancet 2022, 399, 1336–1346. [Google Scholar] [CrossRef]
- Frey, T.K. Neurological aspects of rubella virus infection. Intervirology 1997, 40, 167–175. [Google Scholar] [CrossRef]
- Connolly, J.H.; Hutchinson, W.M.; Allen, I.V.; Lyttle, J.A.; Swallow, M.W.; Dermott, E.; Thomsom, D. Carotid artery thrombosis, encephalitis, myelitis and optic neuritis associated with rubella virus infections. Brain A J. Neurol. 1975, 98, 583–594. [Google Scholar] [CrossRef]
- Cremer, N.E.; Oshiro, L.S.; Weil, M.L.; Lennette, E.H.; Itabashi, H.H.; Carnay, L. Isolation of rubella virus from brain in chronic progressive panencephalitis. J. Gen. Virol. 1975, 29, 143–153. [Google Scholar] [CrossRef]
- Bechar, M.; Davidovich, S.; Goldhammer, G.; Machtey, I.; Gadoth, N. Neurological complications following rubella infection. J. Neurol. 1982, 226, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Townsend, J.J.; Baringer, J.R.; Wolinsky, J.S.; Malamud, N.; Mednick, J.P.; Panitch, H.S.; Scott, R.A.; Oshiro, L.; Cremer, N.E. Progressive rubella panencephalitis. Late onset after congenital rubella. N. Engl. J. Med. 1975, 292, 990–993. [Google Scholar] [CrossRef] [PubMed]
- WHO. Rubella Vaccines: WHO Position Paper—July 2020. Weekly Epidemiological Record; World Health Organization: Geneva, Switzerland, 2020; Volume 95, pp. 306–324.
- Sugita, K.; Ando, M.; Makino, M.; Takanashi, J.; Fujimoto, N.; Niimi, H. Magnetic resonance imaging of the brain in congenital rubella virus and cytomegalovirus infections. Neuroradiology 1991, 33, 239–242. [Google Scholar] [CrossRef]
- Namiki, T.; Takano, C.; Aoki, R.; Trinh, Q.D.; Morioka, I.; Hayakawa, S. Parenchymal calcification is associated with the neurological prognosis in patients with congenital rubella syndrome. Congenit. Anom. 2022, 62, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Sawlani, V.; Shankar, J.J.; White, C. Magnetic resonance imaging findings in a case of congenital rubella encephalitis. Can. J. Infect. Dis. Med. Microbiol. 2013, 24, e122–e123. [Google Scholar] [CrossRef]
- Adamo, M.P.; Zapata, M.; Frey, T.K. Analysis of gene expression in fetal and adult cells infected with rubella virus. Virology 2008, 370, 1–11. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Reis e Sousa, C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef]
- Gitlin, L.; Benoit, L.; Song, C.; Cella, M.; Gilfillan, S.; Holtzman, M.J.; Colonna, M. Melanoma differentiation-associated gene 5 (MDA5) is involved in the innate immune response to Paramyxoviridae infection in vivo. PLoS Pathog 2010, 6, e1000734. [Google Scholar] [CrossRef]
- Roth-Cross, J.K.; Bender, S.J.; Weiss, S.R. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J. Virol. 2008, 82, 9829–9838. [Google Scholar] [CrossRef] [PubMed]
- McCartney, S.A.; Thackray, L.B.; Gitlin, L.; Gilfillan, S.; Virgin, H.W.; Colonna, M. MDA-5 recognition of a murine norovirus. PLoS Pathog 2008, 4, e1000108. [Google Scholar] [CrossRef]
- Plumet, S.; Herschke, F.; Bourhis, J.M.; Valentin, H.; Longhi, S.; Gerlier, D. Cytosolic 5’-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response. PLoS ONE 2007, 2, e279. [Google Scholar] [CrossRef] [PubMed]
- Habjan, M.; Andersson, I.; Klingstrom, J.; Schumann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Muhlberger, E.; et al. Processing of genome 5’ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE 2008, 3, e2032. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, W.B.; Loo, Y.M.; Gale, M., Jr.; Hartman, A.L.; Kimberlin, C.R.; Martinez-Sobrido, L.; Saphire, E.O.; Basler, C.F. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol. 2006, 80, 5168–5178. [Google Scholar] [CrossRef]
- Sumpter, R., Jr.; Loo, Y.M.; Foy, E.; Li, K.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale, M., Jr. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 2005, 79, 2689–2699. [Google Scholar] [CrossRef]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef]
- Gern, O.L.; Mulenge, F.; Pavlou, A.; Ghita, L.; Steffen, I.; Stangel, M.; Kalinke, U. Toll-like Receptors in Viral Encephalitis. Viruses 2021, 13, 2065. [Google Scholar] [CrossRef]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef]
- Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential Regulation of Type I and Type III Interferon Signaling. Int. J. Mol. Sci. 2019, 20, 1445. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Ikegame, S.; Takeda, M.; Ohno, S.; Nakatsu, Y.; Nakanishi, Y.; Yanagi, Y. Both RIG-I and MDA5 RNA helicases contribute to the induction of alpha/beta interferon in measles virus-infected human cells. J. Virol. 2010, 84, 372–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakuragi, S.; Liao, H.; Yajima, K.; Fujiwara, S.; Nakamura, H. Rubella Virus Triggers Type I Interferon Antiviral Response in Cultured Human Neural Cells: Involvement in the Control of Viral Gene Expression and Infectious Progeny Production. Int. J. Mol. Sci. 2022, 23, 9799. https://doi.org/10.3390/ijms23179799
Sakuragi S, Liao H, Yajima K, Fujiwara S, Nakamura H. Rubella Virus Triggers Type I Interferon Antiviral Response in Cultured Human Neural Cells: Involvement in the Control of Viral Gene Expression and Infectious Progeny Production. International Journal of Molecular Sciences. 2022; 23(17):9799. https://doi.org/10.3390/ijms23179799
Chicago/Turabian StyleSakuragi, Sayuri, Huanan Liao, Kodai Yajima, Shigeyoshi Fujiwara, and Hiroyuki Nakamura. 2022. "Rubella Virus Triggers Type I Interferon Antiviral Response in Cultured Human Neural Cells: Involvement in the Control of Viral Gene Expression and Infectious Progeny Production" International Journal of Molecular Sciences 23, no. 17: 9799. https://doi.org/10.3390/ijms23179799
APA StyleSakuragi, S., Liao, H., Yajima, K., Fujiwara, S., & Nakamura, H. (2022). Rubella Virus Triggers Type I Interferon Antiviral Response in Cultured Human Neural Cells: Involvement in the Control of Viral Gene Expression and Infectious Progeny Production. International Journal of Molecular Sciences, 23(17), 9799. https://doi.org/10.3390/ijms23179799