
Downstream Effects of Mutations in SOD1 and TARDBP Converge on Gene Expression Impairment in Patient-Derived Motor Neurons


Abstract
:
1. Introduction
2. Results
2.1. Motor Neuron Differentiation in iPSC-Derived Cell Lines
2.2. RNA-Seq Profiling and Identification of DEGs between SOD1- and TDP43-ALS
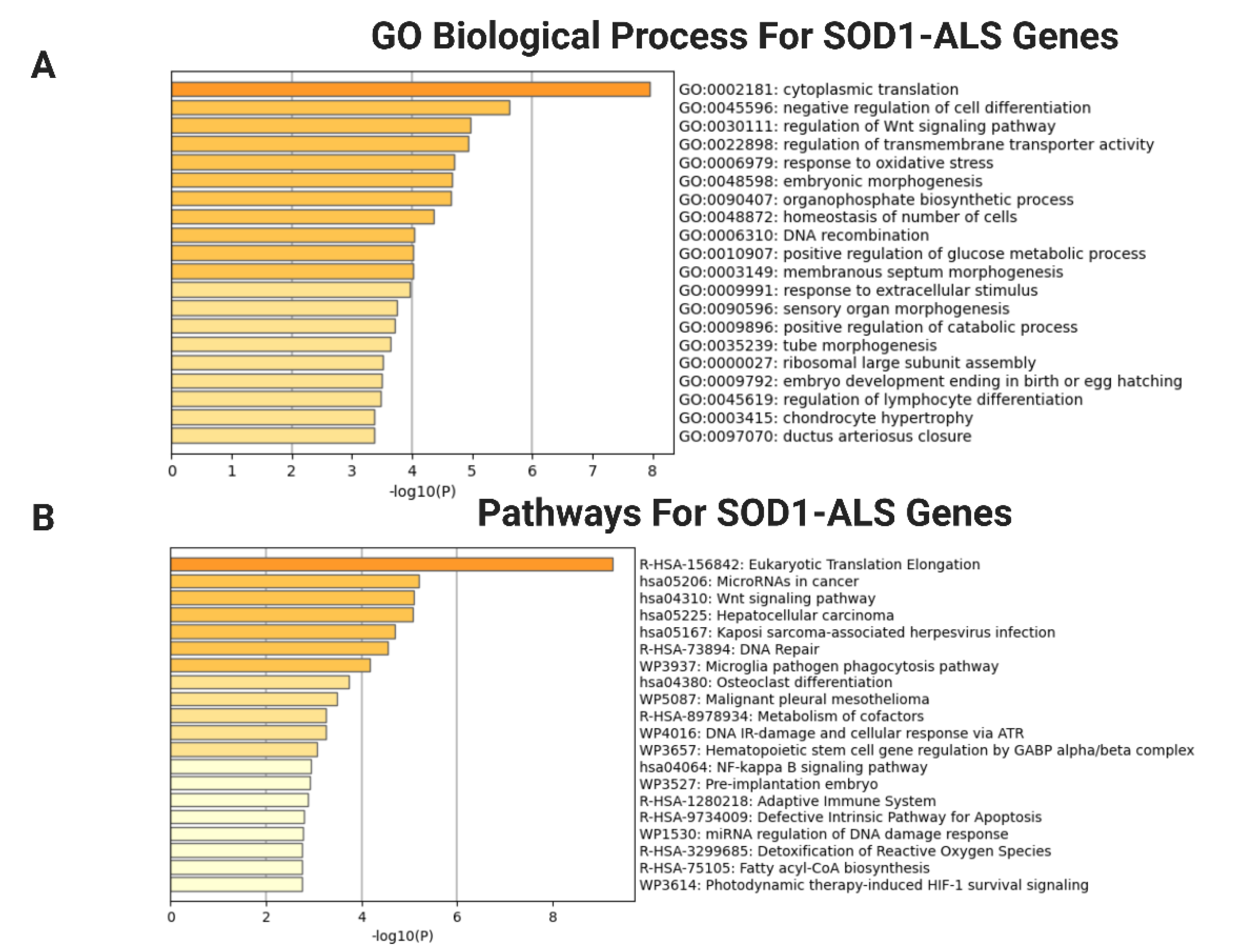
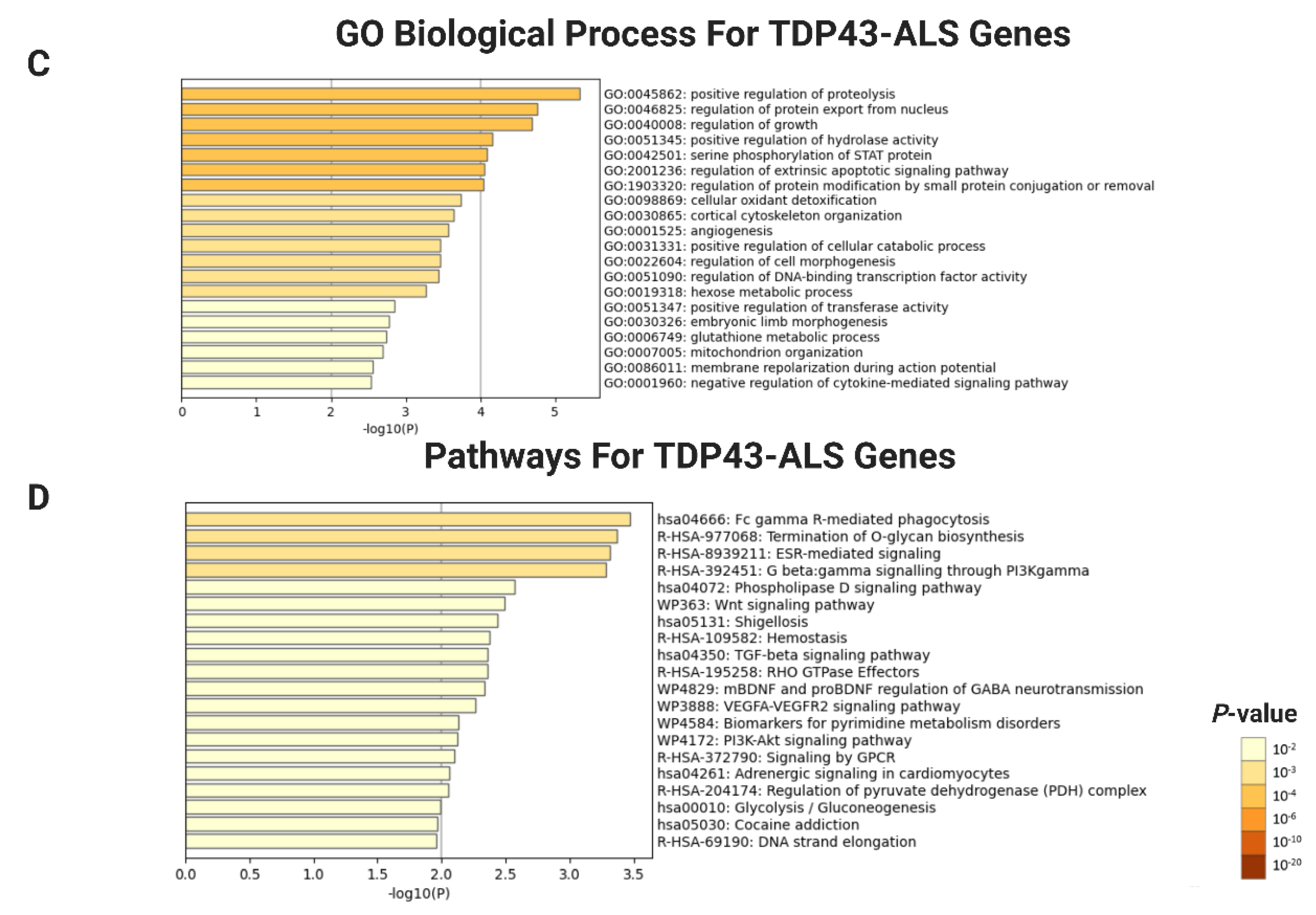
2.3. Functional and Pathway Enrichment Aanalysis of DEGs
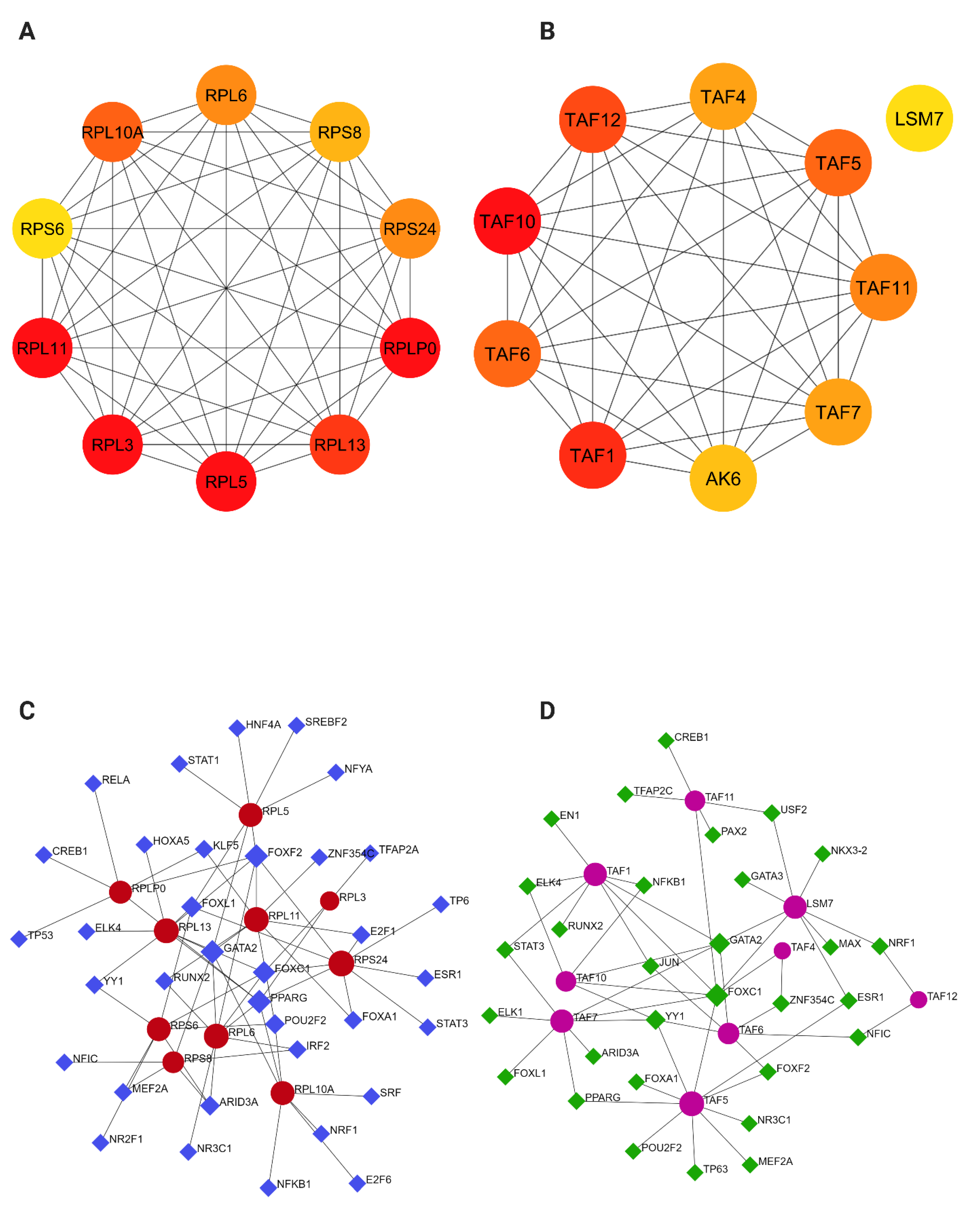
2.4. PPI Network Construction and Module Analysis
2.5. Identification of Hub Genes and Transcription Factor Regulatory Network Analysis
3. Discussion
4. Materials and Methods
4.1. Patient Characteristics
4.2. Generation and Expansion of Cell Lines
4.3. RNA Isolation and Transcriptome Analysis
4.4. Functional Annotation of DEGs by GO and KEGG Analysis
4.5. PPI Network Construction and Identification of Hub Genes
4.6. Module Identification and Enrichment Analysis
4.7. Gene-Transcription Factor Interaction Analysis
4.8. Statistical Analysis
4.9. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| ALS | Amyotrophic lateral sclerosis |
| fALS | Familial ALS |
| sALS | Sporadic ALS |
| MN | Motor neuron |
| FUS | Fused in sarcoma |
| SOD1 | Superoxide dismutase 1 |
| C9ORF72 | Chromosome 9 open reading frame 72 |
| TARDBP | TAR DNA-binding protein 43 |
| iPSC | Human patient-derived induced pluripotent stem cell |
| NPC | Neural progenitor cells |
| DEG | Differentially expressed genes |
| GEO | Gene expression omnibus |
| NGS | Next generation sequencing |
| ECM | Extracellular matrix |
| KEGG | Kyto encyclopedia of genes and genomes |
| STRING | The search tool for the retrieval of interacting genes/proteins |
| DAVID | The Database for Annotaion, Visualization, and Integrated Discovery |
| PGS | Partek genomic suite (Partek Inc) |
| GO | Gene ontology |
| ENCODE | Encyclopedia of DNA elements |
| MAPK | Mitogen activated protein kinase |
| PPI | Protein-protein interactome analysis |
| MCODE | Molecular Complex Detection |
| TF | Transcription factors |
| PPARG | Peroxisome proliferator activated receptor gamma |
| OXPHOS | Oxidative phosphorylation |
| GATA2 | GATA-binding factor 2 |
| snRNAs | Small nuclear ribonucleoproteins |
| hnRNAs | Heterogeneous nuclear ribonucleoproteins |
| FOXC1 | Forkhead Box C1 |
References
- Peters, O.M.; Ghasemi, M.; Brown, R.H., Jr. Emerging mechanisms of molecular pathology in ALS Find the latest version: Emerging mechanisms of molecular pathology in ALS. J. Clin. Investig. 2015, 125, 1767–1779. [Google Scholar] [CrossRef] [PubMed]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Van Broeckhoven, C.; van der Zee, J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 2018, 34, 404–423. [Google Scholar] [CrossRef] [PubMed]
- Magrané, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 2010, 7, 420–429. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Briese, M.; Saal-Bauernschubert, L.; Lüningschrör, P.; Moradi, M.; Dombert, B.; Surrey, V.; Appenzeller, S.; Deng, C.; Jablonka, S.; Sendtner, M. Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol. Commun. 2020, 8, 116. [Google Scholar] [CrossRef]
- Saxena, S.; Cabuy, E.; Caroni, P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef]
- Hoffman, E.K.; Wilcox, H.M.; Scott, R.W.; Siman, R. Proteasome inhibition enhances the stability of mouse Cu/Zn superoxide dismutase with mutations linked to familial amyotrophic lateral sclerosis. J. Neurol. Sci. 1996, 139, 15–20. [Google Scholar] [CrossRef]
- Lamas, N.J.; Roybon, L. Harnessing the Potential of Human Pluripotent Stem Cell-Derived Motor Neurons for Drug Discovery in Amyotrophic Lateral Sclerosis: From the Clinic to the Laboratory and Back to the Patient. Front. Drug Discov. 2021, 1, 1–26. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Serio, A.; Bilican, B.; Barmada, S.J.; Ando, D.M.; Zhao, C.; Siller, R.; Burr, K.; Haghi, G.; Story, D.; Nishimura, A.L.; et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA. 2013, 110, 4697–4702. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Goparaju, S.K.; Tooi, N.; Inoue, H.; Takahashi, R.; Nakatsuji, N.; Aiba, K. Amyotrophic Lateral Sclerosis Model Derived from Human Embryonic Stem Cells Overexpressing Mutant Superoxide Dismutase 1. Stem Cells Transl. Med. 2012, 1, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Dash, B.P.; Naumann, M.; Sterneckert, J.; Hermann, A. Genome wide analysis points towards subtype-specific diseases in different genetic forms of amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2020, 21, 6938. [Google Scholar] [CrossRef] [PubMed]
- Heath, P.R.; Kirby, J.; Shaw, P.J. Investigating cell death mechanisms in amyotrophic lateral sclerosis using transcriptomics. Front. Cell. Neurosci. 2013, 7, 259. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Kirby, J.; Ferraiuolo, L.; Heath, P.R.; Rattray, M.; Shaw, P.J. Gene expression profiling in human neurodegenerative disease. Nat. Rev. Neurol. 2012, 8, 518–530. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef]
- Fernández-Beltrán, L.C.; Godoy-Corchuelo, J.M.; Losa-Fontangordo, M.; Williams, D.; Matias-Guiu, J.; Corrochano, S. A transcriptomic meta-analysis shows lipid metabolism dysregulation as an early pathological mechanism in the spinal cord of sod1 mice. Int. J. Mol. Sci. 2021, 22, 9553. [Google Scholar] [CrossRef]
- Butti, Z.; Patten, S.A. RNA dysregulation in amyotrophic lateral sclerosis. Front. Genet. 2019, 9, 712. [Google Scholar] [CrossRef] [Green Version]
- Lehmkuhl, E.M.; Zarnescu, D.C. Lost in translation: Evidence for protein synthesis deficits in ALS/FTD and related neurodegenerative diseases. Adv. Neurobiol. 2018, 20, 283–301. [Google Scholar] [CrossRef]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Bennett, C.F.; Cleveland, D.W.; Yeo, G.W. Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res. 2012, 1462, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Barmada, S.J. Linking RNA Dysfunction and Neurodegeneration in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Ratti, A.; Buratti, E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 2016, 138, 95–111. [Google Scholar] [CrossRef]
- Cohen, T.J.; Lee, V.M.Y.; Trojanowski, J.Q. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol. Med. 2011, 17, 659–667. [Google Scholar] [CrossRef]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Župunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in als and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Conlon, E.G.; Fagegaltier, D.; Agius, P.; Davis-Porada, J.; Gregory, J.; Hubbard, I.; Kang, K.; Kim, D.; Phatnani, H.; Shneider, N.A.; et al. Unexpected similarities between C9ORF72 and sporadic forms of ALS/FTD suggest a common disease mechanism. Elife 2018, 7, e37754. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Cotney, J.; Nagy, M.; Oh, S.; Leng, J.; Mahajan, M.; Mane, S.; Fenton, W.A.; Noonan, J.P.; Horwich, A.L. RNA-Seq Profiling of Spinal Cord Motor Neurons from a Presymptomatic SOD1 ALS Mouse. PLoS ONE 2013, 8, e53575. [Google Scholar] [CrossRef] [Green Version]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J. The evidence for altered RNA metabolism in amyotrophic lateral sclerosis (ALS). J. Neurol. Sci. 2010, 288, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, E.L.; Burgess, R.W. Accumulating evidence for axonal translation in neuronal homeostasis. Front. Neurosci. 2017, 11, 312. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Bassell, G.J. MRNA localization: An orchestration of assembly, traffic and synthesis. Traffic 2012, 14, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Verheijen, M.H.G.; Peviani, M.; Hendricusdottir, R.; Bell, E.M.; Lammens, M.; Smit, A.B.; Bendotti, C.; Van Minnen, J. Increased axonal ribosome numbers is an early event in the pathogenesis of amyotrophic lateral sclerosis. PLoS ONE 2014, 9, e87255. [Google Scholar] [CrossRef]
- Riva, N.; Clarelli, F.; Domi, T.; Cerri, F.; Gallia, F.; Trimarco, A.; Brambilla, P.; Lunetta, C.; Lazzerini, A.; Lauria, G.; et al. Unraveling gene expression profiles in peripheral motor nerve from amyotrophic lateral sclerosis patients: Insights into pathogenesis. Sci. Rep. 2016, 6, 39297. [Google Scholar] [CrossRef]
- Briese, M.; Saal, L.; Appenzeller, S.; Moradi, M.; Baluapuri, A.; Sendtner, M. Whole transcriptome profiling reveals the RNA content of motor axons. Nucleic Acids Res. 2015, 44, e33. [Google Scholar] [CrossRef]
- Rotem, N.; Magen, I.; Ionescu, A.; Gershoni-Emek, N.; Altman, T.; Costa, C.J.; Gradus, T.; Pasmanik-Chor, M.; Willis, D.E.; Ben-Dov, I.Z.; et al. ALS along the Axons-Expression of coding and noncoding RNA differs in axons of ALS models. Sci. Rep. 2017, 7, 44500. [Google Scholar] [CrossRef]
- Nijssen, J.; Aguila, J.; Hoogstraaten, R.; Kee, N.; Hedlund, E. Axon-Seq Decodes the Motor Axon Transcriptome and Its Modulation in Response to ALS. Stem Cell Rep. 2018, 11, 1565–1578. [Google Scholar] [CrossRef]
- Namboori, S.C.; Thomas, P.; Ames, R.; Hawkins, S.; Garrett, L.O.; Willis, C.R.G.; Rosa, A.; Stanton, L.W.; Bhinge, A. Single-cell transcriptomics identifies master regulators of neurodegeneration in SOD1 ALS iPSC-derived motor neurons. Stem Cell Rep. 2021, 16, 3020–3035. [Google Scholar] [CrossRef]
- Andersen, P.M.; Forsgren, L.; Binzer, M.; Nilsson, P.; Ala-Hurula, V.; Keränen, M.L.; Bergmark, L.; Saarinen, A.; Haltia, T.; Tarvainen, I.; et al. Autosomal recessive adult-onset amyohophic lateral sclerosis associated with homozygosity for Asp90A1a CuZn-superoxide dismutase mutation A clinical and genealogical study of 36 patients. Brain 1996, 119, 1153–1172. [Google Scholar] [CrossRef] [PubMed]
- Günther, R.; Pal, A.; Williams, C.; Zimyanin, V.L.; Liehr, M.; von Neubeck, C.; Krause, M.; Parab, M.G.; Petri, S.; Kalmbach, N.; et al. Alteration of Mitochondrial Integrity as Upstream Event in the Pathophysiology of SOD1-ALS. Cells 2022, 11, 1246. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Höing, S.; Moritz, S.; Parga, J.A.; Wagner, L.; Bruder, J.M.; et al. Derivation and Expansion Using Only Small Molecules of Human Neural Progenitors for Neurodegenerative Disease Modeling. PLoS ONE 2013, 8, e59252. [Google Scholar] [CrossRef]
- Bursch, F.; Kalmbach, N.; Naujock, M.; Staege, S.; Eggenschwiler, R.; Abo-Rady, M.; Japtok, J.; Guo, W.; Hensel, N.; Reinhardt, P.; et al. Altered calcium dynamics and glutamate receptor properties in iPSC-derived motor neurons from ALS patients with C9orf72, FUS, SOD1 or TDP43 mutations. Hum. Mol. Genet. 2019, 28, 2835–2850. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Günther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef]
- Naujock, M.; Stanslowsky, N.; Bufler, S.; Naumann, M.; Reinhardt, P.; Sterneckert, J.; Petri, S. 4-aminopyridine induced activity rescues hypoexcitable Motor Neurons from Amyotrophic Lateral Sclerosis Patient-Derived Induced. Stem Cells 2016, 34, 1563–1575. [Google Scholar] [CrossRef]
- Kreiter, N.; Pal, A.; Lojewski, X.; Corcia, P.; Naujock, M.; Reinhardt, P.; Sterneckert, J.; Petri, S.; Wegner, F.; Storch, A.; et al. Age-dependent neurodegeneration and organelle transport deficiencies in mutant TDP43 patient-derived neurons are independent of TDP43 aggregation. Neurobiol. Dis. 2018, 115, 167–181. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2010, 27, 431–432. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 2003, 4, 2–27. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar]
- Tcherkezian, J.; Brittis, P.A.; Thomas, F.; Roux, P.P.; Flanagan, J.G. Transmembrane Receptor DCC Associates with Protein Synthesis Machinery and Regulates Translation. Cell 2010, 141, 632–644. [Google Scholar] [CrossRef]
- Nagano, S.; Jinno, J.; Abdelhamid, R.F.; Jin, Y.; Shibata, M.; Watanabe, S.; Hirokawa, S.; Nishizawa, M.; Sakimura, K.; Onodera, O.; et al. TDP-43 transports ribosomal protein mRNA to regulate axonal local translation in neuronal axons. Acta Neuropathol. 2020, 140, 695–713. [Google Scholar] [CrossRef]
- Lu, J.X.; Wang, Y.; Zhang, Y.J.; Shen, M.F.; Li, H.Y.; Yu, Z.Q.; Chen, G. Axonal mRNA localization and local translation in neurodegenerative disease. Neural Regen. Res. 2021, 16, 1950–1957. [Google Scholar]
- Cestra, G.; Rossi, S.; Di Salvio, M.; Cozzolino, M. Control of mRNA translation in ALS proteinopathy. Front. Mol. Neurosci. 2017, 10, 85. [Google Scholar] [CrossRef]
- Kirby, J.; Halligan, E.; Baptista, M.J.; Allen, S.; Heath, P.R.; Holden, H.; Barber, S.C.; Loynes, C.A.; Wood-Allum, C.A.; Lunec, J.; et al. Mutant SOD1 alters the motor neuronal transcriptome: Implications for familial ALS. Brain 2005, 128, 1686–1706. [Google Scholar] [CrossRef]
- Hedlund, E.; Karlsson, M.; Osborn, T.; Ludwig, W.; Isacson, O. Global gene expression profiling of somatic motor neuron populations with different vulnerability identify molecules and pathways of degeneration and protection. Brain 2010, 133, 2313–2330. [Google Scholar] [CrossRef]
- Kumimoto, E.L.; Fore, T.R.; Zhang, B. Transcriptome profiling following neuronal and glial expression of ALS-linked SOD1 in drosophila. G3 Genes Genomes Genet. 2013, 3, 695–708. [Google Scholar] [CrossRef]
- D’Erchia, A.M.; Gallo, A.; Manzari, C.; Raho, S.; Horner, D.S.; Chiara, M.; Valletti, A.; Aiello, I.; Mastropasqua, F.; Ciaccia, L.; et al. Massive transcriptome sequencing of human spinal cord tissues provides new insights into motor neuron degeneration in ALS. Sci. Rep. 2017, 7, 10046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.; Yoon, B.C.; Holt, C.E. Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat. Rev. Neurosci. 2012, 13, 308–324. [Google Scholar] [PubMed]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar]
- Karbowski, M.; Neutzner, A. Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol. 2011, 123, 157–171. [Google Scholar]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Sephton, C.F.; Cenik, C.; Kucukural, A.; Dammer, E.B.; Cenik, B.; Han, Y.H.; Dewey, C.M.; Roth, F.P.; Herz, J.; Peng, J.; et al. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J. Biol. Chem. 2011, 286, 1204–1215. [Google Scholar] [CrossRef]
- Shiga, A.; Ishihara, T.; Miyashita, A.; Kuwabara, M.; Kato, T.; Watanabe, N.; Yamahira, A.; Kondo, C.; Yokoseki, A.; Takahashi, M.; et al. Alteration of POLDIP3 splicing associated with loss of function of TDP-43 in tissues affected with ALS. PLoS ONE 2012, 7, e43120. [Google Scholar] [CrossRef]
- Yang, C.; Wang, H.; Qiao, T.; Yang, B.; Aliaga, L.; Qiu, L.; Tan, W.; Salameh, J.; McKenna-Yasek, D.M.; Smith, T.; et al. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, E1121–E1129. [Google Scholar] [CrossRef]
- Mroczek, S.; Dziembowski, A. U6 RNA biogenesis and disease association. Wiley Interdiscip. Rev. RNA 2013, 4, 581–592. [Google Scholar] [CrossRef]
- Tsuiji, H.; Iguchi, Y.; Furuya, A.; Kataoka, A.; Hatsuta, H.; Atsuta, N.; Tanaka, F.; Hashizume, Y.; Akatsu, H.; Murayama, S.; et al. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol. Med. 2013, 5, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, T.; Ariizumi, Y.; Shiga, A.; Kato, T.; Tan, C.F.; Sato, T.; Miki, Y.; Yokoo, M.; Fujino, T.; Koyama, A.; et al. Decreased number of gemini of coiled bodies and U12 snRNA level in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 4136–4147. [Google Scholar] [CrossRef] [PubMed]
- Yahara, M.; Kitamura, A.; Kinjo, M. U6 snRNA expression prevents toxicity in TDP-43-knockdown cells. PLoS ONE 2017, 12, e0187813. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Curle, A.J.; Haider, A.M.; Balmus, G. The role of DNA damage response in amyotrophic lateral sclerosis. Essays Biochem. 2020, 64, 847–861. [Google Scholar]
- Wang, H.; Dharmalingam, P.; Vasquez, V.; Mitra, J.; Boldogh, I.; Rao, K.S.; Kent, T.A.; Mitra, S.; Hegde, M.L. Chronic oxidative damage together with genome repair deficiency in the neurons is a double whammy for neurodegeneration: Is damage response signaling a potential therapeutic target? Mech. Ageing Dev. 2017, 161, 163–176. [Google Scholar]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta- Mol. Basis Dis. 2006, 1762, 1051–1067. [Google Scholar]
- Hill, S.J.; Mordes, D.A.; Cameron, L.A.; Neuberg, D.S.; Landini, S.; Eggan, K.; Livingston, D.M. Two familial ALS proteins function in prevention/repair of transcription-associated DNA damage. Proc. Natl. Acad. Sci. USA 2016, 113, E7701–E7709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Paul Taylor, J.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef]
- Guerrero, E.N.; Mitra, J.; Wang, H.; Rangaswamy, S.; Hegde, P.M.; Basu, P.; Rao, K.S.; Hegde, M.L. Amyotrophic lateral sclerosis-associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage-mediated neuronal apoptosis. Hum. Mol. Genet. 2019, 28, 2459–2476. [Google Scholar] [CrossRef]
- Pal, A.; Glaß, H.; Naumann, M.; Kreiter, N.; Japtok, J.; Sczech, R.; Hermann, A. High content organelle trafficking enables disease state profiling as powerful tool for disease modelling. Sci. Data 2018, 5, 180241. [Google Scholar] [CrossRef]
- Lv, J.; Jiang, S.; Yang, Z.; Hu, W.; Wang, Z.; Li, T.; Yang, Y. PGC-1α sparks the fire of neuroprotection against neurodegenerative disorders. Ageing Res. Rev. 2018, 44, 8–21. [Google Scholar] [PubMed]
- Prashantha Kumar, B.R.; Kumar, A.P.; Jose, J.A.; Prabitha, P.; Yuvaraj, S.; Chipurupalli, S.; Jeyarani, V.; Manisha, C.; Banerjee, S.; Jeyabalan, J.B.; et al. Minutes of PPAR-γ agonism and neuroprotection. Neurochem. Int. 2020, 140, 104814. [Google Scholar]
- Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Beal, M.F. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2005, 191, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Joardar, A.; Menzl, J.; Podolsky, T.C.; Manzo, E.; Estes, P.S.; Ashford, S.; Zarnescu, D.C. PPAR gamma activation is neuroprotective in a Drosophila model of ALS based on TDP-43. Hum. Mol. Genet. 2014, 24, 1741–1754. [Google Scholar] [CrossRef]
- Rodríguez-Cueto, C.; Santos-García, I.; García-Toscano, L.; Espejo-Porras, F.; Bellido, M.; Fernández-Ruiz, J.; Muñoz, E.; de Lago, E. Neuroprotective effects of the cannabigerol quinone derivative VCE-003.2 in SOD1G93A transgenic mice, an experimental model of amyotrophic lateral sclerosis. Biochem. Pharmacol. 2018, 157, 217–226. [Google Scholar] [CrossRef]
- Rahman, M.R.; Islam, T.; Turanli, B.; Zaman, T.; Faruquee, H.M.; Rahman, M.M.; Mollah, M.N.H.; Nanda, R.K.; Arga, K.Y.; Gov, E.; et al. Network-based approach to identify molecular signatures and therapeutic agents in Alzheimer’s disease. Comput. Biol. Chem. 2019, 78, 431–439. [Google Scholar] [CrossRef]
- Rahman, M.R.; Islam, T.; Zaman, T.; Shahjaman, M.; Karim, M.R.; Huq, F.; Quinn, J.M.W.; Holsinger, R.M.D.; Gov, E.; Moni, M.A. Identification of molecular signatures and pathways to identify novel therapeutic targets in Alzheimer’s disease: Insights from a systems biomedicine perspective. Genomics 2020, 112, 1290–1299. [Google Scholar] [CrossRef]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a lrrk2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Yu, T.; Wu, Y.N.; Roy, M.; Kim, J.; Lee, C. An expectation-maximization algorithm for probabilistic reconstructions of full-length isoforms from splice graphs. Nucleic Acids Res. 2006, 34, 3150–3160. [Google Scholar] [CrossRef]
- Casula, M.; Iyer, A.M.; Spliet, W.G.M.; Anink, J.J.; Steentjes, K.; Sta, M.; Troost, D.; Aronica, E. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience 2011, 179, 233–243. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [PubMed]
- Castro-Mondragon, J.A.; Riudavets-Puig, R.; Rauluseviciute, I.; Berhanu Lemma, R.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Perez, N.M.; et al. JASPAR 2022: The 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2021, 50, D165–D173. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Sex | Age at Biopsy (Years) | Mutation | Family History | Age of Disease Onset | ALS Type | Clinical Characteristics | Disease Duration (Months) | Clones | DIV |
|---|---|---|---|---|---|---|---|---|---|---|
| Controls | ||||||||||
| Female | 53 | - | - | - | - | - | 1 | 30 | ||
| Male | 60 | - | - | - | - | - | 1 | 30 | ||
| Female | 45 | - | - | - | - | - | 1 | 30 | ||
| TDP43-ALS | ||||||||||
| Female | 85 | p.S393L | Pos. for ALS | 85 | Bulbar | Progressive anarthria, LMND, no clinical symptoms of FTD | 48 | 1 | 30 | |
| Male | 46 | p.G294V | neg for ALS | 37 | Spinal | Early onset ALS (37 years), monomelic right leg amyotrophy, no clinical symptoms of FTD | >120 (alive) | 2 | 30 | |
| SOD1-ALS | ||||||||||
| Male | 59 | p.R115G | Pos. for ALS (Mother and Brother) | n.d | Spinal | n.d | n.d | 1 | 30 | |
| Female | 46 | p.D90A | Pos. for ALS (Brother) | 41 | Spinal | Slowly progressive classical spinal ALS, no cognitive impairment | 204 | 1 | 30 |
| Cellular Model | TFs | Target Genes | Count |
|---|---|---|---|
| SOD1-ALS | PPARG | RPL6, RPLP0, RPL3, RPS24, RPL13, RPL10A | 6 |
| GATA2 | RPL11, RPS8, RPL6, RPL13, RPL10A, RPL5 | 6 | |
| FOXF2 | RPL11, RPL6, RPLP0, RPS24, RPL13, RPL5 | 6 | |
| FOXC1 | RPS6, RPL6, RPL3, RPS24, RPL13 | 5 | |
| TDP43-ALS | FOXC1 | TAF4, TAF7, LSM7, TAF1, TAF10, TAF11, TAF5 | 7 |
| GATA2 | TAF6, TAF7, LSM7, TAF1, TAF10 | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dash, B.P.; Freischmidt, A.; Weishaupt, J.H.; Hermann, A. Downstream Effects of Mutations in SOD1 and TARDBP Converge on Gene Expression Impairment in Patient-Derived Motor Neurons. Int. J. Mol. Sci. 2022, 23, 9652. https://doi.org/10.3390/ijms23179652
Dash BP, Freischmidt A, Weishaupt JH, Hermann A. Downstream Effects of Mutations in SOD1 and TARDBP Converge on Gene Expression Impairment in Patient-Derived Motor Neurons. International Journal of Molecular Sciences. 2022; 23(17):9652. https://doi.org/10.3390/ijms23179652
Chicago/Turabian StyleDash, Banaja P., Axel Freischmidt, Jochen H. Weishaupt, and Andreas Hermann. 2022. "Downstream Effects of Mutations in SOD1 and TARDBP Converge on Gene Expression Impairment in Patient-Derived Motor Neurons" International Journal of Molecular Sciences 23, no. 17: 9652. https://doi.org/10.3390/ijms23179652
APA StyleDash, B. P., Freischmidt, A., Weishaupt, J. H., & Hermann, A. (2022). Downstream Effects of Mutations in SOD1 and TARDBP Converge on Gene Expression Impairment in Patient-Derived Motor Neurons. International Journal of Molecular Sciences, 23(17), 9652. https://doi.org/10.3390/ijms23179652