
Uncovering Oncogenic Mechanisms of Tumor Suppressor Genes in Breast Cancer Multi-Omics Data


Abstract
:1. Introduction
2. Results
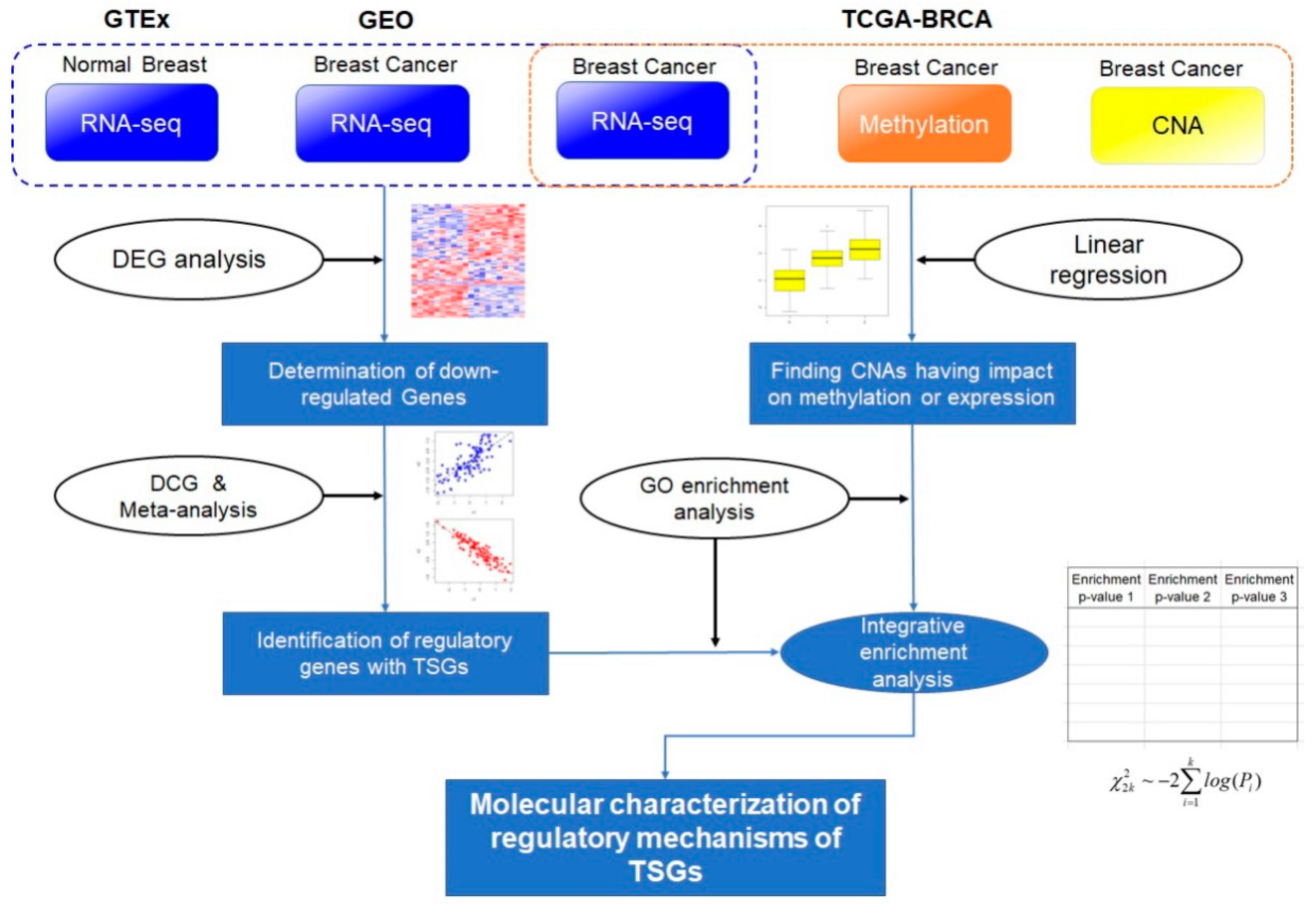
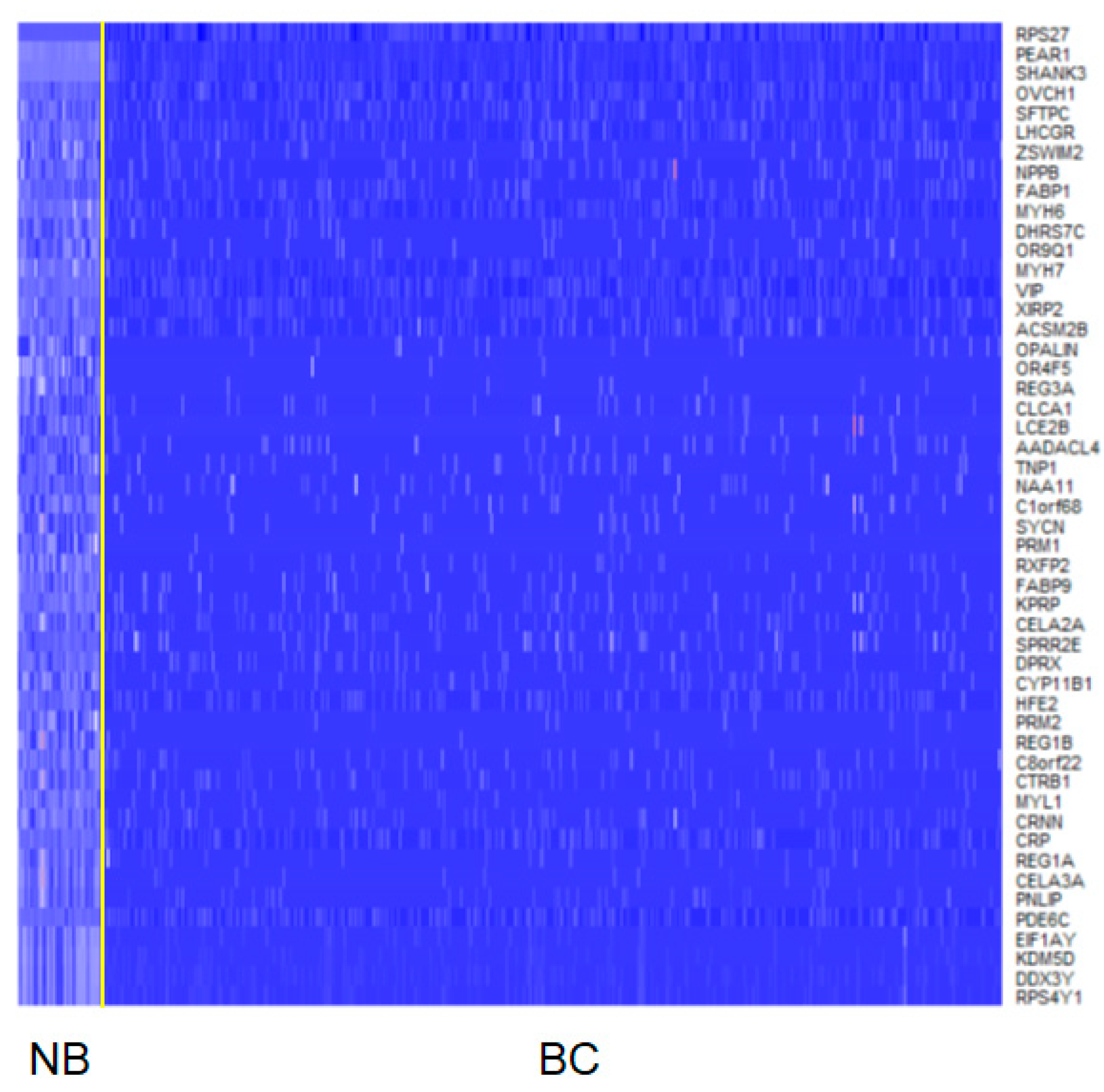
2.1. Data Preprocessing and Differentially Expressed Gene Analysis
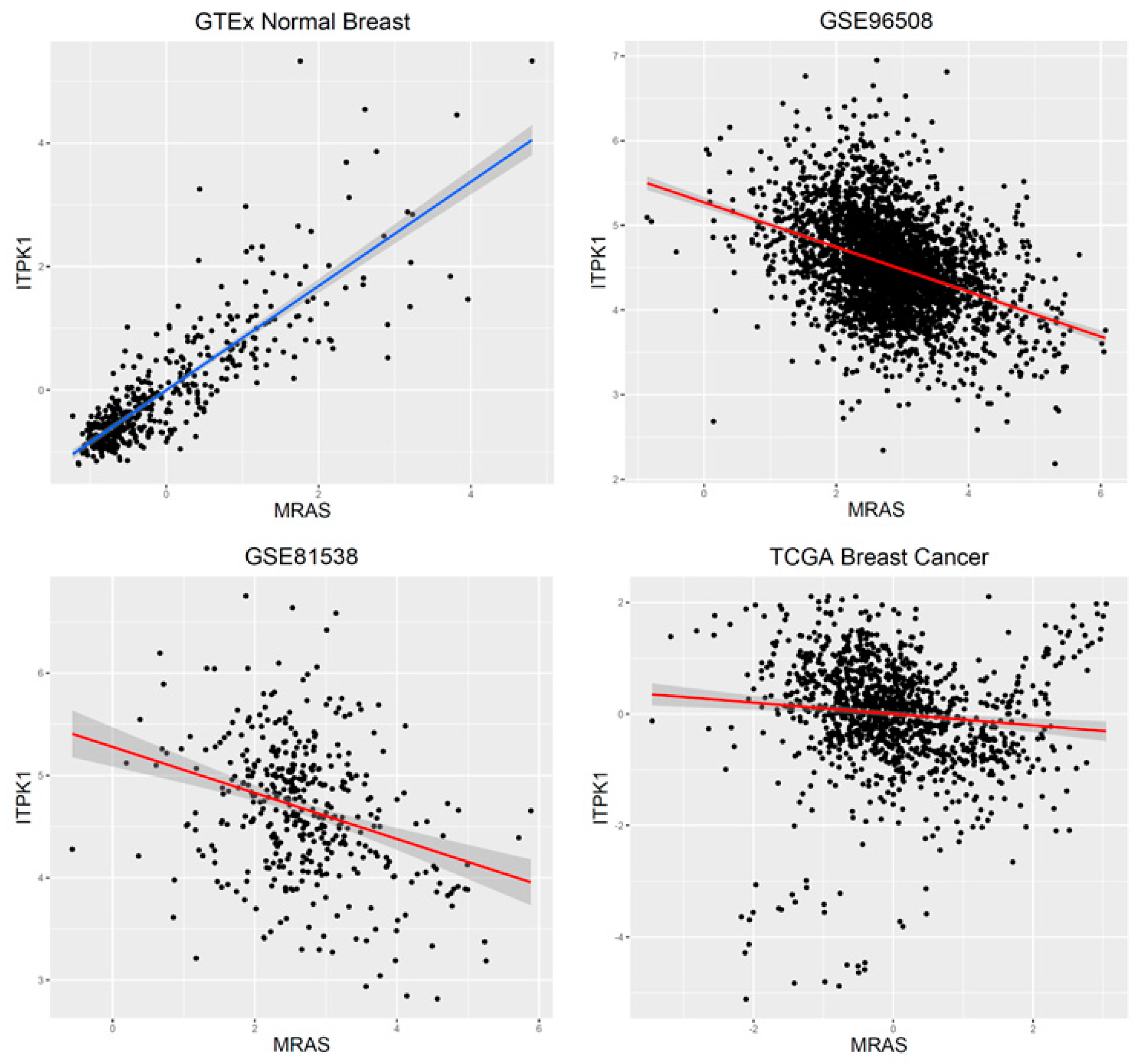
2.2. Differential Coexpression Reveals Genetic Regulatory Network of Tsgs in Breast Cancer
2.3. Identification of Copy Number Alterations Having an Impact on Methylations and Gene Expressions
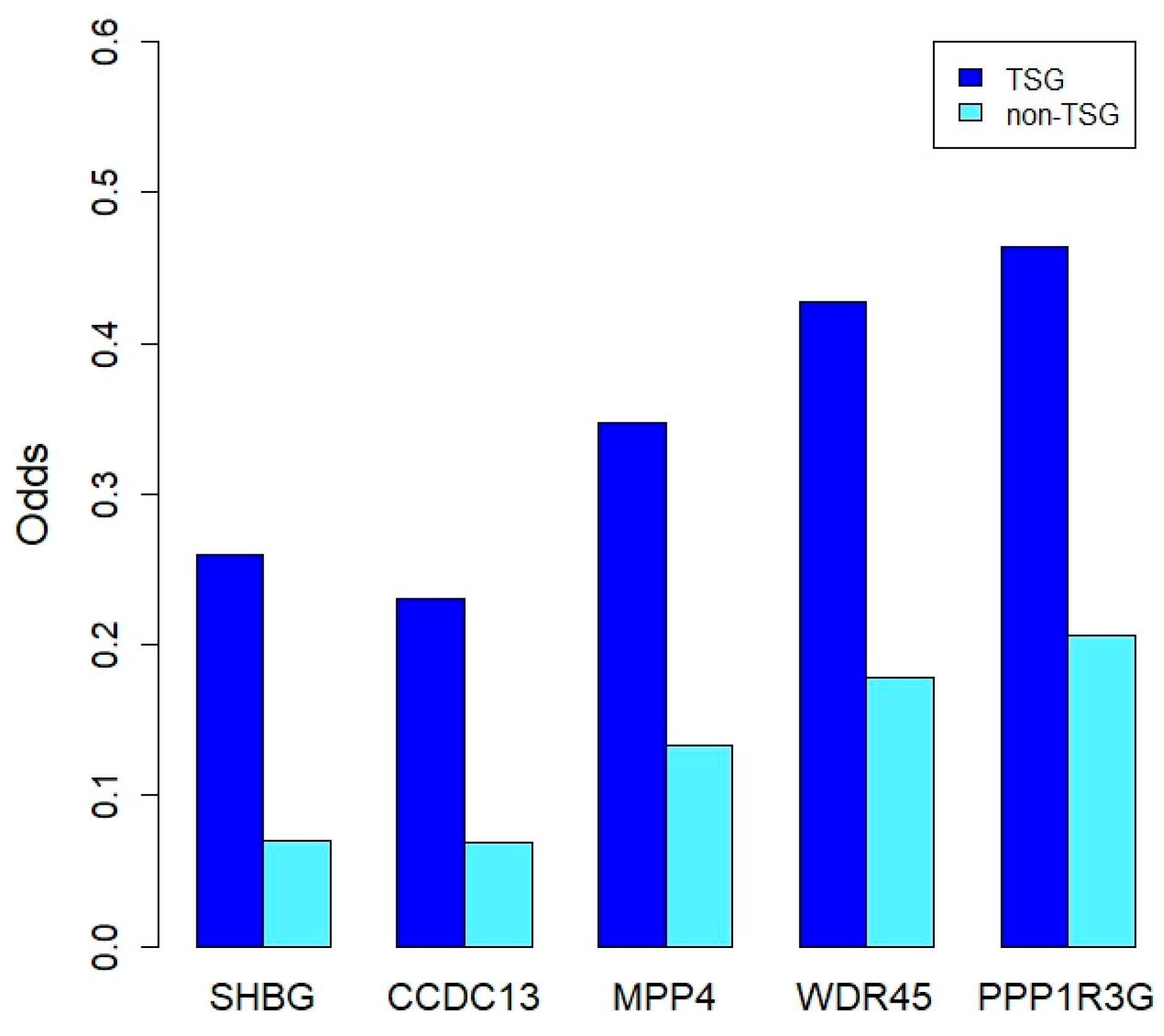
2.4. Prediction of TSGs Using Results of DCG and Multi-Omics Data Analysis
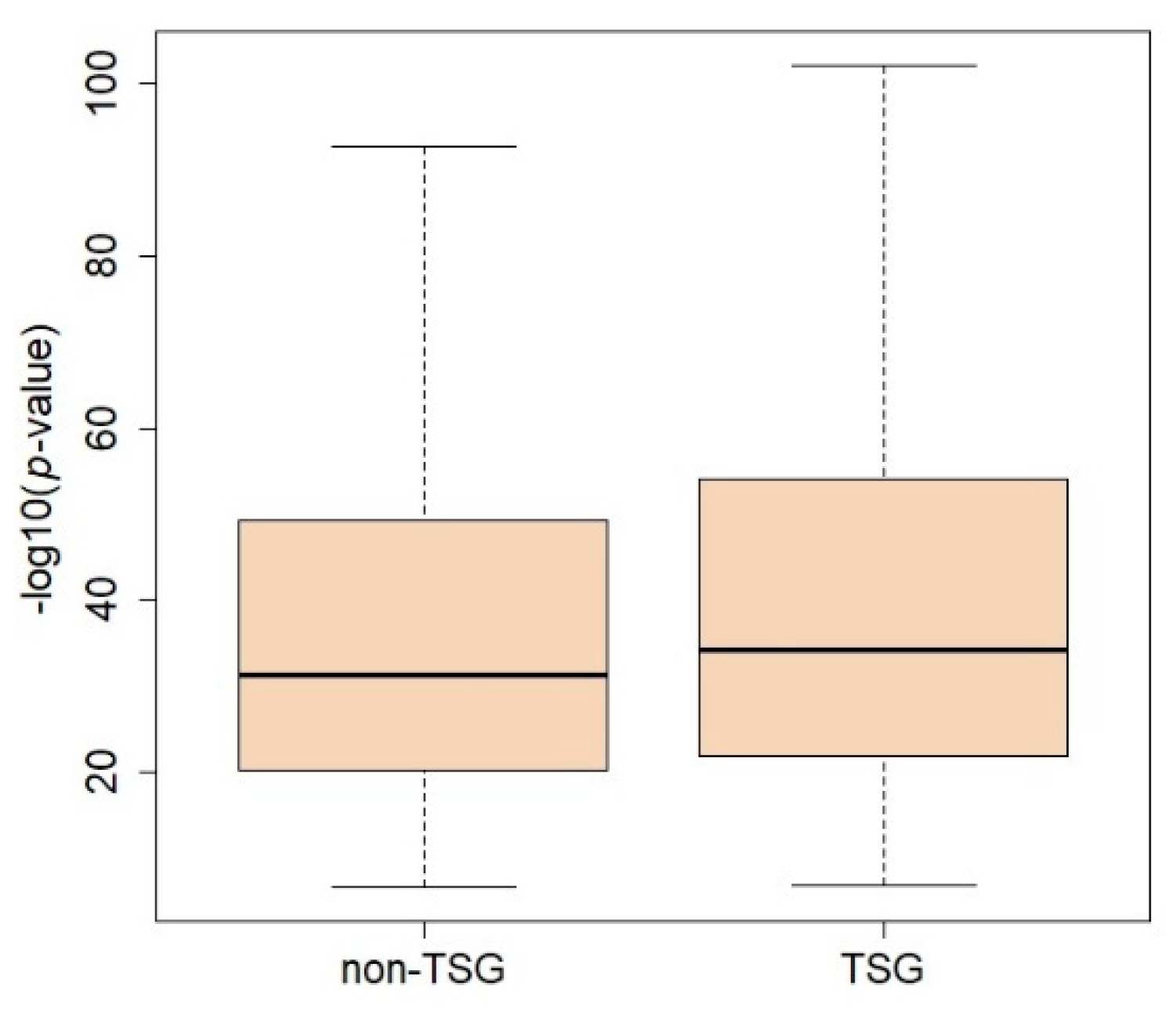
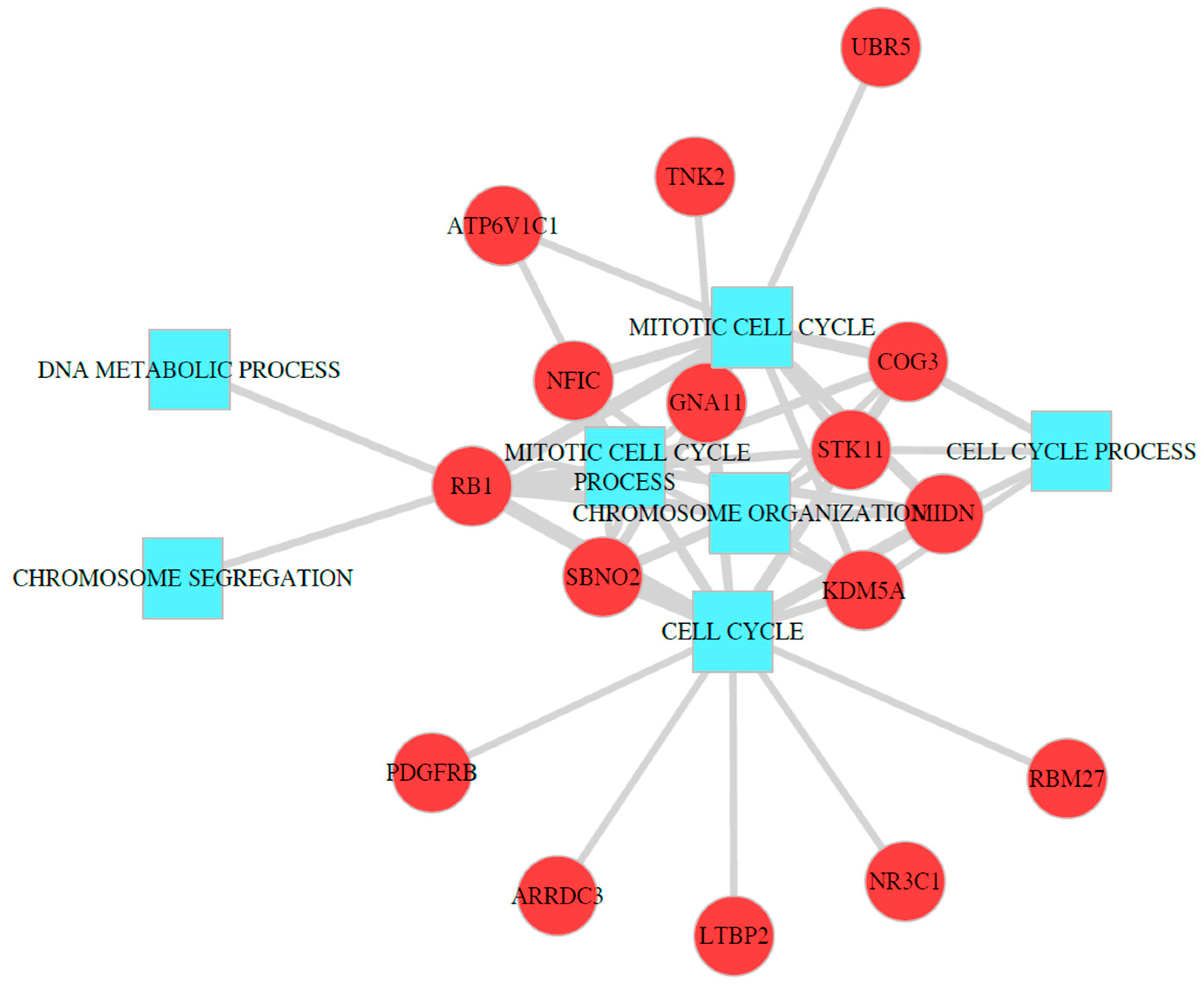
2.5. Identification of Molecular Mechanism of TSGs Using Functional Enrichment Analysis
3. Discussion
4. Materials and Methods
4.1. Breast Cancer Multi-Omics Data
4.2. Identification of Differentially Expressed Genes and Co-Expressed Genes
4.3. Meta-Analysis Using p-Value Combination
4.4. Functional Enrichment Analysis with Gene Ontology and Pathway Information
4.5. Multi-Omics Data Analysis with Linear Model
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Sherr, C.J. Principles of Tumor Suppression. Cell 2004, 116, 235–246. [Google Scholar] [CrossRef]
- Wang, L.-H.; Wu, C.-F.; Rajasekaran, N.; Shin, Y.K. Loss of Tumor Suppressor Gene Function in Human Cancer: An Overview. Cell. Physiol. Biochem. 2018, 51, 2647–2693. [Google Scholar] [CrossRef] [PubMed]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef]
- Kitagawa, K.; Kotake, Y.; Kitagawa, M. Ubiquitin-mediated control of oncogene and tumor suppressor gene products. Cancer Sci. 2009, 100, 1374–1381. [Google Scholar] [CrossRef]
- Krug, U.; Ganser, A.; Koeffler, H.P. Tumor suppressor genes in normal and malignant hematopoiesis. Oncogene 2002, 21, 3475–3495. [Google Scholar] [CrossRef]
- Payne, S.R.; Kemp, C.J. Tumor suppressor genetics. Carcinogenesis 2005, 26, 2031–2045. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Dupont, J.; Yakar, S.; Karas, M.; LeRoith, D. PTEN inhibits cell proliferation and induces apoptosis by downregulating cell surface IGF-IR expression in prostate cancer cells. Oncogene 2004, 23, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-X.; Cao, L.-Y.; Chen, X.; Xiao, J.; Zou, Y.; Chen, Q. PTEN Inhibits Cell Proliferation, Promotes Cell Apoptosis, and Induces Cell Cycle Arrest via Downregulating the PI3K/AKT/hTERT Pathway in Lung Adenocarcinoma A549 Cells. BioMed Res. Int. 2016, 2016, 2476842. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yang, J. Functional Mechanisms for Human Tumor Suppressors. J. Cancer 2010, 1, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Benchabane, H.; Ahmed, Y. The Adenomatous Polyposis Coli Tumor Suppressor and Wnt Signaling in the Regulation of Apoptosis. Adv. Exp. Med. Biol. 2009, 656, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef]
- Su, C.-W.; Lin, C.-W.; Yang, W.-E.; Yang, S.-F. TIMP-3 as a therapeutic target for cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919864247. [Google Scholar] [CrossRef]
- Khan, I.; Steeg, P.S. Metastasis suppressors: Functional pathways. Lab. Investig. 2018, 98, 198–210. [Google Scholar] [CrossRef]
- Gonzalez-Perez, A.; Lopez-Bigas, N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012, 40, e169. [Google Scholar] [CrossRef]
- Dees, N.D.; Zhang, Q.; Kandoth, C.; Wendl, M.C.; Schierding, W.; Koboldt, D.C.; Mooney, T.B.; Callaway, M.B.; Dooling, D.; Mardis, E.R.; et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012, 22, 1589–1598. [Google Scholar] [CrossRef]
- Dietlein, F.; Weghorn, D.; Taylor-Weiner, A.; Richters, A.; Reardon, B.; Liu, D.; Lander, E.S.; Van Allen, E.M.; Sunyaev, S.R. Identification of cancer driver genes based on nucleotide context. Nat. Genet. 2020, 52, 208–218. [Google Scholar] [CrossRef]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. Cumulative Haploinsufficiency and Triplosensitivity Drive Aneuploidy Patterns and Shape the Cancer Genome. Cell 2013, 155, 948–962. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Bader, G.D. Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol. Syst. Biol. 2013, 9, 637. [Google Scholar] [CrossRef] [PubMed]
- Mularoni, L.; Sabarinathan, R.; Deu-Pons, J.; Gonzalez-Perez, A.; López-Bigas, N. OncodriveFML: A general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 2016, 17, 128. [Google Scholar] [CrossRef]
- Wang, Z.; Ng, K.-S.; Chen, T.; Kim, T.-B.; Wang, F.; Shaw, K.; Scott, K.L.; Meric-Bernstam, F.; Mills, G.B.; Chen, K. Cancer driver mutation prediction through Bayesian integration of multi-omic data. PLoS ONE 2018, 13, e0196939. [Google Scholar] [CrossRef]
- Nguyen, Q.-H.; Le, D.-H. Improving existing analysis pipeline to identify and analyze cancer driver genes using multi-omics data. Sci. Rep. 2020, 10, 20521. [Google Scholar] [CrossRef] [PubMed]
- Andrysik, Z.; Bender, H.; Galbraith, M.D.; Espinosa, J.M. Multi-omics analysis reveals contextual tumor suppressive and oncogenic gene modules within the acute hypoxic response. Nat. Commun. 2021, 12, 1375. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas (TCGA). Available online: https://www.cancer.gov/tcga (accessed on 15 January 2022).
- Halaburkova, A.; Cahais, V.; Novoloaca, A.; da Silva Araujo Araujo, M.G.; Khoueiry, R.; Ghantous, A.; Herceg, Z. Pan-cancer multi-omics analysis and orthogonal experimental assessment of epigenetic driver genes. Genome Res. 2020, 30, 1517–1532. [Google Scholar] [CrossRef]
- Zhao, M.; Zhao, Z. Concordance of copy number loss and down-regulation of tumor suppressor genes: A pan-cancer study. BMC Genom. 2016, 17 (Suppl. 7), 532. [Google Scholar] [CrossRef]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef]
- Lyu, J.; Li, J.J.; Su, J.; Peng, F.; Chen, Y.E.; Ge, X.; Li, W. DORGE: Discovery of Oncogenes and tumoR suppressor genes using Genetic and Epigenetic features. Sci. Adv. 2020, 6, eaba6784. [Google Scholar] [CrossRef]
- Kumar, N.; Prasad, P.; Jash, E.; Jayasundar, S.; Singh, I.; Alam, N.; Murmu, N.; Somashekhar, S.P.; Goldman, A.; Sehrawat, S. cAMP regulated EPAC1 supports microvascular density, angiogenic and metastatic properties in a model of triple negative breast cancer. Carcinogenesis 2018, 39, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.R.; Liu, S.; Xiang, M.; Nicolais, M.; Hatzi, K.; Giannopoulou, E.; Elemento, O.; Cerchietti, L.; Melnick, A.; Frank, D.A. The transcriptional modulator BCL6 as a molecular target for breast cancer therapy. Oncogene 2015, 34, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Arshad, Z.; McDonald, J.F. Changes in gene-gene interactions associated with cancer onset and progression are largely independent of changes in gene expression. iScience 2021, 24, 103522. [Google Scholar] [CrossRef] [PubMed]
- Poole, W.; Gibbs, D.L.; Shmulevich, I.; Bernard, B.; Knijnenburg, T.A. Combining dependent P-values with an empirical adaptation of Brown’s method. Bioinformatics 2016, 32, i430–i436. [Google Scholar] [CrossRef]
- cBioPortal. Available online: https://www.cbioportal.org/ (accessed on 20 January 2022).
- Tong, X.; Feng, Y.; Li, J.J. Neyman-Pearson classification algorithms and NP receiver operating characteristics. Sci. Adv. 2018, 4, eaao1659. [Google Scholar] [CrossRef]
- Troyanskaya, O.G.; Cantor, M.; Sherlock, G.; Brown, P.O.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing value estimation methods for DNA microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar] [CrossRef]
- Gialeli, C.; Tuysuz, E.C.; Staaf, J.; Guleed, S.; Paciorek, V.; Mörgelin, M.; Papadakos, K.S.; Blom, A.M. Complement inhibitor CSMD1 modulates epidermal growth factor receptor oncogenic signaling and sensitizes breast cancer cells to chemotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 258. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; The ENCODE Project Consortium; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Anglani, R.; Creanza, T.M.; Liuzzi, V.C.; Piepoli, A.; Panza, A.; Andriulli, A.; Ancona, N. Loss of Connectivity in Cancer Co-Expression Networks. PLoS ONE 2014, 9, e87075. [Google Scholar] [CrossRef]
- Belin, S.; Beghin, A.; Solano-Gonzàlez, E.; Bezin, L.; Brunet-Manquat, S.; Textoris, J.; Prats, A.-C.; Mertani, H.C.; Dumontet, C.; Diaz, J.-J. Dysregulation of Ribosome Biogenesis and Translational Capacity Is Associated with Tumor Progression of Human Breast Cancer Cells. PLoS ONE 2009, 4, e7147. [Google Scholar] [CrossRef] [PubMed]
- Harold, C.; Buhagiar, A.; Cheng, Y.; Baserga, S. Ribosomal RNA Transcription Regulation in Breast Cancer. Genes 2021, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- Bastide, A.; David, A. The ribosome, (slow) beating heart of cancer (stem) cell. Oncogenesis 2018, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Janin, M.; Coll-SanMartin, L.; Esteller, M. Disruption of the RNA modifications that target the ribosome translation machinery in human cancer. Mol. Cancer 2020, 19, 70. [Google Scholar] [CrossRef]
- Goudarzi, K.M.; Lindström, M.S. Role of ribosomal protein mutations in tumor development (Review). Int. J. Oncol. 2016, 48, 1313–1324. [Google Scholar] [CrossRef]
- Li, X.; Gao, Y.; Li, J.; Zhang, K.; Han, J.; Li, W.; Hao, Q.; Zhang, W.; Wang, S.; Zeng, C.; et al. FOXP3 inhibits angiogenesis by downregulating VEGF in breast cancer. Cell Death Dis. 2018, 9, 744. [Google Scholar] [CrossRef]
- Zhao, A.; Li, D.; Mao, X.; Yang, M.; Deng, W.; Hu, W.; Chen, C.; Yang, G.; Li, L. GNG2 acts as a tumor suppressor in breast cancer through stimulating MRAS signaling. Cell Death Dis. 2022, 13, 260. [Google Scholar] [CrossRef]
- Humphries, B.; Wang, Z.; Yang, C. Rho GTPases: Big Players in Breast Cancer Initiation, Metastasis and Therapeutic Responses. Cells 2020, 9, 2167. [Google Scholar] [CrossRef]
- Ray, S.K.; Mukherjee, S. Altered Expression of TRIM Proteins-Inimical Outcome and Inimitable Oncogenic Function in Breast Cancer with Diverse Carcinogenic Hallmarks. Curr. Mol. Med. 2022, 23, 44–53. [Google Scholar] [CrossRef]
- Zhang, Y.; Dho, S.E.; Othman, K.; Simpson, C.D.; Lapierre, J.; Bondoc, A.; McGlade, C.J. Numb exon 9 inclusion regulates Integrinbeta5 surface expression and promotes breast cancer metastasis. Oncogene 2022, 41, 2079–2094. [Google Scholar] [CrossRef]
- Chen, J.; Sun, J.; Wang, Q.; Du, Y.; Cheng, J.; Yi, J.; Xie, B.; Jin, S.; Chen, G.; Wang, L.; et al. Systemic Deficiency of PTEN Accelerates Breast Cancer Growth and Metastasis. Front. Oncol. 2022, 12, 825484. [Google Scholar] [CrossRef]
- Shan, W.; Jiang, Y.; Yu, H.; Huang, Q.; Liu, L.; Guo, X.; Li, L.; Mi, Q.; Zhang, K.; Yang, Z. HDAC2 overexpression correlates with aggressive clinicopathological features and DNA-damage response pathway of breast cancer. Am. J. Cancer Res. 2017, 7, 1213–1226. [Google Scholar] [PubMed]
- Zhao, H.; Yu, Z.; Zhao, L.; He, M.; Ren, J.; Wu, H.; Chen, Q.; Yao, W.; Wei, M. HDAC2 overexpression is a poor prognostic factor of breast cancer patients with increased multidrug resistance-associated protein expression who received anthracyclines therapy. Jpn. J. Clin. Oncol. 2016, 46, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Young, L.C.; Rodriguez-Viciana, P. MRAS: A Close but Understudied Member of the RAS Family. Cold Spring Harb. Perspect. Med. 2018, 8, a033621. [Google Scholar] [CrossRef] [PubMed]
- Saiardi, A.; Cockcroft, S. Human ITPK1: A Reversible Inositol Phosphate Kinase/Phosphatase That Links Receptor-Dependent Phospholipase C to Ca 2+-Activated Chloride Channels. Sci. Signal. 2008, 1, pe5. [Google Scholar] [CrossRef] [PubMed]
- Desfougères, Y.; Wilson, M.S.C.; Laha, D.; Miller, G.J.; Saiardi, A. ITPK1 mediates the lipid-independent synthesis of inositol phosphates controlled by metabolism. Proc. Natl. Acad. Sci. USA 2019, 116, 24551–24561. [Google Scholar] [CrossRef]
- Tantivejkul, K.; Vucenik, I.; Eiseman, J.; Shamsuddin, A.M. Inositol hexaphosphate (IP6) enhances the anti-proliferative effects of adriamycin and tamoxifen in breast cancer. Breast Cancer Res. Treat. 2003, 79, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ke, Q.; Shao, Y.; Zhu, G.; Li, Y.; Geng, N.; Jin, F.; Li, F. GATA1 induces epithelial-mesenchymal transition in breast cancer cells through PAK5 oncogenic signaling. Oncotarget 2015, 6, 4345–4356. [Google Scholar] [CrossRef]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Suvorova, E.S.; Kurten, R.C.; Lupashin, V.V. Identification of a Human Orthologue of Sec34p as a Component of the cis-Golgi Vesicle Tethering Machinery. J. Biol. Chem. 2001, 276, 22810–22818. [Google Scholar] [CrossRef] [Green Version]
- The Genotype-Tissue Expression (GTEx) Project. Available online: https://gtexportal.org (accessed on 30 January 2022).
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, J.; Domingues, D.; Martins Lopes, F. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef]
- Cui, X.; Churchill, G.A. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 2003, 4, 210. [Google Scholar] [CrossRef]
- Leek, J.T.; Scharpf, R.B.; Bravo, H.C.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef] [Green Version]
- Gene Set Enrichment Analysis (GSEA). Available online: https://www.gsea-msigdb.org (accessed on 10 February 2022).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene1 | Gene2 | GTEx | GSE90538 | GSE81555 | TCGA | p-Value |
|---|---|---|---|---|---|---|
| MRAS | ITPK1 | 0.84 | –0.38 | –0.31 | –0.10 | 1.09 × 10–298 |
| ESYT3 | IFT140 | 0.80 | –0.33 | –0.39 | –0.35 | 1.80 × 10–290 |
| CDK5RAP3 | ZNF667 | 0.87 | –0.18 | –0.11 | –0.12 | 7.42 × 10–287 |
| PLA2R1 | PDE7A | 0.86 | –0.20 | –0.21 | –0.13 | 8.47 × 10–287 |
| ESYT3 | CMYA5 | 0.84 | –0.27 | –0.34 | –0.10 | 1.06 × 10–283 |
| PEX19 | LDHB | 0.83 | –0.39 | –0.30 | –0.05 | 7.00 × 10–282 |
| ACACB | ABCD1 | 0.84 | –0.32 | –0.19 | –0.10 | 4.63 × 10–278 |
| TTLL4 | JMJD7 | 0.80 | –0.33 | –0.47 | –0.22 | 3.49 × 10–276 |
| ESYT3 | DTX3 | 0.83 | –0.25 | –0.32 | –0.23 | 3.40 × 10–275 |
| SFT2D2 | RAD50 | 0.83 | –0.28 | –0.32 | –0.12 | 3.36 × 10–272 |
| Gene Name | GO BP | p-Value |
|---|---|---|
| CSMD1 | mRNA PROCESSING | 8.96 × 10–36 |
| GATA1 | mRNA METABOLIC PROCESS | 6.38 × 10–34 |
| CSMD1 | mRNA METABOLIC PROCESS | 1.07 × 10–33 |
| GATA1 | mRNA PROCESSING | 6.33 × 10–32 |
| BNC2 | mRNA METABOLIC PROCESS | 1.45 × 10–31 |
| BNC2 | mRNA PROCESSING | 2.04 × 10–30 |
| BNC2 | RNA PROCESSING | 6.50 × 10–29 |
| GATA1 | RNA PROCESSING | 2.40 × 10–28 |
| BNC2 | RNA SPLICING | 9.30 × 10–28 |
| PTPRD | mRNA METABOLIC PROCESS | 1.96 × 10–27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, S.B. Uncovering Oncogenic Mechanisms of Tumor Suppressor Genes in Breast Cancer Multi-Omics Data. Int. J. Mol. Sci. 2022, 23, 9624. https://doi.org/10.3390/ijms23179624
Cho SB. Uncovering Oncogenic Mechanisms of Tumor Suppressor Genes in Breast Cancer Multi-Omics Data. International Journal of Molecular Sciences. 2022; 23(17):9624. https://doi.org/10.3390/ijms23179624
Chicago/Turabian StyleCho, Seong Beom. 2022. "Uncovering Oncogenic Mechanisms of Tumor Suppressor Genes in Breast Cancer Multi-Omics Data" International Journal of Molecular Sciences 23, no. 17: 9624. https://doi.org/10.3390/ijms23179624
APA StyleCho, S. B. (2022). Uncovering Oncogenic Mechanisms of Tumor Suppressor Genes in Breast Cancer Multi-Omics Data. International Journal of Molecular Sciences, 23(17), 9624. https://doi.org/10.3390/ijms23179624