
Characterization of Philadelphia-like Pre-B Acute Lymphoblastic Leukemia: Experiences in Mexican Pediatric Patients

 , , ,
, , ,  , ,
, ,  and
and 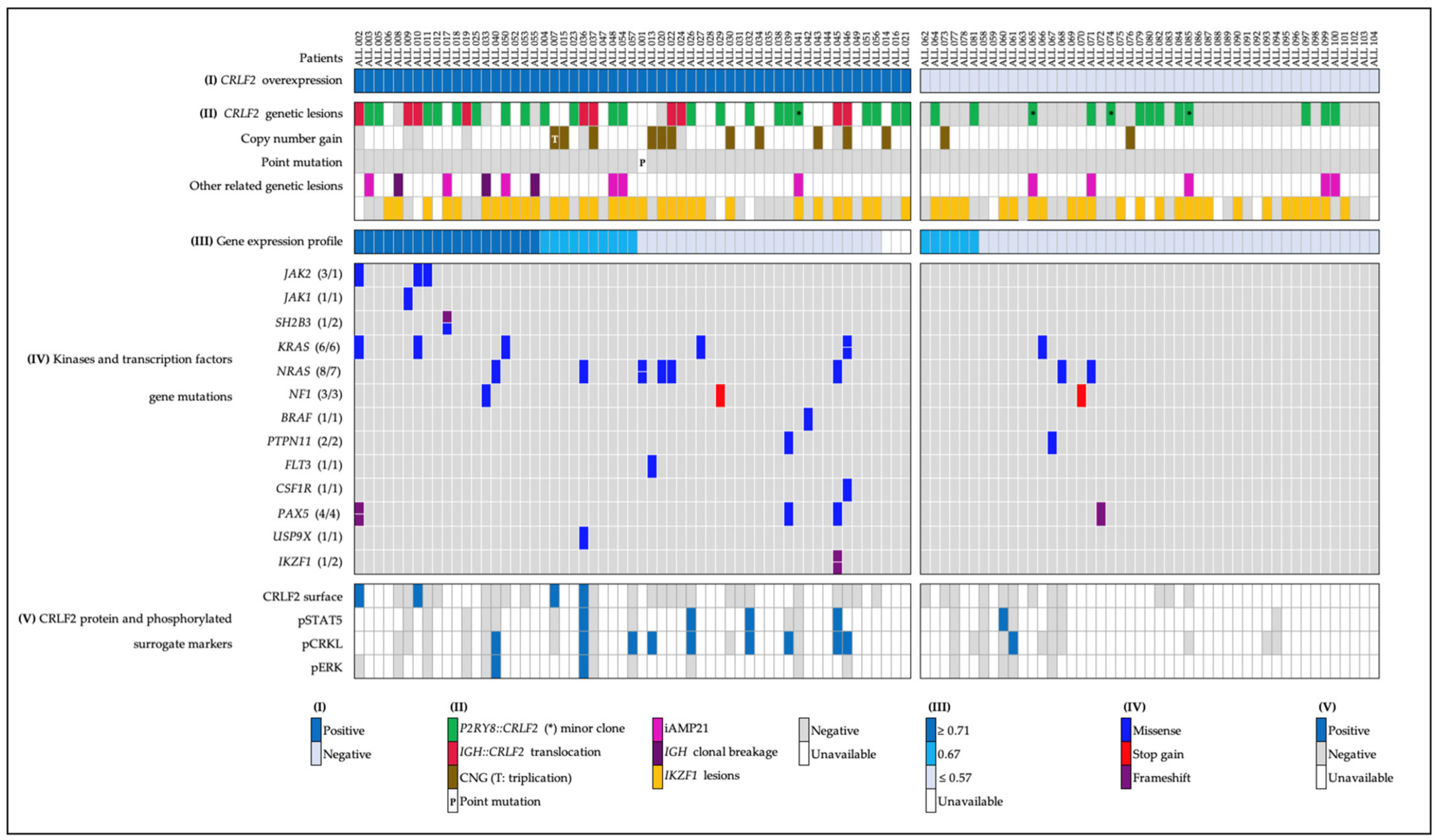{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
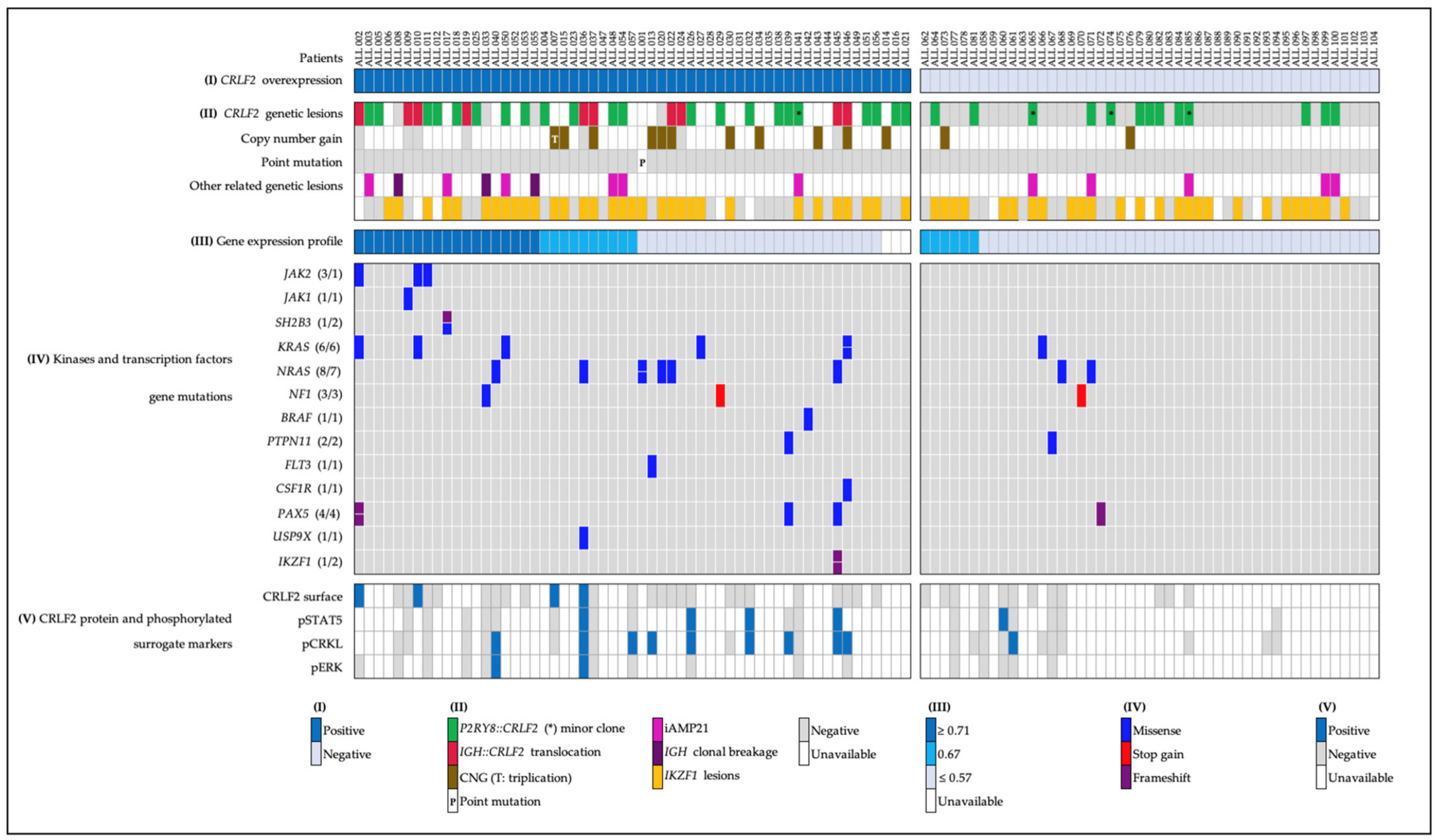
2.1. CRLF2 Expression and Genetic Lesions
- (a)
- Forty-five patients with CRLF2 rearrangements, thirty-five of them showed the P2RY8::CRLF2 deletion, and ten cases with IGH::CRLF2 translocation. Three of them showed two different cell clones, one with extra copies of the IGH and CRLF2 loci as a result of hyperdiploidy, and one presented the coexistence of hyperdiploidy and IGH::CRLF2 translocation (Figure 2A). Unfortunately, twenty-one patients with high expressions of CRLF2, including hyperdiploid cases, were not analyzed for IGH::CRLF2 since cell samples were not available.
- (b)
- Nine patients presented increased copy numbers of CRLF2 as a result of hyperdiploidy involving sex chromosomes, and the array-CGH analysis showed one case with a CRLF2 triplication without trisomy of sexual chromosomes (Figure 2B).
- (c)
- The CRLF2 c.695T > G p.F23 activating point mutation was detected in one patient (Figure 2C).

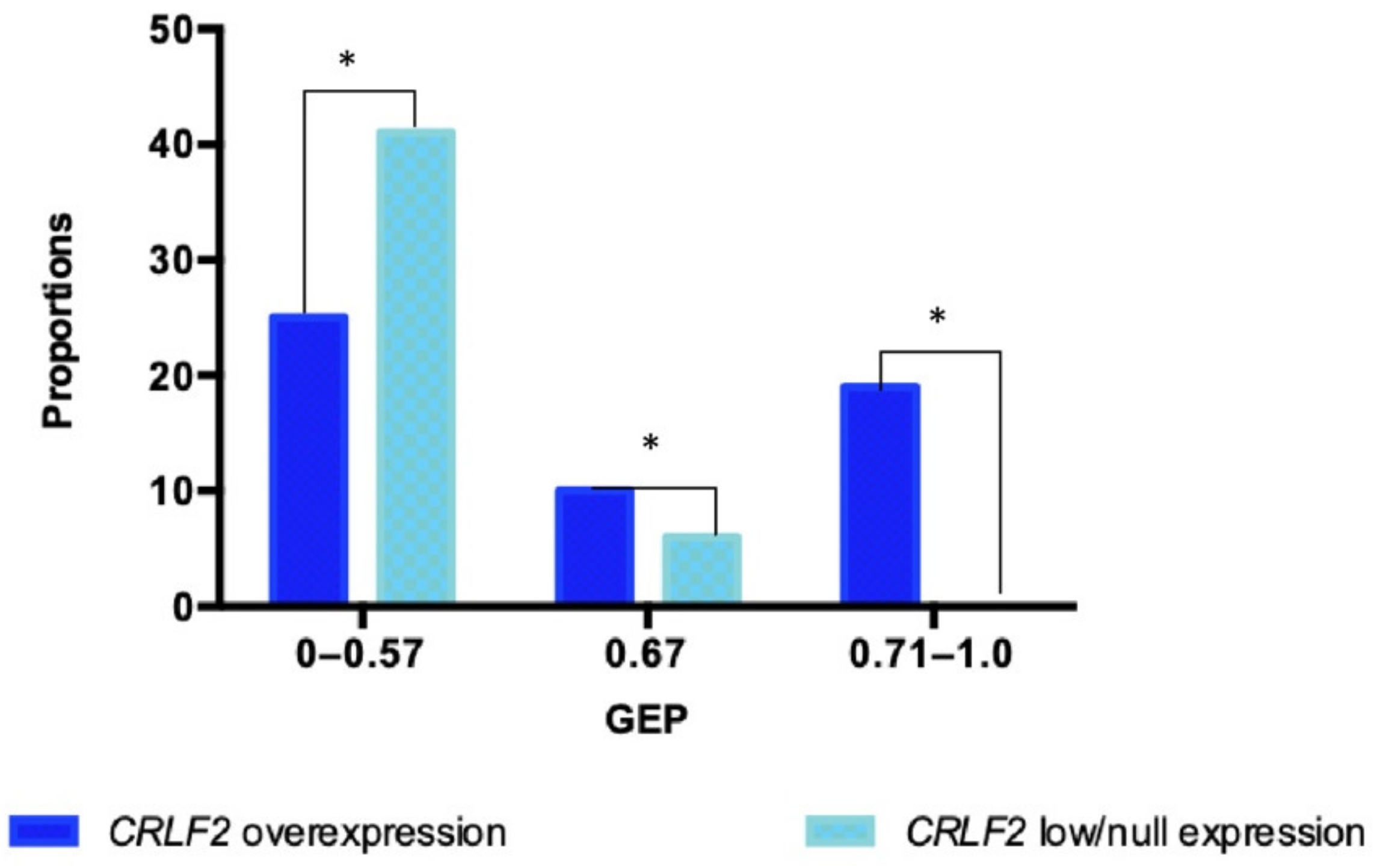
2.2. Gene Expression Profile (GEP)
2.3. IKZF1 Abnormalities
2.4. Pathogenic and Likely Pathogenic Mutations in Kinase and Transcription Factor Genes
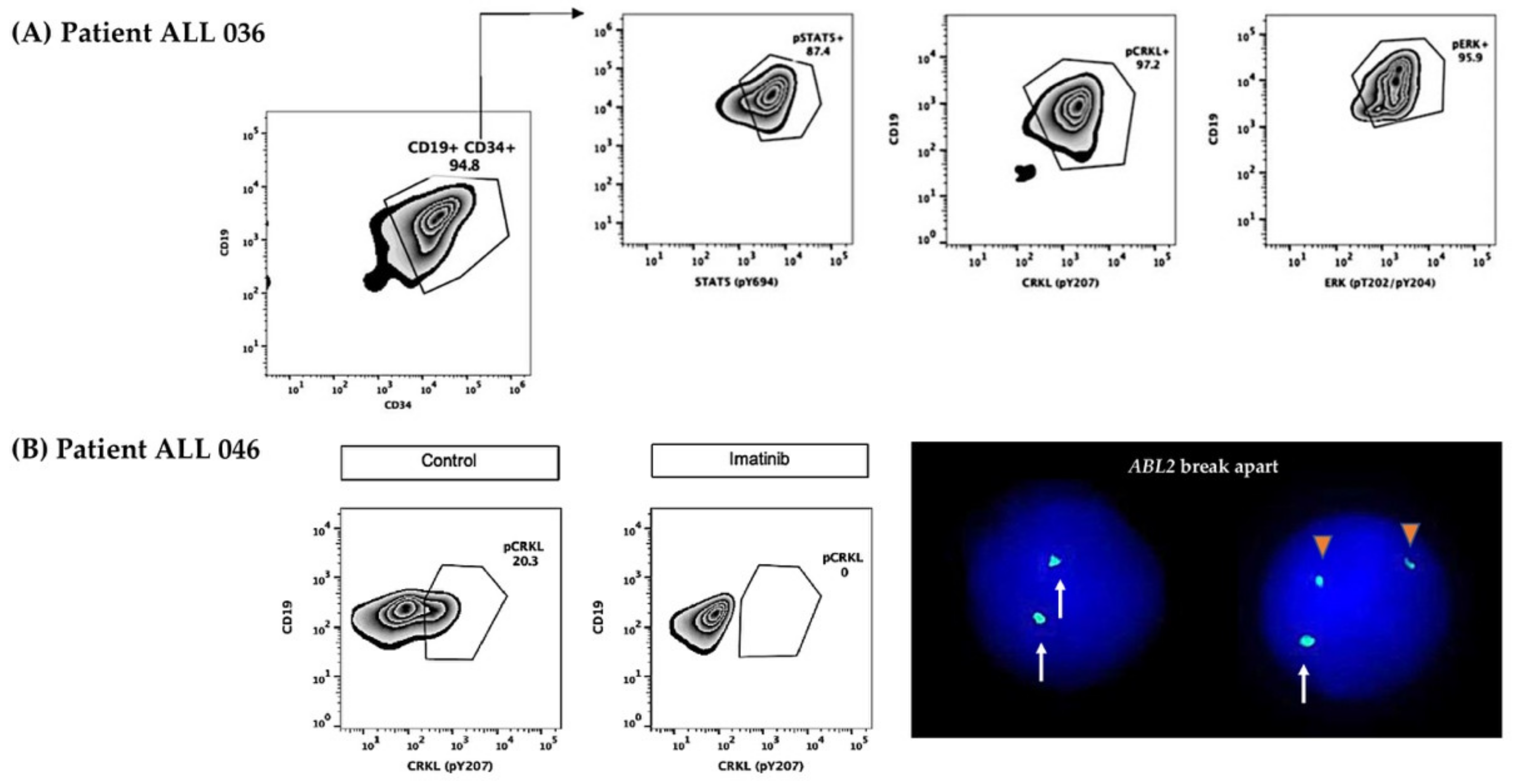
2.5. CRLF2 Protein Surface, Phosphorylated Surrogate Markers, and Inhibition Analysis
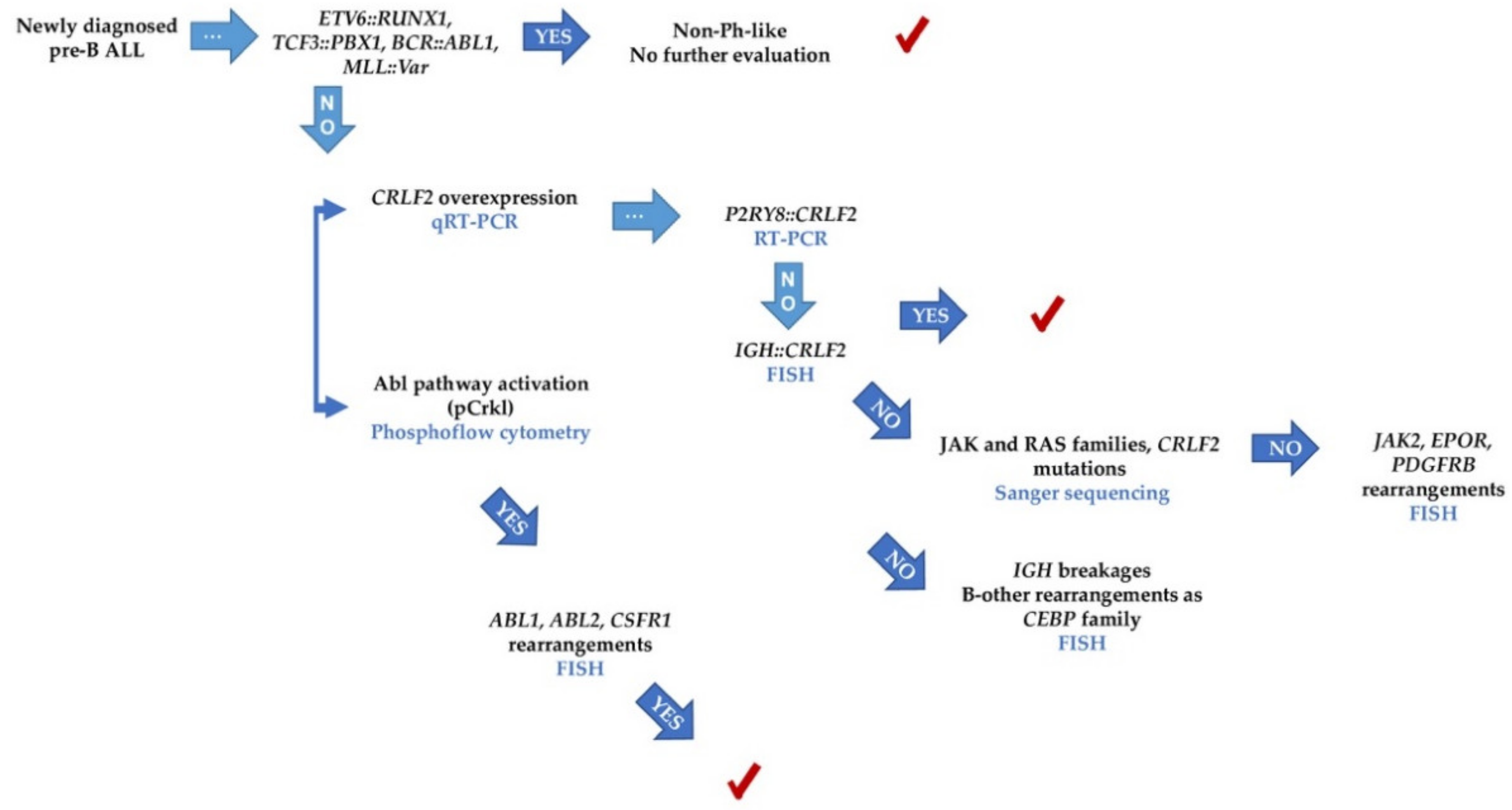
2.6. Patients with Features Associated with Ph-like and with B-Other Subtypes
- (a)
- All patients with IGH::CRLF2, cases with P2RY8::CRLF2 deletion, and the single patient with triplication of CRLF2 [13].
- (b)
- (c)
- All patients with GEP coefficients of 7.1–1.0, and the four cases who matched with the Reshmi et al. 2017 positive profile [12] were included in this subset.
- (d)
- All patients who were positive for the surrogate markers pStat5, pCrkl, and pErk [10]. Two more patients (ALL 060 and ALL 061) presented phosphorylation of surrogate markers; thus, they were also considered part of the Ph-like subgroup, although did not present CRLF2 overexpression
- (a)
- For the Jak2-Stat5 class, the presence of CRLF2 overexpression and rearrangements, JAK family mutations, and the Jak2-Stat5 activation pathway.
- (b)
- For ABL class, the presence of the pCrkl surrogate marker and the following detection of ABL1 or ABL2 rearrangements.
- (c)
- Despite few patients being analyzed with the pErk, those cases tested for the Ras pathway and mutations showed consistent results.

3. Discussion
4. Materials and Methods
4.1. Editorial Policies and Ethical Considerations
4.2. Patients and Samples
4.3. Analysis of GEP
4.4. Determination of the CRLF2 Protein
4.5. Detection of P2RY8::CRLF2 Deletion
4.6. Detection of the IGH::CRLF2 Rearrangement and ABL1 and ABL2 Breakages
4.7. Analysis of Ik6 Transcript
4.8. Target Specific NGS and Sanger Sequencing
4.9. Array-CGH Analysis
4.10. Kinase Activation and Inhibition Assay by Phosphoflow Cytometry
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roberts, K.G. The biology of Philadelphia chromosome-like ALL. Best Pract. Res. Clin. Haematol. 2017, 30, 212–221. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Aldoss, I.; Yang, D.; Tomasian, V.; Mokhtari, S.; Jackson, R.; Gu, Z.; Telatar, M.; Yew, H.; Al Malki, M.M.; Salhotra, A.; et al. Outcomes of Allogeneic Hematopoietic Cell Transplantation in Adults with Fusions Associated with Ph-like ALL. Blood Adv. 2022, in press. [Google Scholar] [CrossRef]
- Jain, N.; Roberts, K.G.; Jabbour, E.; Patel, K.; Eterovic, A.K.; Chen, K.; Zweidler-McKay, P.; Lu, X.; Fawcett, G.; Wang, S.A.; et al. Ph-like acute lymphoblastic leukemia: A high-risk subtype in adults. Blood 2017, 129, 572–581. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.G.; Gu, Z.; Payne-Turner, D.; McCastlain, K.; Harvey, R.; Chen, I.-M.; Pei, D.; Iacobucci, I.; Valentine, M.; Pounds, S.B.; et al. High Frequency and Poor Outcome of Philadelphia Chromosome–Like Acute Lymphoblastic Leukemia in Adults. J. Clin. Oncol. 2017, 35, 394–401. [Google Scholar] [CrossRef]
- Roberts, K.G.; Reshmi, S.C.; Harvey, R.; Chen, I.-M.; Patel, K.; Stonerock, E.; Jenkins, H.; Dai, Y.; Valentine, M.; Gu, Z.; et al. Genomic and outcome analyses of Ph-like ALL in NCI standard-risk patients: A report from the Children’s Oncology Group. Blood 2018, 132, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Harvey, R.; Mullighan, C.G.; Chen, I.-M.; Wharton, W.; Mikhail, F.M.; Carroll, A.J.; Kang, H.; Liu, W.; Dobbin, K.K.; Smith, M.A.; et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 2010, 115, 5312–5321. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Luna, R.; Velasco-Hidalgo, L.; Zapata-Tarrés, M.; Cárdenas-Cardos, R.; Aguilar-Ortiz, M.R. Current outlook of childhood cancer epidemiology in a middle-income country under a public health insurance program. Pediatr. Hematol. Oncol. 2017, 34, 43–50. [Google Scholar] [CrossRef]
- Muñoz-Aguirre, P.; Huerta-Gutierrez, R.; Zamora, S.; Mohar, A.; Vega-Vega, L.; Hernández-Ávila, J.E.; Morales-Carmona, E.; Zapata-Tarres, M.; Bautista-Arredondo, S.; Perez-Cuevas, R.; et al. Acute Lymphoblastic Leukaemia Survival in Children Covered by Seguro Popular in Mexico: A National Comprehensive Analysis 2005–2017. Health Syst. Reform 2021, 7, e1914897. [Google Scholar] [CrossRef]
- Roberts, K.G.; Morin, R.D.; Zhang, J.; Hirst, M.; Zhao, Y.; Su, X.; Chen, S.-C.; Payne-Turner, D.; Churchman, M.L.; Harvey, R.C.; et al. Genetic Alterations Activating Kinase and Cytokine Receptor Signaling in High-Risk Acute Lymphoblastic Leukemia. Cancer Cell 2012, 22, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Ofran, Y. Activated kinases in ALL: Time to act. Blood 2017, 129, 3280–3282. [Google Scholar] [CrossRef]
- Reshmi, S.C.; Harvey, R.C.; Roberts, K.G.; Stonerock, E.; Smith, A.; Jenkins, H.; Chen, I.-M.; Valentine, M.; Liu, Y.; Li, Y.; et al. Targetable kinase gene fusions in high-risk B-ALL: A study from the Children’s Oncology Group. Blood 2017, 129, 3352–3361. [Google Scholar] [CrossRef] [Green Version]
- Herold, T.; Schneider, S.; Metzeler, K.H.; Neumann, M.; Hartmann, L.; Roberts, K.G.; Konstandin, N.P.; Greif, P.A.; Bräundl, K.; Ksienzyk, B.; et al. Adults with Philadelphia chromosome–like acute lymphoblastic leukemia frequently have IGH-CRLF2 and JAK2 mutations, persistence of minimal residual disease and poor prognosis. Haematologica 2017, 102, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Den Boer, M.L.; van Slegtenhorst, M.; De Menezes, R.X.; Cheok, M.H.; Buijs-Gladdines, J.G.; Peters, S.T.; Van Zutven, L.J.; Beverloo, H.B.; Van der Spek, P.J.; Escherich, G.; et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 2009, 10, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Mullighan, C.G.; Su, X.; Zhang, J.; Radtke, I.; Phillips, L.A.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C.; Schulman, B.A.; et al. Deletion ofIKZF1and Prognosis in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef]
- Harvey, R.; Mullighan, C.; Wang, X.; Dobbin, K.K.; Davidson, G.S.; Bedrick, E.J.; Chen, I.-M.; Atlas, S.R.; Kang, H.; Ar, K.; et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: Correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood 2010, 116, 4874–4884. [Google Scholar] [CrossRef]
- Boer, J.M.; Marchante, J.R.M.; Evans, W.E.; Horstmann, M.A.; Escherich, G.; Pieters, R.; Boer, M.L.D. BCR-ABL1-like cases in pediatric acute lymphoblastic leukemia: A comparison between DCOG/Erasmus MC and COG/St. Jude signatures. Haematologica 2015, 100, e354–e357. [Google Scholar] [CrossRef] [Green Version]
- Harvey, R.C.; Kang, H.; Roberts, K.G.; Chen, D.I.-M.L.; Atlas, S.R.; Bedrick, E.J.; Gastier-Foster, J.M.; Zhang, J.; Gerhard, D.S.; Smith, M.A.; et al. Development and Validation Of a Highly Sensitive and Specific Gene Expression Classifier To Prospectively Screen and Identify B-Precursor Acute Lymphoblastic Leukemia (ALL) Patients With a Philadelphia Chromosome-Like (“Ph-like” or “BCR-ABL1-Like”) Signature For Therapeutic Targeting and Clinical Intervention. Blood 2013, 122, 826. [Google Scholar] [CrossRef]
- Conant, J.L.; Czuchlewski, D.R. BCR-ABL1-like B-lymphoblastic leukemia/lymphoma: Review of the entity and detection methodologies. Int. J. Lab. Hematol. 2019, 41 (Suppl. S1), 126–130. [Google Scholar] [CrossRef] [Green Version]
- Raca, G.; Abdel-Azim, H.; Yue, F.; Broach, J.; Payne, J.L.; Reeves, M.E.; Gowda, C.; Schramm, J.; Desai, D.; Dovat, E.; et al. Increased Incidence of IKZF1 deletions and IGH-CRLF2 translocations in B-ALL of Hispanic/Latino children—A novel health disparity. Leukemia 2021, 35, 2399–2402. [Google Scholar] [CrossRef]
- Tran, T.H.; Langlois, S.; Meloche, C.; Caron, M.; Saint-Onge, P.; Rouette, A.; Bataille, A.R.; Jimenez-Cortes, C.; Sontag, T.; Bittencourt, H.; et al. Whole-transcriptome analysis in acute lymphoblastic leukemia: A report from the DFCI ALL Consortium Protocol 16-001. Blood Adv. 2022, 6, 1329–1341. [Google Scholar] [CrossRef]
- Juárez-Velázquez, M.D.R.; Moreno-Lorenzana, D.L.; Anaya, D.A.M.; Monterde, E.A.H.; Aguilar-Hernández, M.M.; Reyes-León, A.; Chávez-González, M.A.; Santiago, N.L.; Tarrés, M.Z.; Villegas, L.J.; et al. High occurrence of CRLF2 abnormalities in Mexican children with B-cell acute lymphoblastic leukemia. Cytokine 2022, 155, 155896. [Google Scholar] [CrossRef]
- Starý, J.; Zuna, J.; Zaliova, M. New biological and genetic classification and therapeutically relevant categories in childhood B-cell precursor acute lymphoblastic leukemia. F1000Research 2018, 7, 1569. [Google Scholar] [CrossRef]
- Harrison, C.J. Blood Spotlight on iAMP21 acute lymphoblastic leukemia (ALL), a high-risk pediatric disease. Blood 2015, 125, 1383–1386. [Google Scholar] [CrossRef] [Green Version]
- Palmi, C.; Vendramini, E.; Silvestri, D.; Longinotti, G.; Frison, D.; Cario, G.; Shochat, C.; Stanulla, M.; Rossi, V.; Di Meglio, A.M.; et al. Poor prognosis for P2RY8-CRLF2 fusion but not for CRLF2 over-expression in children with intermediate risk B-cell precursor acute lymphoblastic leukemia. Leukemia 2012, 26, 2245–2253. [Google Scholar] [CrossRef]
- Panzer-Grümayer, R.; Köhrer, S.; Haas, O.A. The enigmatic role(s) of P2RY8-CRLF2. Oncotarget 2017, 8, 96466–96467. [Google Scholar] [CrossRef]
- Passet, M.; Boissel, N.; Sigaux, F.; Saillard, C.; Bargetzi, M.; Ba, I.; Thomas, X.; Graux, C.; Chalandon, Y.; Leguay, T.; et al. PAX5 P80R mutation identifies a novel subtype of B-cell precursor acute lymphoblastic leukemia with favorable outcome. Blood 2019, 133, 280–284. [Google Scholar] [CrossRef]
- Ribera, J.-M. Philadelphia chromosome-like acute lymphoblastic leukemia. Still a pending matter. Haematologica 2020, 106, 1514–1516. [Google Scholar] [CrossRef]
- Tanasi, I.; Ba, I.; Sirvent, N.; Braun, T.; Cuccuini, W.; Ballerini, P.; Duployez, N.; Tanguy-Schmidt, A.; Tamburini, J.; Maury, S.; et al. Efficacy of tyrosine kinase inhibitors in Ph-like acute lymphoblastic leukemia harboring ABL-class rearrangements. Blood 2019, 134, 1351–1355. [Google Scholar] [CrossRef]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [Green Version]
- Jain, N.; Lu, X.; Daver, N.; Thakral, B.; Wang, S.A.; Konoplev, S.; Patel, K.; Kanagal-Shamanna, R.; Valentine, M.; Tang, G.; et al. Co-occurrence of CRLF2-rearranged and Ph+ acute lymphoblastic leukemia: A report of four patients. Haematologica 2017, 102, e514–e517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldoss, I.; Kamal, M.O.; Forman, S.J.; Pullarkat, V. Adults with Philadelphia Chromosome—Like Acute Lymphoblastic Leukemia: Considerations for Allogeneic Hematopoietic Cell Transplantation in First Complete Remission. Biol. Blood Marrow Transplant. 2019, 25, e41–e45. [Google Scholar] [CrossRef] [Green Version]
- Tasian, S.K.; Hurtz, C.; Wertheim, G.B.; Bailey, N.G.; Lim, M.S.; Harvey, R.C.; Chen, I.-M.; Willman, C.L.; Astles, R.; Zebrowski, A.; et al. High incidence of Philadelphia chromosome-like acute lymphoblastic leukemia in older adults with B-ALL. Leukemia 2017, 31, 981–984. [Google Scholar] [CrossRef] [Green Version]
- Imamura, T.; Kiyokawa, N.; Kato, M.; Imai, C.; Okamoto, Y.; Yano, M.; Ohki, K.; Yamashita, Y.; Kodama, Y.; Saito, A.; et al. Characterization of pediatric Philadelphia-negative B-cell precursor acute lymphoblastic leukemia with kinase fusions in Japan. Blood Cancer J. 2016, 6, e419. [Google Scholar] [CrossRef]
- Pérez-Vera, P.; Montero-Ruíz, O.; Frías, S.; Rivera-Luna, R.; Valladares, A.; Arenas, D.; Paredes-Aguilera, R.; Carnevale, A. Multiple copies of RUNX1: Description of 14 new patients, follow-up, and a review of the literature. Cancer Genet. Cytogenet. 2008, 180, 129–134. [Google Scholar] [CrossRef]
- Lorenzana, D.M.; Velázquez, M.d.R.J.; León, A.R.; Anaya, D.M.; Monterde, A.H.; Labadía, C.S.; Meneses, M.d.P.N.; Tarrés, M.Z.; Villegas, L.J.; Ramírez, B.J.; et al. CRLF2 and IKZF1 abnormalities in Mexican children with acute lymphoblastic leukemia and recurrent gene fusions: Exploring surrogate markers of signaling pathways. J. Pathol. Clin. Res. 2021, 7, 410–421. [Google Scholar] [CrossRef]
- Kang, H.; Chen, I.-M.; Wilson, C.S.; Bedrick, E.J.; Harvey, R.; Atlas, S.R.; Devidas, M.; Mullighan, C.; Wang, X.; Murphy, M.; et al. Gene expression classifiers for relapse-free survival and minimal residual disease improve risk classification and outcome prediction in pediatric B-precursor acute lymphoblastic leukemia. Blood 2010, 115, 1394–1405. [Google Scholar] [CrossRef]
- Russell, L.J.; Jones, L.; Enshaei, A.; Tonin, S.; Ryan, S.L.; Eswaran, J.; Nakjang, S.; Papaemmanuil, E.; Tubio, J.M.C.; Fielding, A.K.; et al. Characterisation of the genomic landscape ofCRLF2-rearranged acute lymphoblastic leukemia. Genes, Chromosom. Cancer 2017, 56, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Zhang, R.; Liu, J.; Li, M.; Song, C.; Dovat, S.; Li, J.; Ge, Z. Characterization of LEF1 High Expression and Novel Mutations in Adult Acute Lymphoblastic Leukemia. PLoS ONE 2015, 10, e0125429. [Google Scholar] [CrossRef]
- Lin, X.; Zou, X.; Wang, Z.; Fang, Q.; Chen, S.; Huang, J.; Zhe, N.; Yu, M.; Zhang, Y.; Wang, J. Targeting of heme oxygenase-1 attenuates the negative impact of Ikaros isoform 6 in adult BCR-ABL1-positive B-ALL. Oncotarget 2016, 7, 53679–53701. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv 2018, 3, 201178. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Yoda, A.; Yoda, Y.; Chiaretti, S.; Bar-Natan, M.; Mani, K.; Rodig, S.J.; West, N.; Xiao, Y.; Brown, J.R.; Mitsiades, C.; et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 252–257. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Anaya, D.; Moreno-Lorenzana, D.; Reyes-León, A.; Juárez-Figueroa, U.; Dean, M.; Aguilar-Hernández, M.M.; Rivera-Sánchez, N.; García-Islas, J.; Vieyra-Fuentes, V.; Zapata-Tarrés, M.; et al. Characterization of Philadelphia-like Pre-B Acute Lymphoblastic Leukemia: Experiences in Mexican Pediatric Patients. Int. J. Mol. Sci. 2022, 23, 9587. https://doi.org/10.3390/ijms23179587
Martínez-Anaya D, Moreno-Lorenzana D, Reyes-León A, Juárez-Figueroa U, Dean M, Aguilar-Hernández MM, Rivera-Sánchez N, García-Islas J, Vieyra-Fuentes V, Zapata-Tarrés M, et al. Characterization of Philadelphia-like Pre-B Acute Lymphoblastic Leukemia: Experiences in Mexican Pediatric Patients. International Journal of Molecular Sciences. 2022; 23(17):9587. https://doi.org/10.3390/ijms23179587
Chicago/Turabian StyleMartínez-Anaya, Daniel, Dafné Moreno-Lorenzana, Adriana Reyes-León, Ulises Juárez-Figueroa, Michael Dean, María Montserrat Aguilar-Hernández, Netzi Rivera-Sánchez, Jessica García-Islas, Victoria Vieyra-Fuentes, Marta Zapata-Tarrés, and et al. 2022. "Characterization of Philadelphia-like Pre-B Acute Lymphoblastic Leukemia: Experiences in Mexican Pediatric Patients" International Journal of Molecular Sciences 23, no. 17: 9587. https://doi.org/10.3390/ijms23179587
APA StyleMartínez-Anaya, D., Moreno-Lorenzana, D., Reyes-León, A., Juárez-Figueroa, U., Dean, M., Aguilar-Hernández, M. M., Rivera-Sánchez, N., García-Islas, J., Vieyra-Fuentes, V., Zapata-Tarrés, M., Juárez-Villegas, L., Paredes-Aguilera, R., Vega-Vega, L., Rivera-Luna, R., Juárez-Velázquez, M. d. R., & Pérez-Vera, P. (2022). Characterization of Philadelphia-like Pre-B Acute Lymphoblastic Leukemia: Experiences in Mexican Pediatric Patients. International Journal of Molecular Sciences, 23(17), 9587. https://doi.org/10.3390/ijms23179587