
HER2 c-Terminal Fragments Are Expressed via Internal Translation of the HER2 mRNA


Abstract
:1. Introduction
2. Results
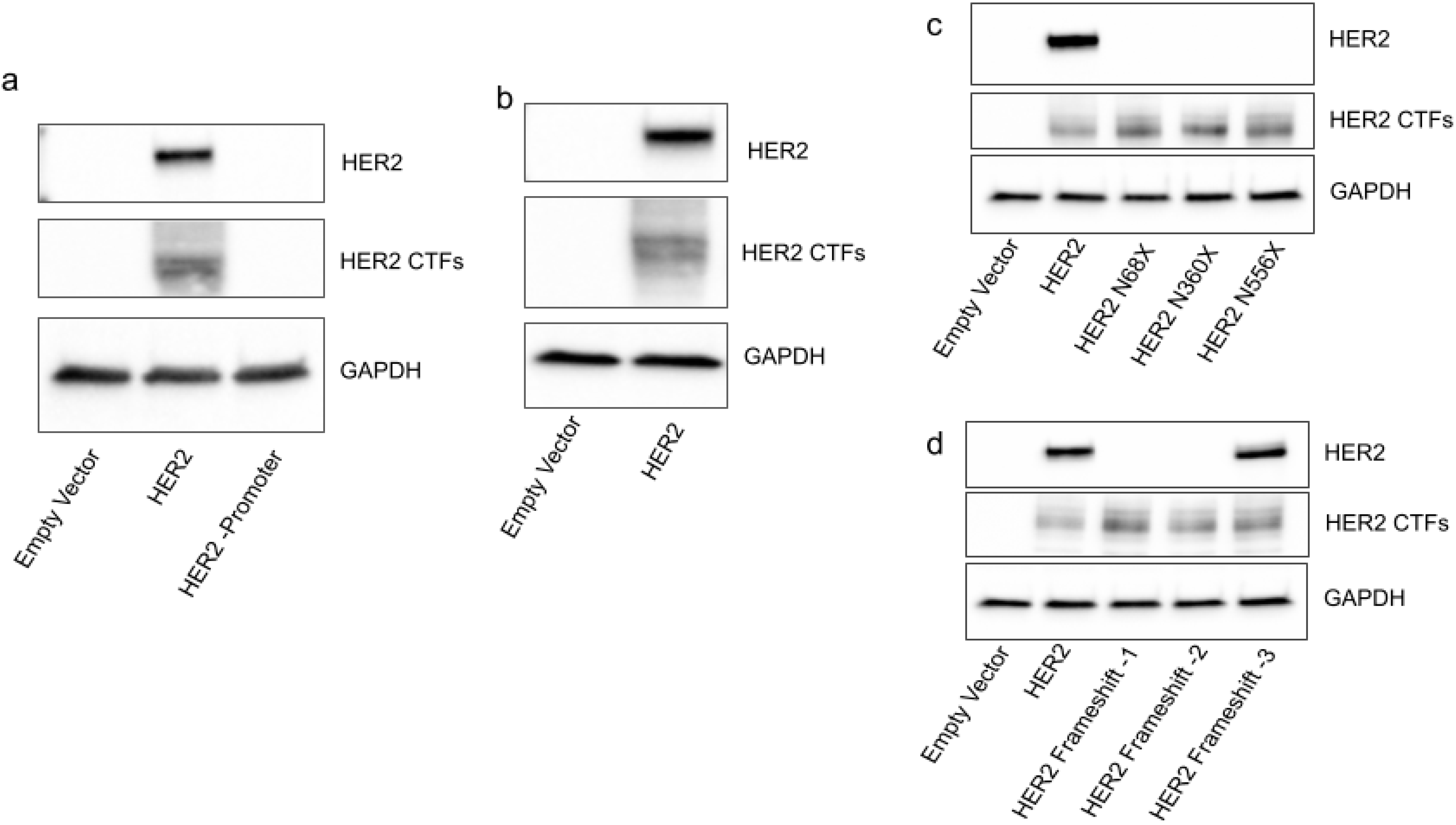
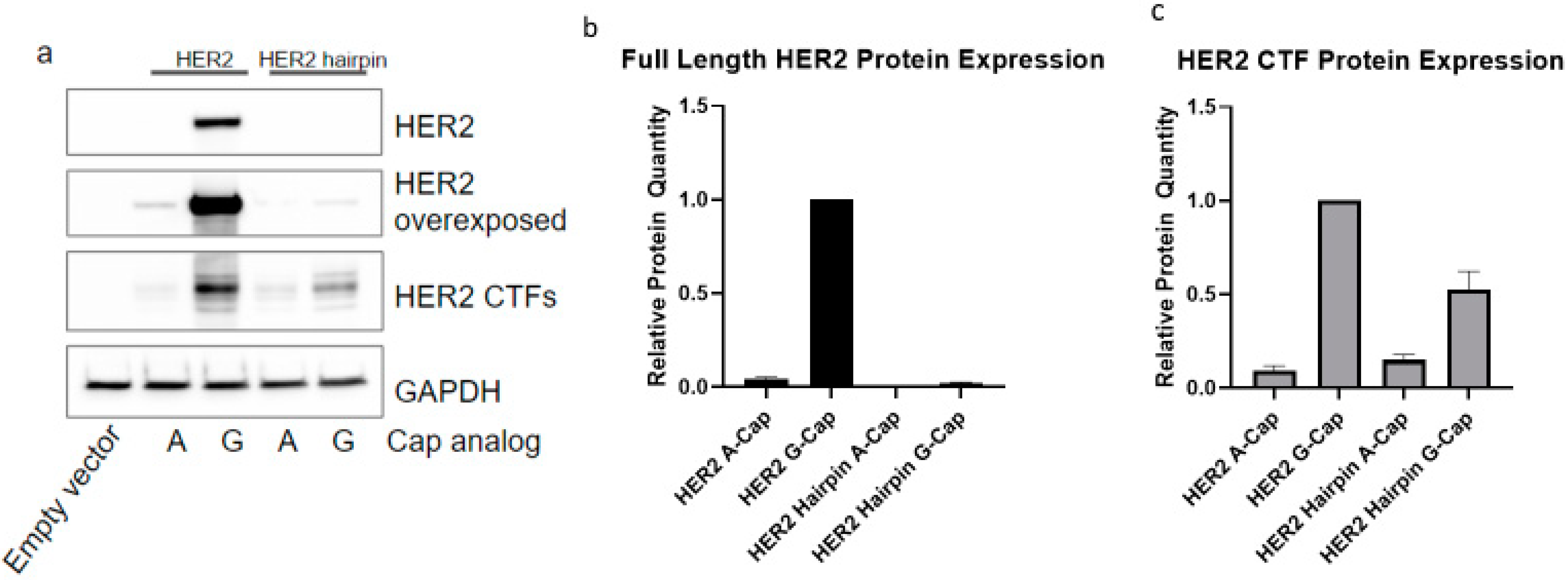
2.1. HER2 CTFs Are Expressed from the Full-Length HER2 mRNA
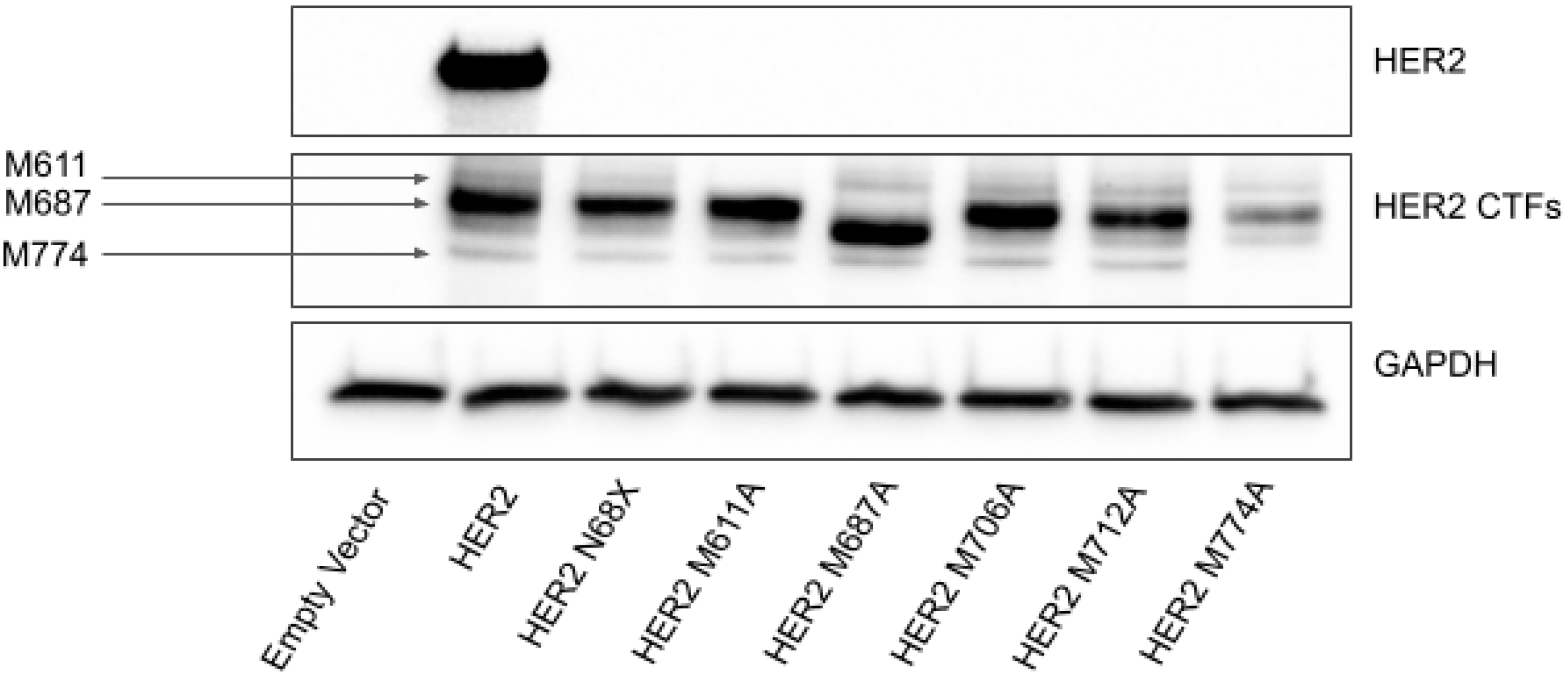
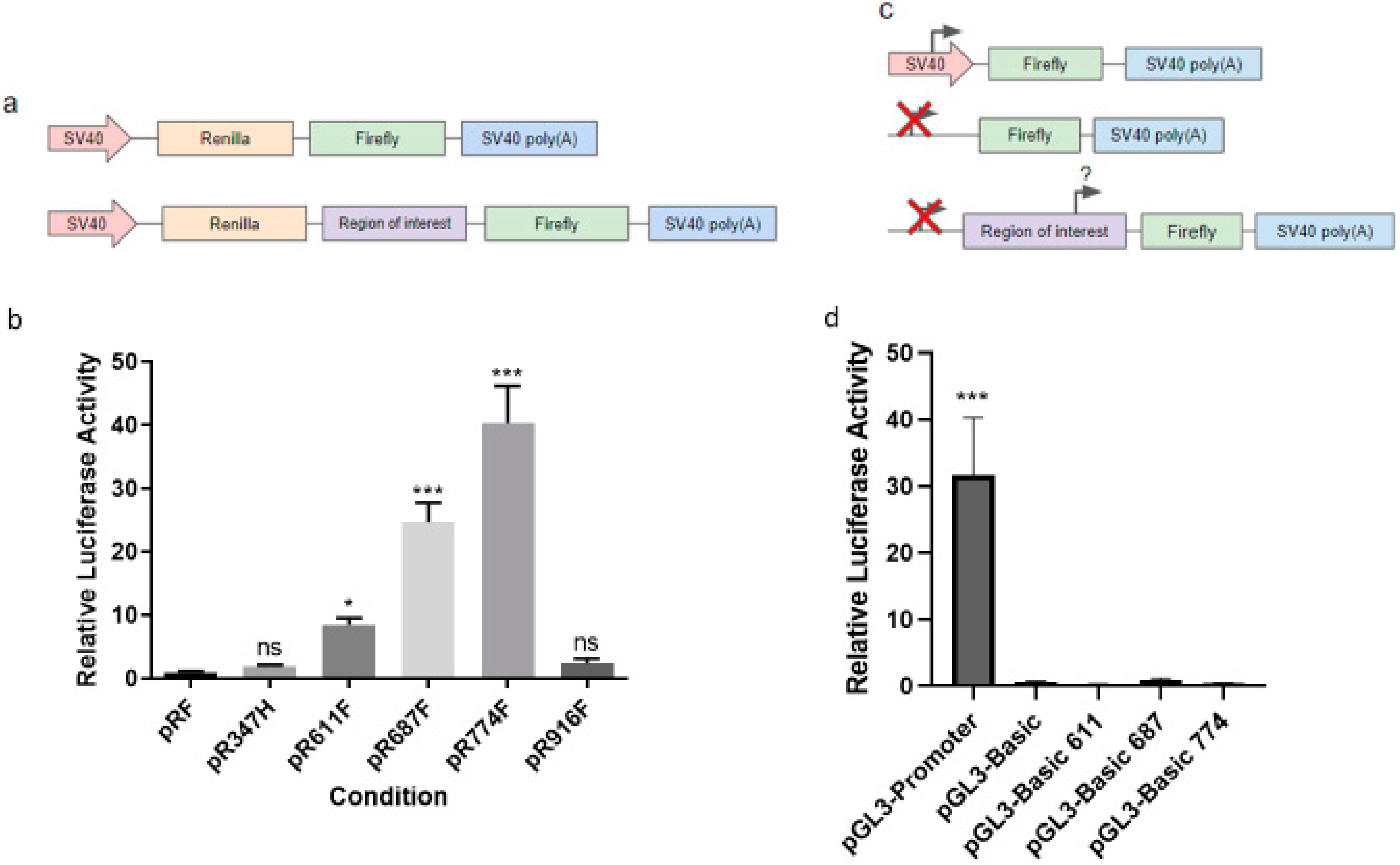
2.2. Translation of HER2 CTFs Are Initiated at a Cluster of Internal Methionine Codons
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Mutagenesis
4.3. In Vitro Transcription
4.4. Transfection
4.5. SDS-PAGE
4.6. Luciferase Assays
4.7. Western Blot Quantification
4.8. Hairpin Construct Cloning
4.9. Kozak Context Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klement, G.L.; Goukassian, D.; Hlatky, L.; Carrozza, J.; Morgan, J.P.; Yan, X. Cancer Therapy Targeting the HER2-PI3K Pathway: Potential Impact on the Heart. Front. Pharmacol. 2012, 3, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Schwaederle, M.; Arguello, D.; Millis, S.Z.; Gatalica, Z.; Kurzrock, R. HER2 expression status in diverse cancers: Review of results from 37,992 patients. Cancer Metastasis Rev. 2015, 34, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbeck, N. Advances in targeting HER2-positive breast cancer. Curr. Opin. Obs. Gynecol. 2018, 30, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Schillaci, R.; Guzmán, P.; Cayrol, F.; Beguelin, W.; Díaz Flaqué, M.C.; Proietti, C.J.; Pineda, V.; Palazzi, J.; Frahm, I.; Charreau, E.H.; et al. Clinical relevance of ErbB-2/HER2 nuclear expression in breast cancer. BMC Cancer 2012, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Redmond, A.M.; Omarjee, S.; Chernukhin, I.; Le Romancer, M.; Carroll, J.S. Analysis of HER2 genomic binding in breast cancer cells identifies a global role in direct gene regulation. PLoS ONE 2019, 14, e0225180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.C.; Lien, H.C.; Xia, W.; Chen, I.F.; Lo, H.W.; Wang, Z.; Ali-Seyed, M.; Lee, D.F.; Bartholomeusz, G.; Ou-Yang, F.; et al. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell 2004, 6, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.C.; Liu, X.; Li, Y.; Covington, M.; Wynn, R.; Huber, R.; Hillman, M.; Yang, G.; Ellis, D.; Marando, C.; et al. Identification of ADAM10 as a major source of HER2 ectodomain sheddase activity in HER2 overexpressing breast cancer cells. Cancer Biol. Ther. 2006, 5, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Yuan, C.X.; Lasut, A.L.; Wynn, R.; Neff, N.T.; Hollis, G.F.; Ramaker, M.L.; Rupar, M.J.; Liu, P.; Meade, R. Purification of Her-2 extracellular domain and identification of its cleavage site. Protein Expr. Purif. 2003, 29, 217–222. [Google Scholar] [CrossRef]
- Zabrecky, J.R.; Lam, T.; McKenzie, S.J.; Carney, W. The extracellular domain of p185/neu is released from the surface of human breast carcinoma cells, SK-BR-3. J. Biol. Chem. 1991, 266, 1716–1720. [Google Scholar] [CrossRef]
- Pedersen, K.; Angelini, P.D.; Laos, S.; Bach-Faig, A.; Cunningham, M.P.; Ferrer-Ramón, C.; Luque-García, A.; García-Castillo, J.; Parra-Palau, J.L.; Scaltriti, M.; et al. A naturally occurring HER2 carboxy-terminal fragment promotes mammary tumor growth and metastasis. Mol. Cell. Biol. 2009, 29, 3319–3331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anido, J.; Scaltriti, M.; Bech Serra, J.J.; Santiago Josefat, B.; Todo, F.R.; Baselga, J.; Arribas, J. Biosynthesis of tumorigenic HER2 C-terminal fragments by alternative initiation of translation. EMBO J. 2006, 25, 3234–3244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Castillo, J.; Pedersen, K.; Angelini, P.D.; Bech-Serra, J.J.; Colomé, N.; Cunningham, M.P.; Parra-Palau, J.L.; Canals, F.; Baselga, J.; Arribas, J. HER2 carboxyl-terminal fragments regulate cell migration and cortactin phosphorylation. J. Biol. Chem. 2009, 284, 25302–25313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, M.A.; Sáez, R.; Ramsey, E.E.; Garcia-Barchino, M.J.; Rojo, F.; Evans, A.J.; Albanell, J.; Keenan, E.J.; Lluch, A.; García-Conde, J.; et al. NH(2)-terminal truncated HER-2 protein but not full-length receptor is associated with nodal metastasis in human breast cancer. Clin. Cancer Res. 2002, 8, 347–353. [Google Scholar] [PubMed]
- Parra-Palau, J.L.; Pedersen, K.; Peg, V.; Scaltriti, M.; Angelini, P.D.; Escorihuela, M.; Mancilla, S.; Sánchez Pla, A. A major role of p95/611-CTF, a carboxy-terminal fragment of HER2, in the down-modulation of the estrogen receptor in HER2-positive breast cancers. Cancer Res. 2010, 70, 8537–8546. [Google Scholar] [CrossRef] [Green Version]
- Tural, D.; Akar, E.; Mutlu, H.; Kilickap, S. P95 HER2 fragments and breast cancer outcome. Expert Rev. Anticancer. Ther. 2014, 9, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Sperinde, J.; Huang, W.; Vehtari, A.; Chenna, A.; Kellokumpu-Lehtinen, P.-L.; Winslow, J.; Bono, P.; Lie, Y.S.; Petropoulos, C.J.; Weidler, J.; et al. p95HER2 Methionine 611 Carboxy-Terminal Fragment Is Predictive of Trastuzumab Adjuvant Treatment Benefit in the FinHer Trial. Clin. Cancer Res. 2018, 24, 3046–3052. [Google Scholar] [CrossRef] [Green Version]
- Scaltriti, M.; Rojo, F.; Ocana, A.; Anido, J.; Guzman, M.; Cortes, J.; Di Cosimo, S.; Matias-Guiu, X.; Ramon y Cajal, S.; Arribas, J.; et al. Expression of p95HER2, a Truncated Form of the HER2 Receptor, and Response to Anti-HER2 Therapies in Breast Cancer. JNCI J. Natl. Cancer Inst. 2007, 99, 628–638. [Google Scholar] [CrossRef] [Green Version]
- Rexer, B.N.; Ghosh, R.; Narasanna, A.; Estrada, M.V.; Chakrabarty, A.; Song, Y.; Engelman, J.A.; Arteaga, C.L. Human breast cancer cells harboring a gatekeeper T798M mutation in HER2 overexpress EGFR ligands and are sensitive to dual inhibition of EGFR and HER2. Clin. Cancer Res. 2013, 19, 5390–5401. [Google Scholar] [CrossRef] [Green Version]
- Jackson, R.; Hellen, C.; Pestova, T. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, Z. IRES-mediated cap-independent translation, a path leading to hidden proteome. J. Mol. Cell Biol. 2019, 11, 911–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komar, A.A.; Hatzoglou, M. Internal ribosome entry sites in cellular mRNAs: Mystery of their existence. J. Biol. Chem. 2005, 280, 23425–23428. [Google Scholar] [CrossRef] [Green Version]
- Karginov, T.A.; Pastor, D.; Semler, B.L.; Gomez, C.M. Mammalian Polycistronic mRNAs and Disease. Trends Genet. TIG 2017, 33, 129–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babendure, J.R.; Babendure, J.L.; Ding, J.H.; Tsien, R.Y. Control of mammalian translation by mRNA structure near caps. RNA 2006, 12, 851–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, A.G.; Nielsen, H. Neural network prediction of translation initiation sites in eukaryotes: Perspectives for EST and genome analysis. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1997, 5, 226–233. [Google Scholar]
- Du, X.; Wang, J.; Zhu, H.; Rinaldo, L.; Lamar, K.M.; Palmenberg, A.C.; Hansel, C.; Gomez, C.M. Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell 2013, 154, 118–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Wei, C.; Hejazi Pastor, D.P.; Rao, E.R.; Li, Y.; Grasselli, G.; Godfrey, J.; Palmenberg, A.C.; Andrade, J.; Hansel, C.; et al. α1ACT Is Essential for Survival and Early Cerebellar Programming in a Critical Neonatal Window. Neuron 2019, 102, 770–785. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Du, X.; Muramatsu, S.; Gomez, C.M. An miRNA-mediated therapy for SCA6 blocks IRES-driven translation of the CACNA1A second cistron. Sci. Transl. Med. 2016, 8, 347ra94. [Google Scholar] [CrossRef] [Green Version]
- Lauring, A.S.; Overbaugh, J. Evidence that an IRES within the Notch2 coding region can direct expression of a nuclear form of the protein. Mol. Cell 2000, 6, 939–945. [Google Scholar] [CrossRef]
- Cornelis, S.; Bruynooghe, Y.; Denecker, G.; Van Huffel, S.; Tinton, S.; Beyaert, R. Identification and characterization of a novel cell cycle-regulated internal ribosome entry site. Mol. Cell 2000, 5, 597–605. [Google Scholar] [CrossRef]
- Yueh, A.; Schneider, R.J. Translation by ribosome shunting on adenovirus and hsp70 mRNAs facilitated by complementarity to 18S rRNA. Genes Dev. 2000, 14, 414–421. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence |
|---|---|
| HER2 N68X Forward | cctacctgcccacctaagccagcctgtcctt |
| HER2 N68X Reverse | aaggacaggctggcttaggtgggcaggtagg |
| HER2 N360X Forward | gggcagttaccagtgcctaaatccaggagtttgctgg |
| HER2 N360X Reverse | ccagcaaactcctggatttaggcactggtaactgccc |
| HER2 N556X Forward | ccccagggagtatgtgtaagccaggcactgtttgc |
| HER2 N556X Reverse | gcaaacagtgcctggcttacacatactccctgggg |
| HER2 M611A Forward | ggaaacttccagatgggcgcgtaggagaggtcaggttt |
| HER2 M611A Reverse | aaacctgacctctcctacgcgcccatctggaagtttcc |
| HER2 M687A Forward | ctgcagcagtctccgcgccgtgtacttccggatc |
| HER2 M687A Reverse | gatccggaagtacacggcgcggagactgctgcag |
| HER2 M706A Forward | cgcctggttgggcgccgctccgctaggt |
| HER2 M706A Reverse | acctagcggagcggcgcccaaccaggcg |
| HER2 M712A Forward | tctttcaggatccgcgcctgcgcctggttggg |
| HER2 M712A Reverse | cccaaccaggcgcaggcgcggatcctgaaaga |
| HER2 M774A Forward | gagcccacaccagccgccacgtatgcttcgtc |
| HER2 M774A Reverse | gacgaagcatacgtggcggctggtgtgggctc |
| HER2 Frameshift 1 Forward | gaggattgtcagagctgacgcgcactgtct |
| HER2 Frameshift 1 Reverse | agacagtgcgcgtcagctctgacaatcctc |
| HER2 Frameshift 2 Forward | gaggattgtcagagcgacgcgcactgtctg |
| HER2 Frameshift 2 Reverse | cagacagtgcgcgtcgctctgacaatcctc |
| HER2 Frameshift 3 Forward | gaggattgtcagagcacgcgcactgtctgt |
| HER2 Frameshift 3 Reverse | acagacagtgcgcgtgctctgacaatcctc |
| Target | Producer | Product Code | Species |
|---|---|---|---|
| FLAG (M2) | Sigma-Aldrich (St. Louis, MO, USA) | F1804 | Mouse |
| GAPDH | Abcam (Cambridge, UK) | ab9485 | Rabbit |
| Primer Name | Sequence |
|---|---|
| pR347F | atgcactagtcgcgagcacccaagtgtg |
| pR347F | atgcccatgggcccagaccatagcacactc |
| pR611F | atgcactagtcagccctggtcacctacaacacagac |
| pR611F | atgcccatgggtaggagaggtcaggtttcacaccgctg |
| pR687F | atgcactagtggcagttaccagtgccaatatc |
| pR687F | atgcccatggcgtgtacttccggatcttctgc |
| pR774F | atgcactagtctactcgctgaccctgcaagggctg |
| pR774F | atgcccatggcacgtatgcttcgtctaagatttctttgttggctttg |
| pR916F | atgcactagtctccacactgccaaccgg |
| pR916F | atgcccatggcagctcccacacagtcacac |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godfrey, J.D.; Hejazi, D.; Du, X.; Wei, C.; Rao, E.; Gomez, C.M. HER2 c-Terminal Fragments Are Expressed via Internal Translation of the HER2 mRNA. Int. J. Mol. Sci. 2022, 23, 9549. https://doi.org/10.3390/ijms23179549
Godfrey JD, Hejazi D, Du X, Wei C, Rao E, Gomez CM. HER2 c-Terminal Fragments Are Expressed via Internal Translation of the HER2 mRNA. International Journal of Molecular Sciences. 2022; 23(17):9549. https://doi.org/10.3390/ijms23179549
Chicago/Turabian StyleGodfrey, Jack D., Daniel Hejazi, Xiaofei Du, Cenfu Wei, Eshaan Rao, and Christopher M. Gomez. 2022. "HER2 c-Terminal Fragments Are Expressed via Internal Translation of the HER2 mRNA" International Journal of Molecular Sciences 23, no. 17: 9549. https://doi.org/10.3390/ijms23179549
APA StyleGodfrey, J. D., Hejazi, D., Du, X., Wei, C., Rao, E., & Gomez, C. M. (2022). HER2 c-Terminal Fragments Are Expressed via Internal Translation of the HER2 mRNA. International Journal of Molecular Sciences, 23(17), 9549. https://doi.org/10.3390/ijms23179549