


The Ambivalence of Connexin43 Gap Peptides in Cardioprotection of the Isolated Heart against Ischemic Injury

 ,
, 
Abstract
:1. Introduction
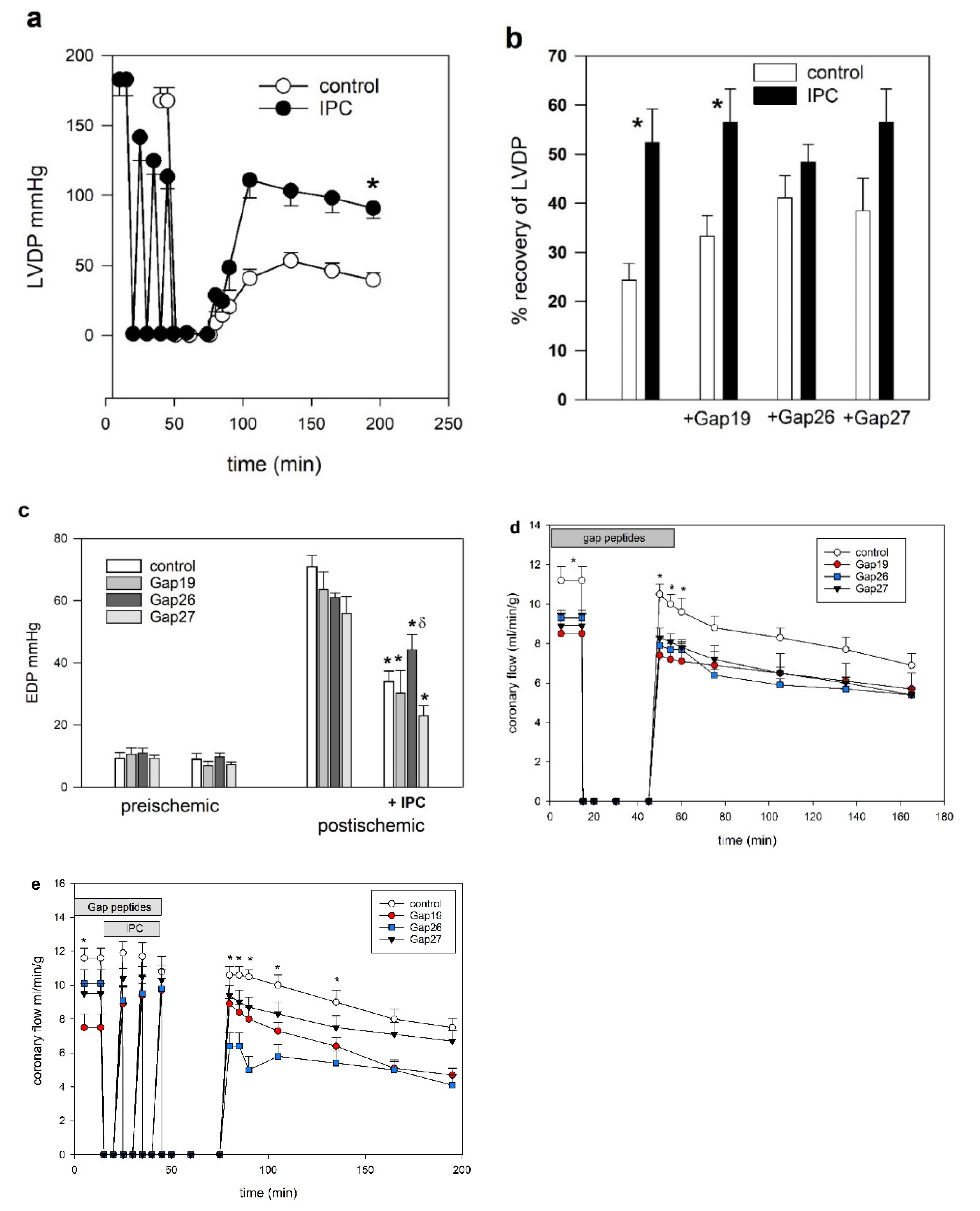
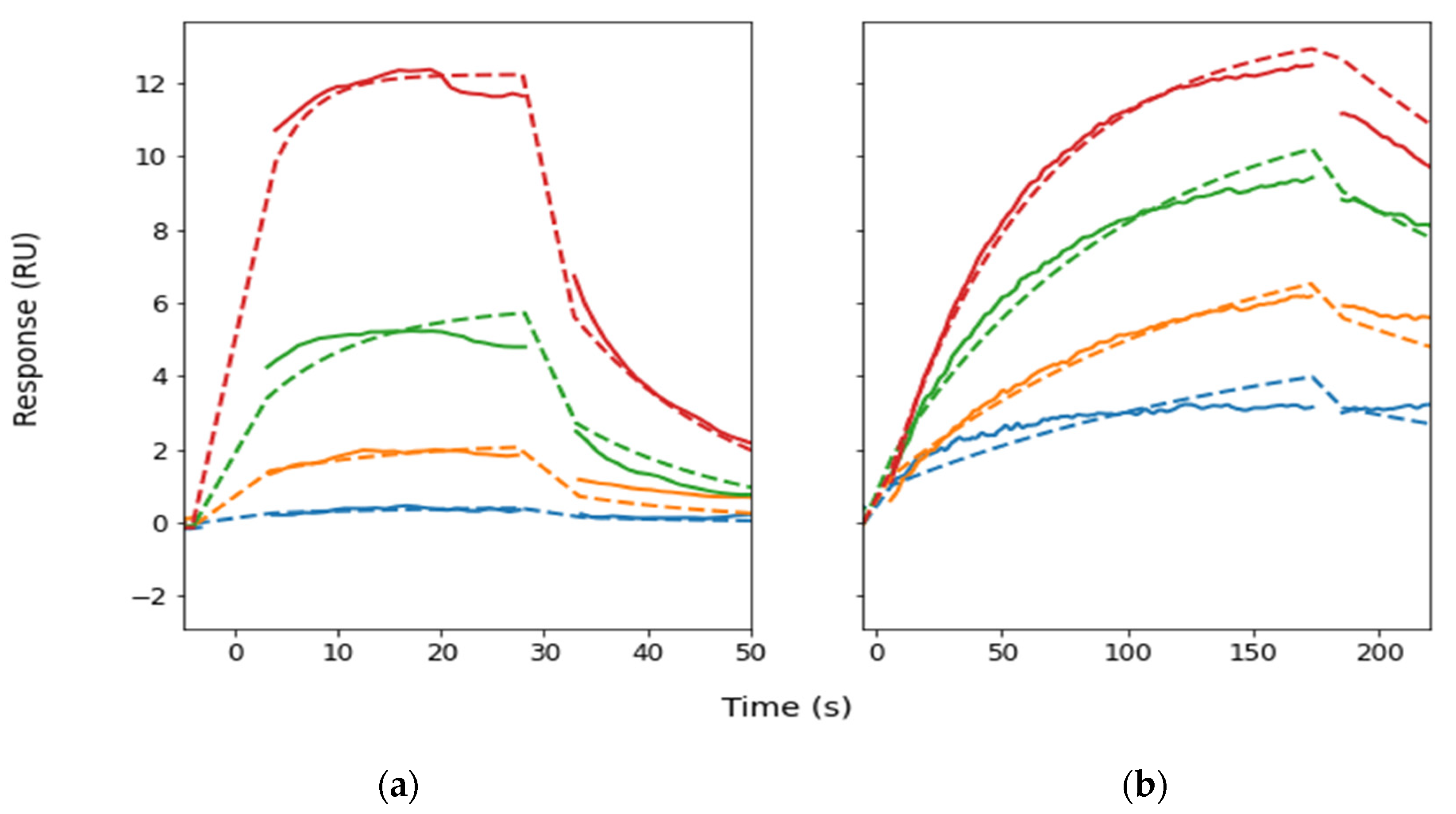
2. Results
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Langendorff Perfusions
4.3. Peptides
4.4. Infarct Size Measurements
4.5. Mitochondrial Isolation and Respiration
4.6. Binding Kinetics
4.7. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.R.; Ferdinandy, P.; Beyer, E.C.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in Cardiovascular and Neurovascular Health and Disease: Pharmacological Implications. Pharmacol. Rev. 2017, 69, 396–478. [Google Scholar] [CrossRef] [PubMed]
- Rusiecka, O.M.; Montgomery, J.; Morel, S.; Batista-Almeida, D.; Van Campenhout, R.; Vinken, M.; Girao, H.; Kwak, B.R. Canonical and Non-Canonical Roles of Connexin 43 in Cardioprotection. Biomolecules 2020, 10, 1225. [Google Scholar] [CrossRef]
- Boengler, K.; Leybaert, L.; Ruiz-Meana, M.; Schulz, R. Connexin 43 in Mitochondria: What Do We Really Know About Its Function? Front. Physiol. 2022, 13, 928934. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.M.; Kells, R.M.; Berthoud, V.M. Degradation of connexins and gap junctions. FEBS Lett. 2014, 588, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Sundset, R.; Cooper, M.; Mikalsen, S.O.; Ytrehus, K. Ischemic preconditioning protects against gap junctional uncoupling in cardiac myofibroblasts. Cell Commun. Adhes. 2004, 11, 51–66. [Google Scholar] [CrossRef]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef]
- Johansen, D.; Cruciani, V.; Sundset, R.; Ytrehus, K.; Mikalsen, S.O. Ischemia induces closure of gap junctional channels and opening of hemichannels in heart-derived cells and tissue. Cell Physiol. Biochem. 2011, 28, 103–114. [Google Scholar] [CrossRef]
- Ytrehus, K.; Liu, Y.; Downey, J.M. Preconditioning protects ischemic rabbit heart by protein kinase C activation. Am. J. Physiol. 1994, 266, H1145–H1152. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Garcia-Dorado, D.; Botker, H.E.; Davidson, S.M.; Downey, J.; Engel, F.B.; Jennings, R.; Lecour, S.; Leor, J.; Madonna, R.; et al. Novel targets and future strategies for acute cardioprotection: Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc. Res. 2017, 113, 564–585. [Google Scholar] [CrossRef]
- Miura, T.; Ohnuma, Y.; Kuno, A.; Tanno, M.; Ichikawa, Y.; Nakamura, Y.; Yano, T.; Miki, T.; Sakamoto, J.; Shimamoto, K. Protective role of gap junctions in preconditioning against myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H214–H221. [Google Scholar] [CrossRef]
- Wang, N.; De Vuyst, E.; Ponsaerts, R.; Boengler, K.; Palacios-Prado, N.; Wauman, J.; Lai, C.P.; De Bock, M.; Decrock, E.; Bol, M.; et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2013, 108, 309. [Google Scholar] [CrossRef]
- Hawat, G.; Benderdour, M.; Rousseau, G.; Baroudi, G. Connexin 43 mimetic peptide Gap26 confers protection to intact heart against myocardial ischemia injury. Pflug. Arch. 2010, 460, 583–592. [Google Scholar] [CrossRef]
- Lemieux, H.; Semsroth, S.; Antretter, H.; Hofer, D.; Gnaiger, E. Mitochondrial respiratory control and early defects of oxidative phosphorylation in the failing human heart. Int. J. Biochem. Cell Biol. 2011, 43, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Green, C.R.; Nicholson, L.F.; Bennet, L.; Gunn, A.J. Connexin hemichannel blockade is neuroprotective after, but not during, global cerebral ischemia in near-term fetal sheep. Exp. Neurol. 2013, 248, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Shintani-Ishida, K.; Uemura, K.; Yoshida, K. Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1714–H1720. [Google Scholar] [CrossRef]
- Pohl, U. Connexins: Key Players in the Control of Vascular Plasticity and Function. Physiol. Rev. 2020, 100, 525–572. [Google Scholar] [CrossRef]
- Saez, J.C.; Contreras-Duarte, S.; Gomez, G.I.; Labra, V.C.; Santibanez, C.A.; Gajardo-Gomez, R.; Avendano, B.C.; Diaz, E.F.; Montero, T.D.; Velarde, V.; et al. Connexin 43 Hemichannel Activity Promoted by Pro-Inflammatory Cytokines and High Glucose Alters Endothelial Cell Function. Front. Immunol. 2018, 9, 1899. [Google Scholar] [CrossRef]
- Hautefort, A.; Pfenniger, A.; Kwak, B.R. Endothelial connexins in vascular function. Vasc. Biol. 2019, 1, H117–H124. [Google Scholar] [CrossRef]
- Kim, Y.; Griffin, J.M.; Harris, P.W.; Chan, S.H.; Nicholson, L.F.; Brimble, M.A.; O’Carroll, S.J.; Green, C.R. Characterizing the mode of action of extracellular Connexin43 channel blocking mimetic peptides in an in vitro ischemia injury model. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 68–78. [Google Scholar] [CrossRef]
- Baines, C.P.; Goto, M.; Downey, J.M. Oxygen radicals released during ischemic preconditioning contribute to cardioprotection in the rabbit myocardium. J. Mol. Cell Cardiol. 1997, 29, 207–216. [Google Scholar] [CrossRef]
- Penna, C.; Mancardi, D.; Rastaldo, R.; Pagliaro, P. Cardioprotection: A radical view Free radicals in pre and postconditioning. Biochim. Biophys. Acta 2009, 1787, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Boardman, N.T.; Falck, A.T.; Lund, T.; Chu, X.; Martin-Armas, M.; Norvik, J.V.; Jenssen, T.G.; Ytrehus, K. Human concentrations of uric acid scavenges adaptive and maladaptive reactive oxygen species in isolated rat hearts subjected to ischemic stress. Can. J. Physiol. Pharmacol. 2020, 98, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, S.B.; Skovsted, G.F.; Berchtold, L.A.; Radziwon-Balicka, A.; Dreisig, K.; Edvinsson, L.; Sheykhzade, M.; Haanes, K.A. Role of pannexin and adenosine triphosphate (ATP) following myocardial ischemia/reperfusion. Scand. Cardiovasc. J. 2018, 52, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Vessey, D.A.; Li, L.; Kelley, M. P2X7 receptor agonists pre- and postcondition the heart against ischemia-reperfusion injury by opening pannexin-1/P2X (7) channels. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H881–H887. [Google Scholar] [CrossRef]
- Andelova, K.; Egan Benova, T.; Szeiffova Bacova, B.; Sykora, M.; Prado, N.J.; Diez, E.R.; Hlivak, P.; Tribulova, N. Cardiac Connexin-43 Hemichannels and Pannexin1 Channels: Provocative Antiarrhythmic Targets. Int. J. Mol. Sci. 2020, 22, 260. [Google Scholar] [CrossRef]
- Dahl, G. Gap junction-mimetic peptides do work, but in unexpected ways. Cell Commun. Adhes. 2007, 14, 259–264. [Google Scholar] [CrossRef]
- Juhaszova, M.; Zorov, D.B.; Kim, S.H.; Pepe, S.; Fu, Q.; Fishbein, K.W.; Ziman, B.D.; Wang, S.; Ytrehus, K.; Antos, C.L.; et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Investig. 2004, 113, 1535–1549. [Google Scholar] [CrossRef]
- Andreadou, I.; Schulz, R.; Papapetropoulos, A.; Turan, B.; Ytrehus, K.; Ferdinandy, P.; Daiber, A.; Di Lisa, F. The role of mitochondrial reactive oxygen species, NO and H2S in ischaemia/reperfusion injury and cardioprotection. J. Cell Mol. Med. 2020, 24, 6510–6522. [Google Scholar] [CrossRef]
- Liem, D.A.; Manintveld, O.C.; Schoonderwoerd, K.; McFalls, E.O.; Heinen, A.; Verdouw, P.D.; Sluiter, W.; Duncker, D.J. Ischemic preconditioning modulates mitochondrial respiration, irrespective of the employed signal transduction pathway. Transl. Res. 2008, 151, 17–26. [Google Scholar] [CrossRef]
- Munch-Ellingsen, J.; Lokebo, J.E.; Bugge, E.; Jonassen, A.K.; Ravingerova, T.; Ytrehus, K. 5-HD abolishes ischemic preconditioning independently of monophasic action potential duration in the heart. Basic Res. Cardiol. 2000, 95, 228–234. [Google Scholar] [CrossRef]
- Garlid, A.O.; Jaburek, M.; Jacobs, J.P.; Garlid, K.D. Mitochondrial reactive oxygen species: Which ROS signals cardioprotection? Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H960–H968. [Google Scholar] [CrossRef]
- Johansen, D.; Sanden, E.; Hagve, M.; Chu, X.; Sundset, R.; Ytrehus, K. Heptanol triggers cardioprotection via mitochondrial mechanisms and mitochondrial potassium channel opening in rat hearts. Acta Physiol. 2011, 201, 435–444. [Google Scholar] [CrossRef]
- Boengler, K.; Stahlhofen, S.; van de Sand, A.; Gres, P.; Ruiz-Meana, M.; Garcia-Dorado, D.; Heusch, G.; Schulz, R. Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res. Cardiol. 2009, 104, 141–147. [Google Scholar] [CrossRef]
- Hirschhauser, C.; Lissoni, A.; Gorge, P.M.; Lampe, P.D.; Heger, J.; Schluter, K.D.; Leybaert, L.; Schulz, R.; Boengler, K. Connexin 43 phosphorylation by casein kinase 1 is essential for the cardioprotection by ischemic preconditioning. Basic Res. Cardiol. 2021, 116, 21. [Google Scholar] [CrossRef] [PubMed]
- Srisakuldee, W.; Makazan, Z.; Nickel, B.E.; Zhang, F.; Thliveris, J.A.; Pasumarthi, K.B.; Kardami, E. The FGF-2-triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin 43-dependent. Cardiovasc. Res. 2014, 103, 72–80. [Google Scholar] [CrossRef] [PubMed]
- da Silva, M.M.; Sartori, A.; Belisle, E.; Kowaltowski, A.J. Ischemic preconditioning inhibits mitochondrial respiration, increases H2O2 release, and enhances K+ transport. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H154–H162. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Ruiz-Meana, M.; Gent, S.; Ungefug, E.; Soetkamp, D.; Miro-Casas, E.; Cabestrero, A.; Fernandez-Sanz, C.; Semenzato, M.; Di Lisa, F.; et al. Mitochondrial connexin 43 impacts on respiratory complex I activity and mitochondrial oxygen consumption. J. Cell Mol. Med. 2012, 16, 1649–1655. [Google Scholar] [CrossRef]
- Wang, M.; Smith, K.; Yu, Q.; Miller, C.; Singh, K.; Sen, C.K. Mitochondrial connexin 43 in sex-dependent myocardial responses and estrogen-mediated cardiac protection following acute ischemia/reperfusion injury. Basic Res. Cardiol. 2019, 115, 1. [Google Scholar] [CrossRef]
- Madonna, R.; Moscato, S.; Polizzi, E.; Pieragostino, D.; Cufaro, M.C.; Del Boccio, P.; Bianchi, F.; De Caterina, R.; Mattii, L. Connexin 43 and Connexin 26 Involvement in the Ponatinib-Induced Cardiomyopathy: Sex-Related Differences in a Murine Model. Int. J. Mol. Sci. 2021, 22, 5815. [Google Scholar] [CrossRef]
- Tribulova, N.; Dupont, E.; Soukup, T.; Okruhlicova, L.; Severs, N.J. Sex differences in connexin-43 expression in left ventricles of aging rats. Physiol. Res. 2005, 54, 705–708. [Google Scholar]
- Genade, S.; Ytrehus, K.; Lochner, A. Melatonin prevents cardioprotection induced by a multi-cycle ischaemic preconditioning protocol in the isolated perfused rat heart. Cardiovasc. J. S. Afr. 2006, 17, 239–244. [Google Scholar] [PubMed]
- Martiouchova, K.; Ytrehus, K. Ischemic and bradykinin stimulated release of prostaglandins may play a role in protection in the isolated rat heart model of ischemic preconditioning (abstract). Circulation 1999, 100 (Suppl. S18), 491. [Google Scholar]
- Martiouchova, K.; Ytrehus, K. Protection by ischemic preconditioning (IP) in the rat heart is dependent on cyclooxygenase activity (abstract). J. Mol. Cell Cardiol. 1999, 31, A96. [Google Scholar]
- Hawat, G.; Helie, P.; Baroudi, G. Single intravenous low-dose injections of connexin 43 mimetic peptides protect ischemic heart in vivo against myocardial infarction. J. Mol. Cell Cardiol. 2012, 53, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.W.; Tandler, B.; Hoppel, C.L. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 1977, 252, 8731–8739. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxygen Flux [nmol O2 (µmol IU CS Activity)−1] | Glutamate + Maleate | +ADP 50 µM | State 4 (LEAK ATP) | OXPHOS Saturating +ADP 2.5 mM | +Oligomycin | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| crt | IPC | crt | IPC | crt | IPC | crt | IPC | crt | IPC | |
| Without peptide | ||||||||||
| 16.1 ± 2.9 | 17.4 ± 3.4 | 69.9 ± 16.0 | 75.1 ± 15.8 | 20.8 ± 3.6 | 22.7 ± 2.5 | 114.5 ± 22.3 | 106.6 ± 18.6 | 42.5 ± 5.0 | 39.0 ± 6.3 | |
| Gap19 (µM) | ||||||||||
| 0.5 | 16.6 ± 5.0 | 15.2 ± 4.2 | 68.9 ± 6.5 | 69.8 ± 28.0 | 23.8 ± 5.2 | 21.2 ± 5.3 | 89.2 ± 35.6 | 92.5 ± 25.1 | 42.8 ± 4.5 | 37.8 ± 7.4 |
| 5 | 16.6 ± 2.6 | 15.9 ± 1.7 | 75.1 ± 11.3 | 71.4 ± 7.5 | 22.1 ± 1.6 | 21.0 ± 2.6 | 113.7 ± 12.9 | 98.3 ± 10.5 | 43.5 ± 2.6 | 39.0 ± 3.7 |
| 50 | 18.2 ± 0.9 | 19.6 ± 6.1 | 78.9 ± 14.0 | 76.8 ± 21.6 | 24.8 ± 3.0 | 24.0 ± 6.1 | 115.3 ± 8.7 | 116.2 ± 37.7 | 44.6 ± 5.1 | 44.2 ± 7.5 |
| Gap26 (µM) | ||||||||||
| 0.5 | 15.0 ± 3.2 | 17.7 ± 4.5 | 63.7 ± 6.7 | 71.6 ± 28.5 | 21.4 ± 1.0 | 22.8 ± 4.4 | 99.2 ± 18.0 | 93.0 ± 20.7 | 41.3 ± 5.1 | 37.2 ± 4.7 |
| 5 | 16.3 ± 3.5 | 16.3 ± 2.6 | 77.1 ± 10.6 | 71.3 ± 7.5 | 21.2 ± 1.7 | 21.3 ± 2.8 | 118.6 ± 25.5 | 98.2 ± 6.1 | 45.0 ± 7.5 | 38.0 ± 6.0 |
| 50 | 16.8 ± 1.9 | 18.8 ± 4.1 | 74.5 ± 10.2 | 80.8 ± 19.9 | 21.6 ± 0.9 | 24.1 ± 3.5 | 100.0 ± 13.1 | 109.5 ± 23.1 | 37.1 ± 10.1 | 41.1 ± 11.4 |
| Gap27 (µM) | ||||||||||
| 0.5 | 16.3 ± 6.2 | 15.1 ± 2.1 | 67.7 ± 11.3 | 66.2 ± 13.2 | 22.7 ± 4.6 | 19.3 ± 1.8 | 103.3 ± 20.4 | 93.6 ± 4.1 | 43.1 ± 4.1 | 35.0 ± 6.7 |
| 5 | 15.8 ± 3.2 | 15.5 ± 3.7 | 68.8 ± 10.3 | 71.0 ± 15.3 | 20.8 ± 1.1 | 20.1 ± 2.5 | 112.9 ± 28.2 | 96.7 ± 11.0 | 41.8 ± 5.8 | 38.8 ± 8.0 |
| 50 | 14.7 ± 1.5 | 17.0 ± 15.2 | 46.4 ± 12.1 | 68.9 ± 17.9 | 21.3 ± 2.8 | 22.5 ± 3.6 | 92.1 ± 12.0 | 94.8 ± 22.7 | 41.6 ± 5.1 | 40.8 ± 7.6 |
| ka (1/ms) | kd (1/s) | KD (µM) | |
|---|---|---|---|
| EL1-G27 | 10,000 (1000) | 0.061 (0.004) | 6 |
| EL2-G26 | 511 (7) | 0.0043 (0.0001) | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falck, A.T.; Lund, B.A.; Johansen, D.; Lund, T.; Ytrehus, K. The Ambivalence of Connexin43 Gap Peptides in Cardioprotection of the Isolated Heart against Ischemic Injury. Int. J. Mol. Sci. 2022, 23, 10197. https://doi.org/10.3390/ijms231710197
Falck AT, Lund BA, Johansen D, Lund T, Ytrehus K. The Ambivalence of Connexin43 Gap Peptides in Cardioprotection of the Isolated Heart against Ischemic Injury. International Journal of Molecular Sciences. 2022; 23(17):10197. https://doi.org/10.3390/ijms231710197
Chicago/Turabian StyleFalck, Aleksander Tank, Bjarte Aarmo Lund, David Johansen, Trine Lund, and Kirsti Ytrehus. 2022. "The Ambivalence of Connexin43 Gap Peptides in Cardioprotection of the Isolated Heart against Ischemic Injury" International Journal of Molecular Sciences 23, no. 17: 10197. https://doi.org/10.3390/ijms231710197
APA StyleFalck, A. T., Lund, B. A., Johansen, D., Lund, T., & Ytrehus, K. (2022). The Ambivalence of Connexin43 Gap Peptides in Cardioprotection of the Isolated Heart against Ischemic Injury. International Journal of Molecular Sciences, 23(17), 10197. https://doi.org/10.3390/ijms231710197