
From Double-Strand Break Recognition to Cell-Cycle Checkpoint Activation: High Content and Resolution Image Cytometry Unmasks 53BP1 Multiple Roles in DNA Damage Response and p53 Action
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
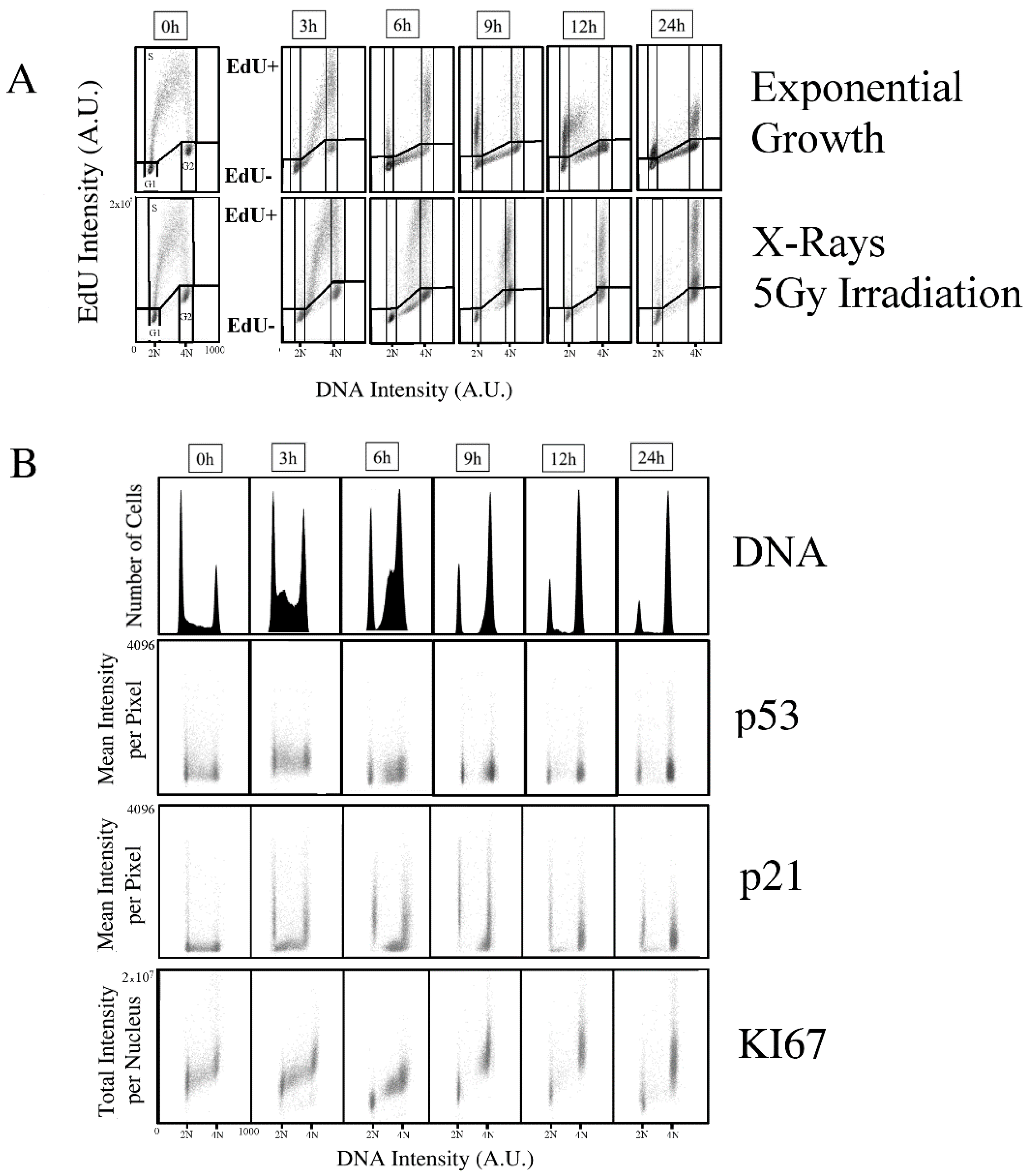
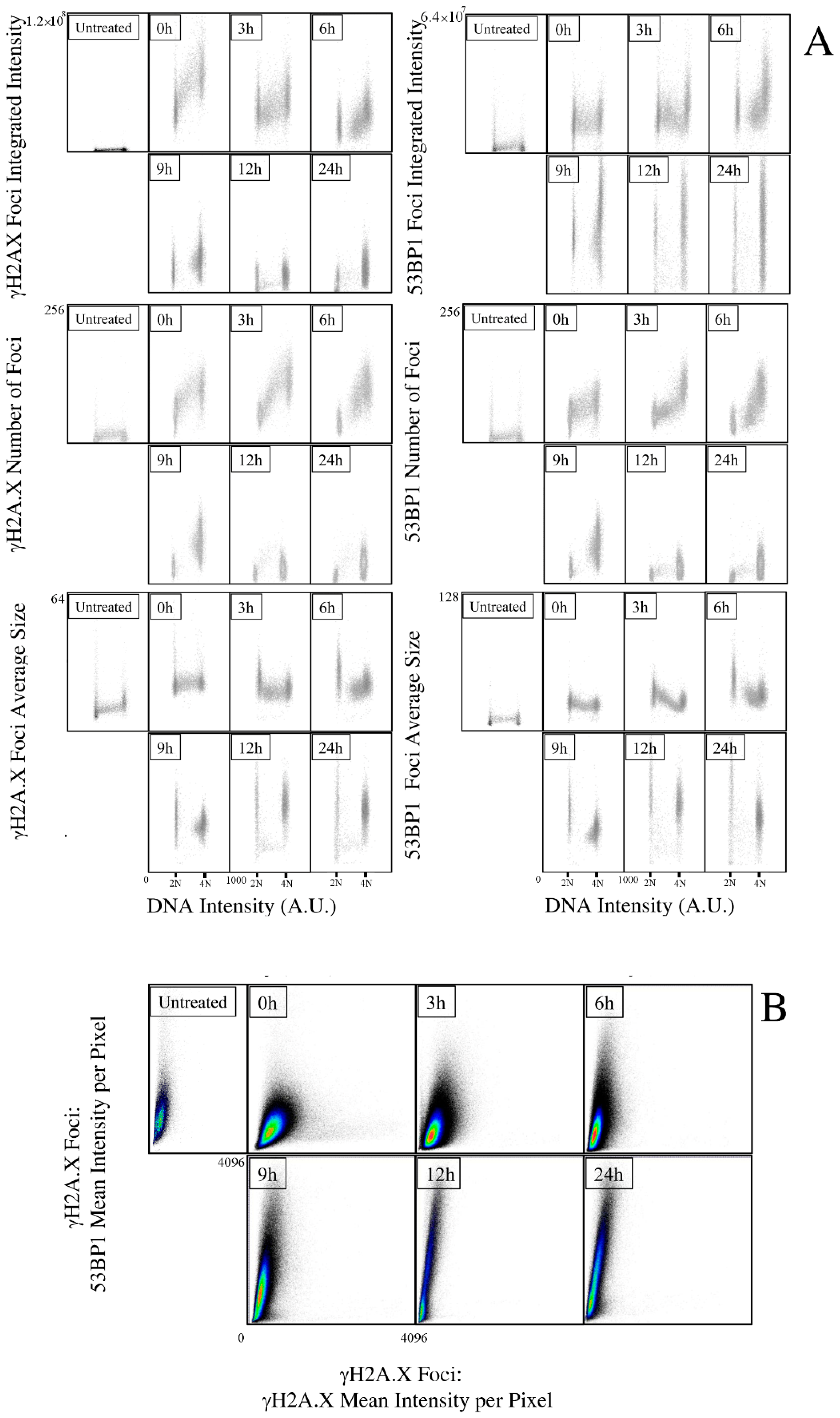
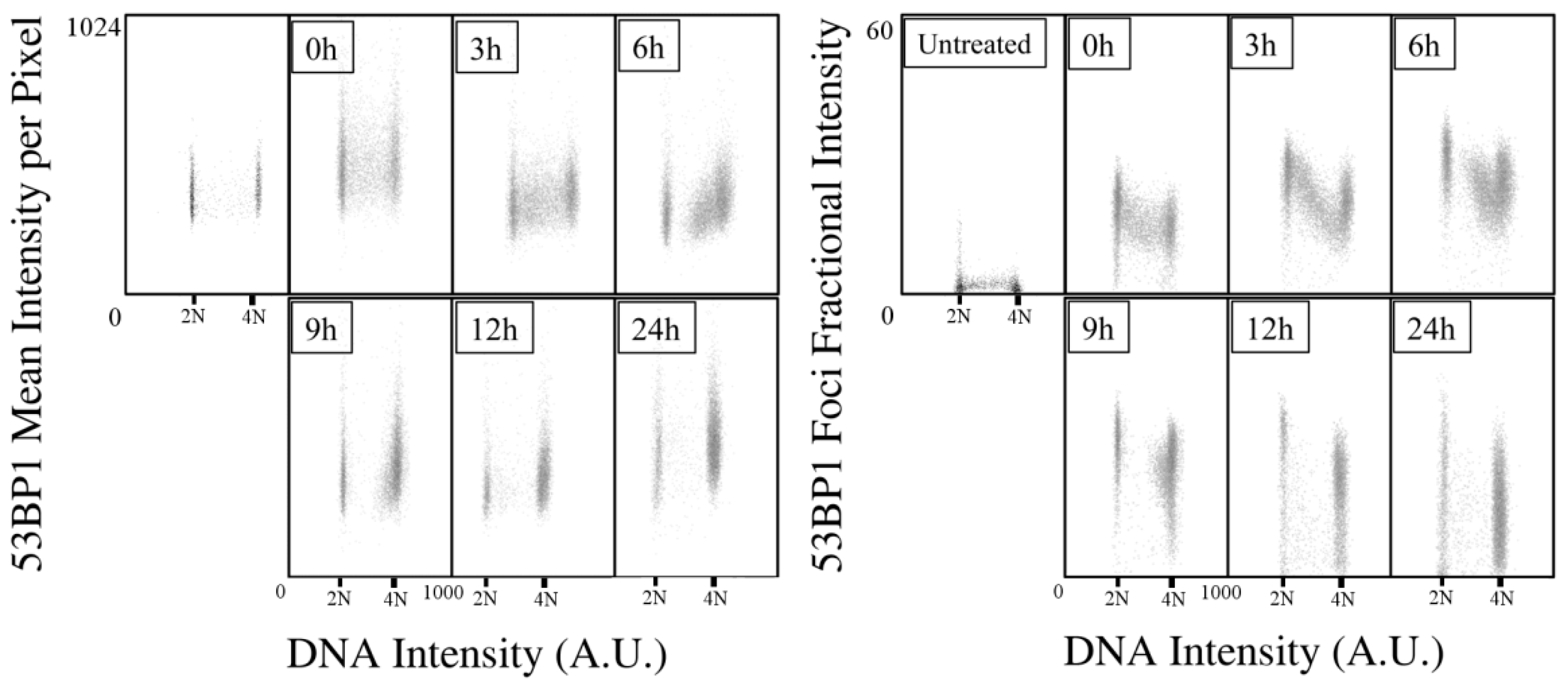
2.1. Kinetics of X-rays Irradiation Induced DNA Damage, Processing, and Cell-Cycle Arrest
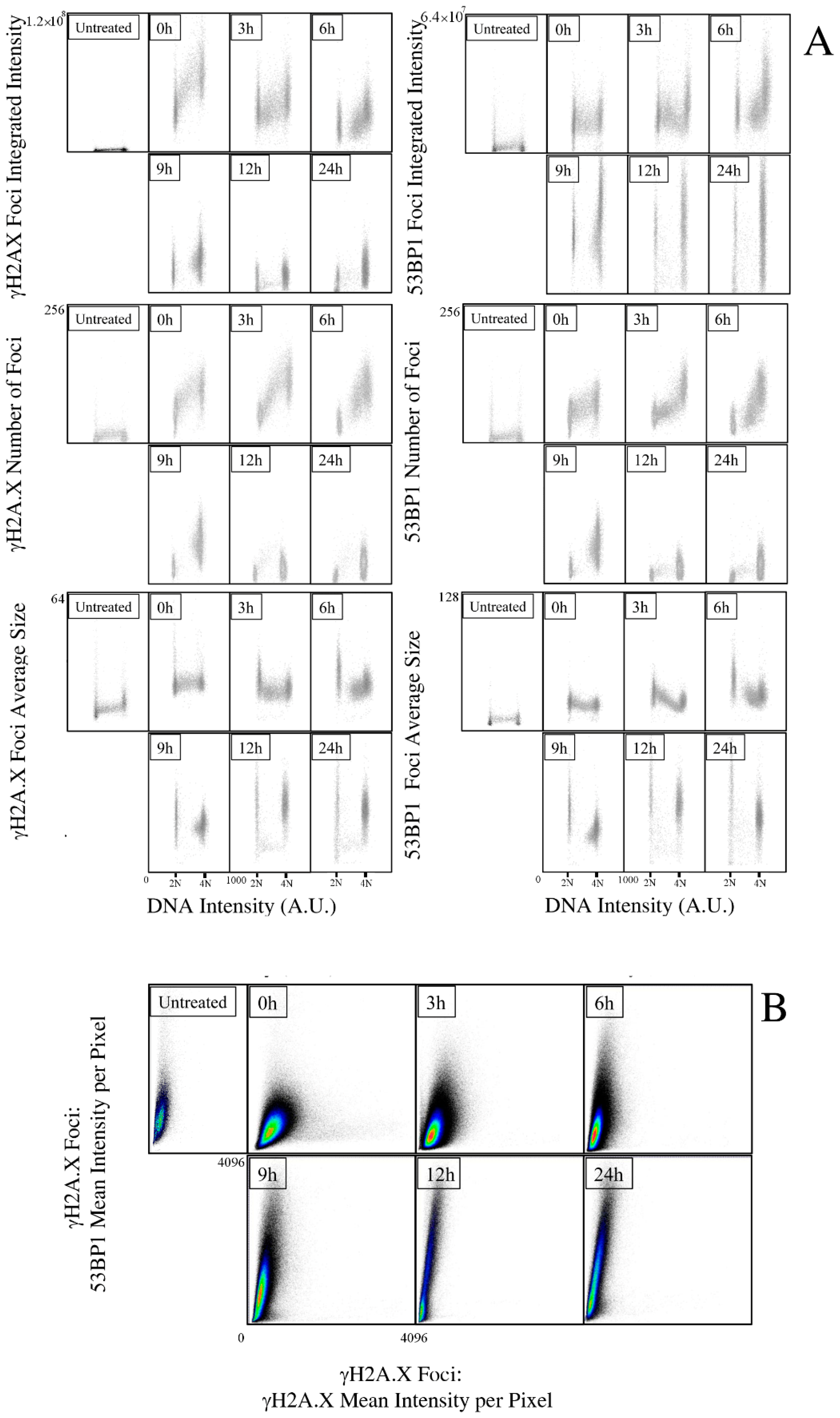
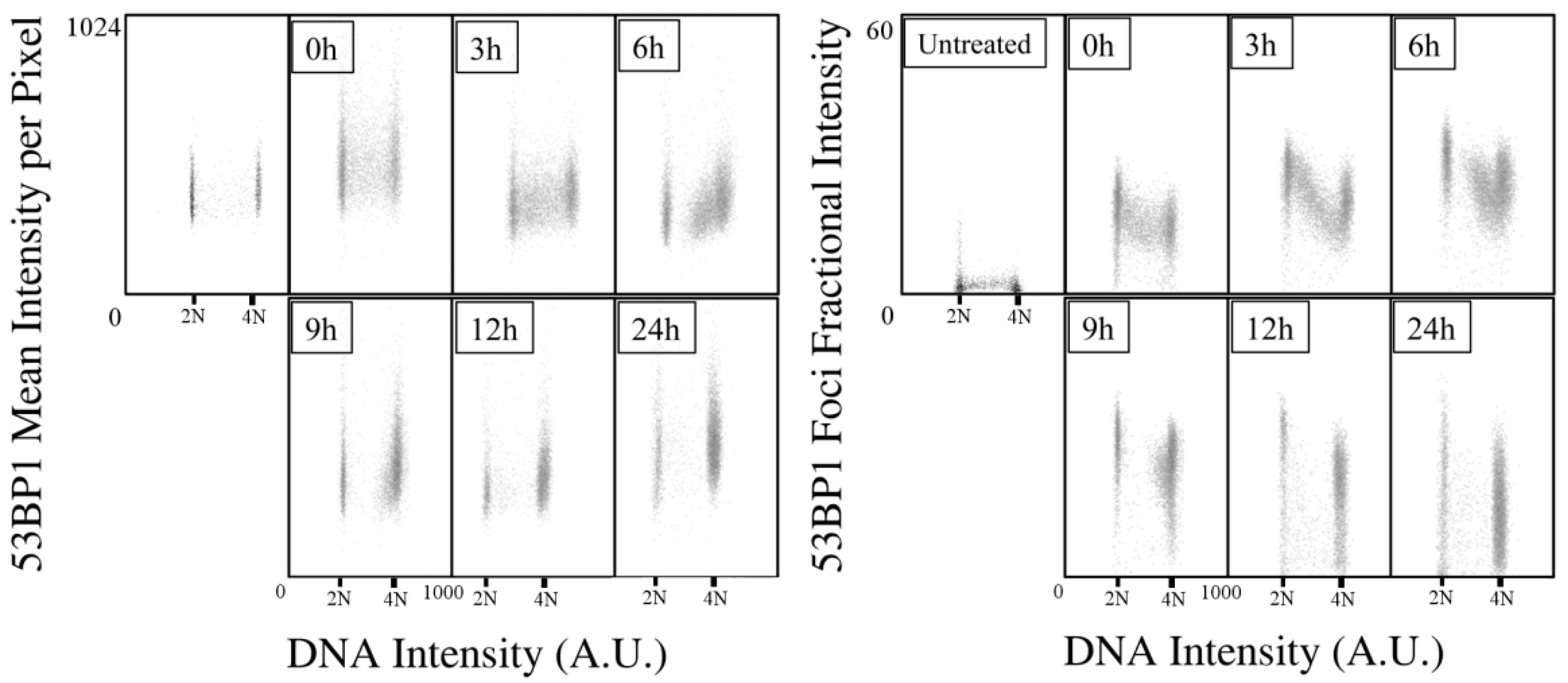
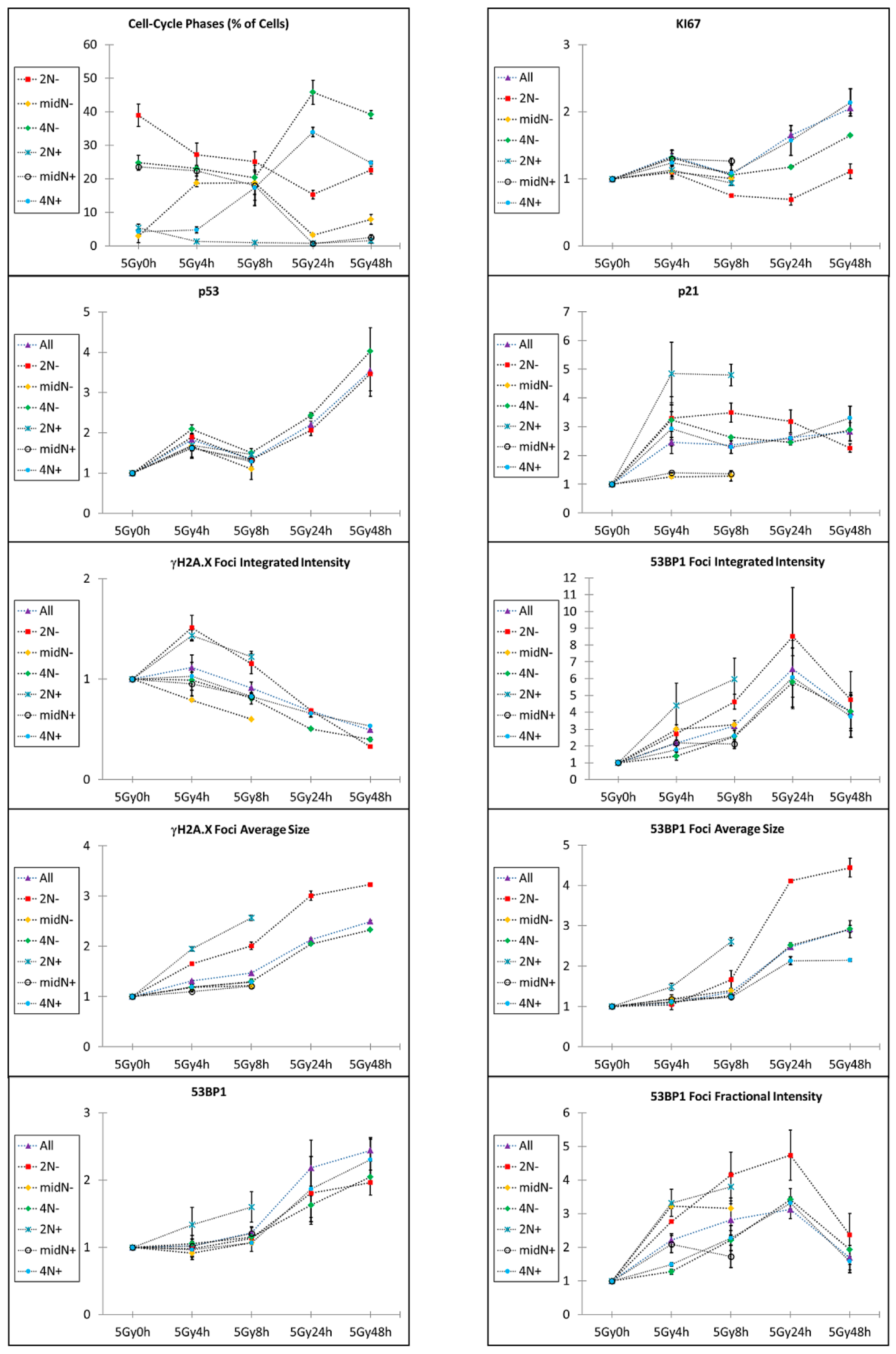
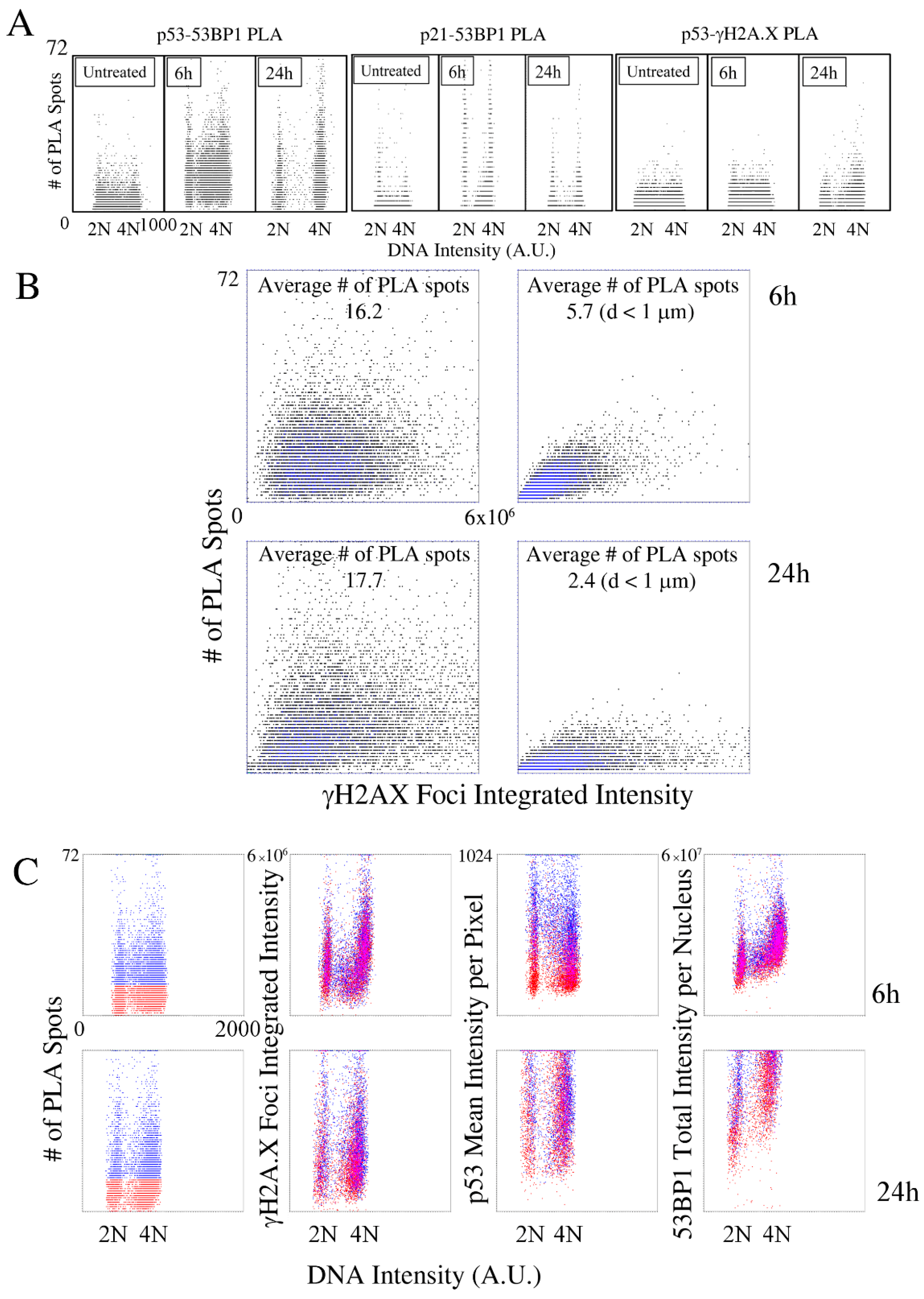
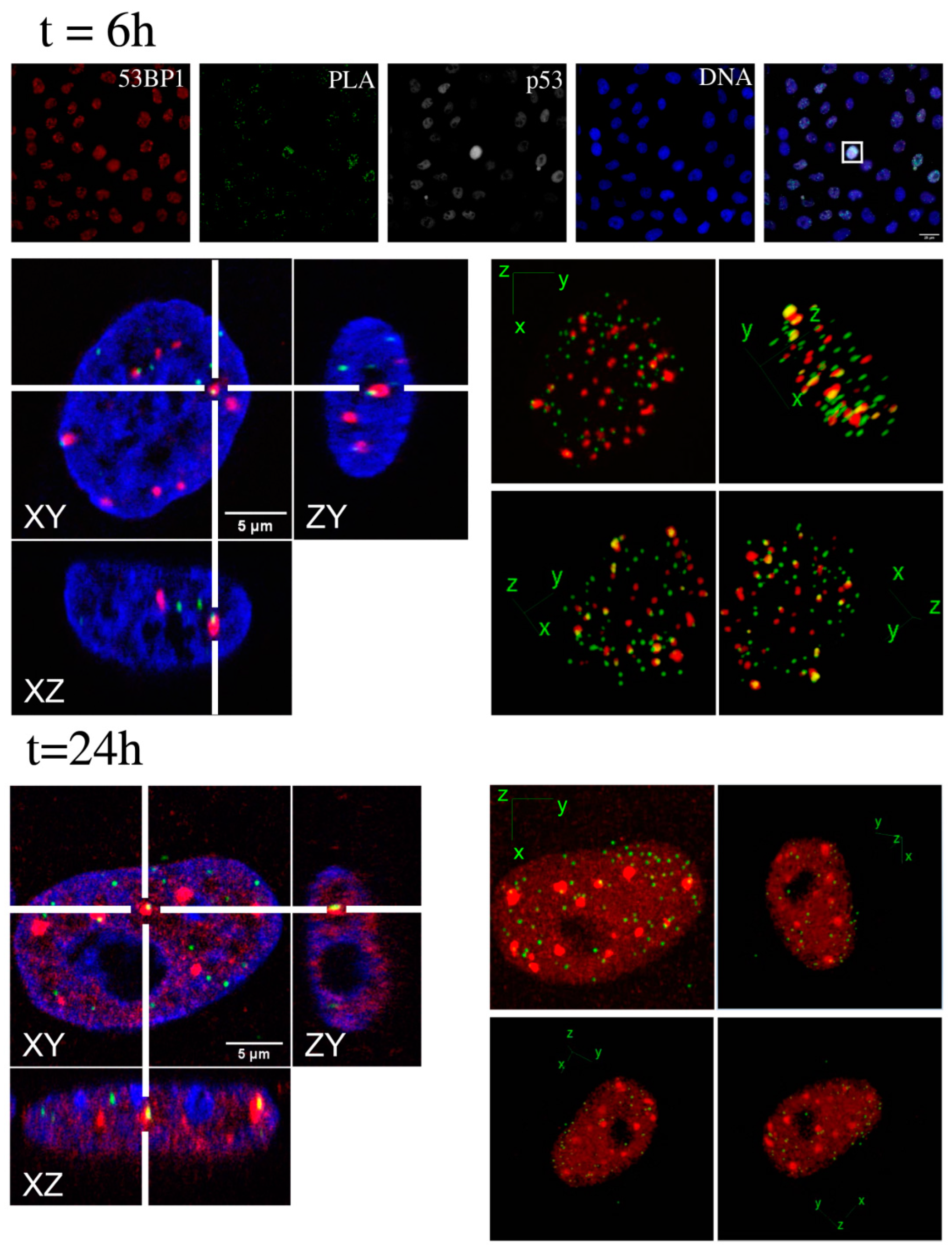
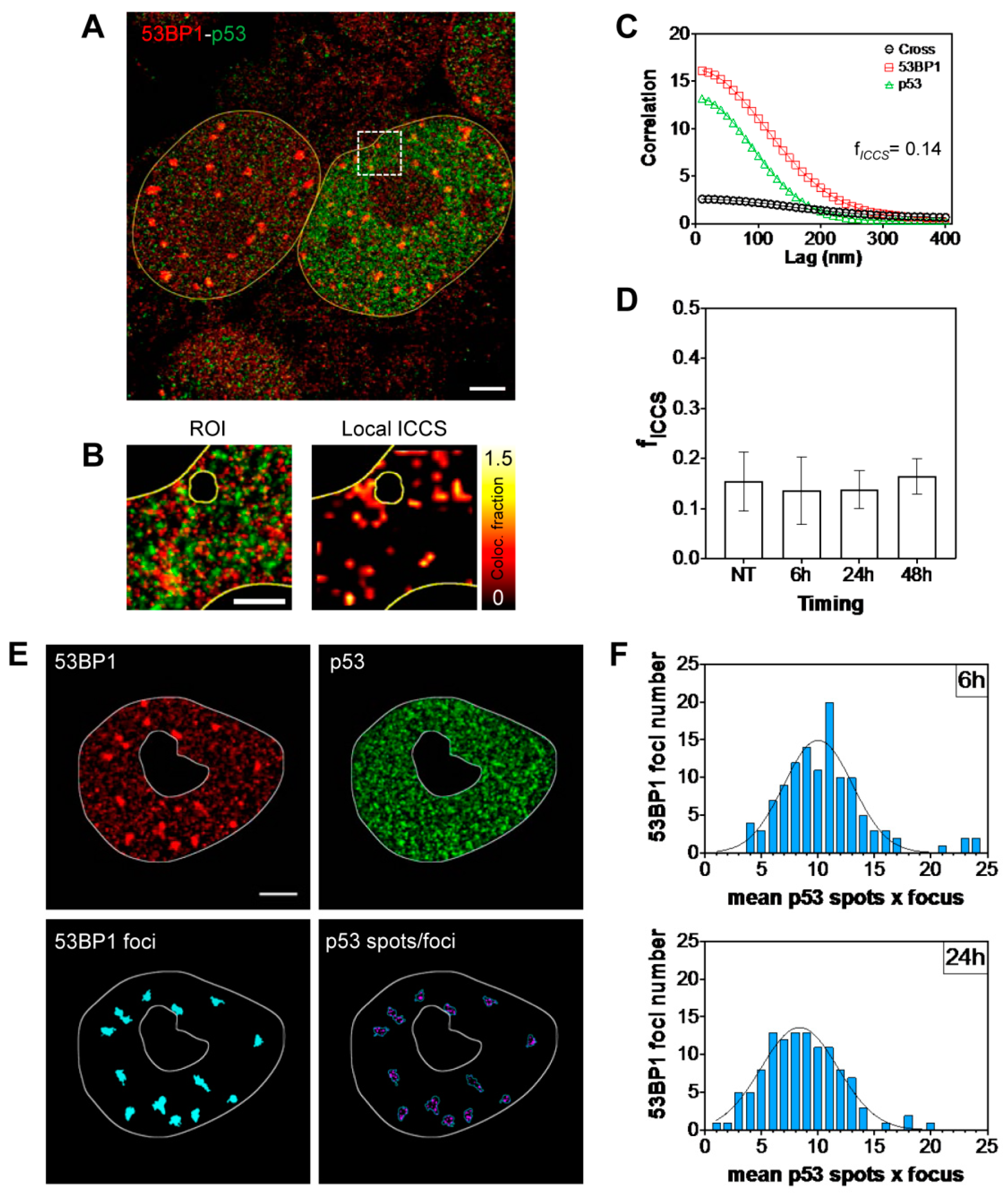
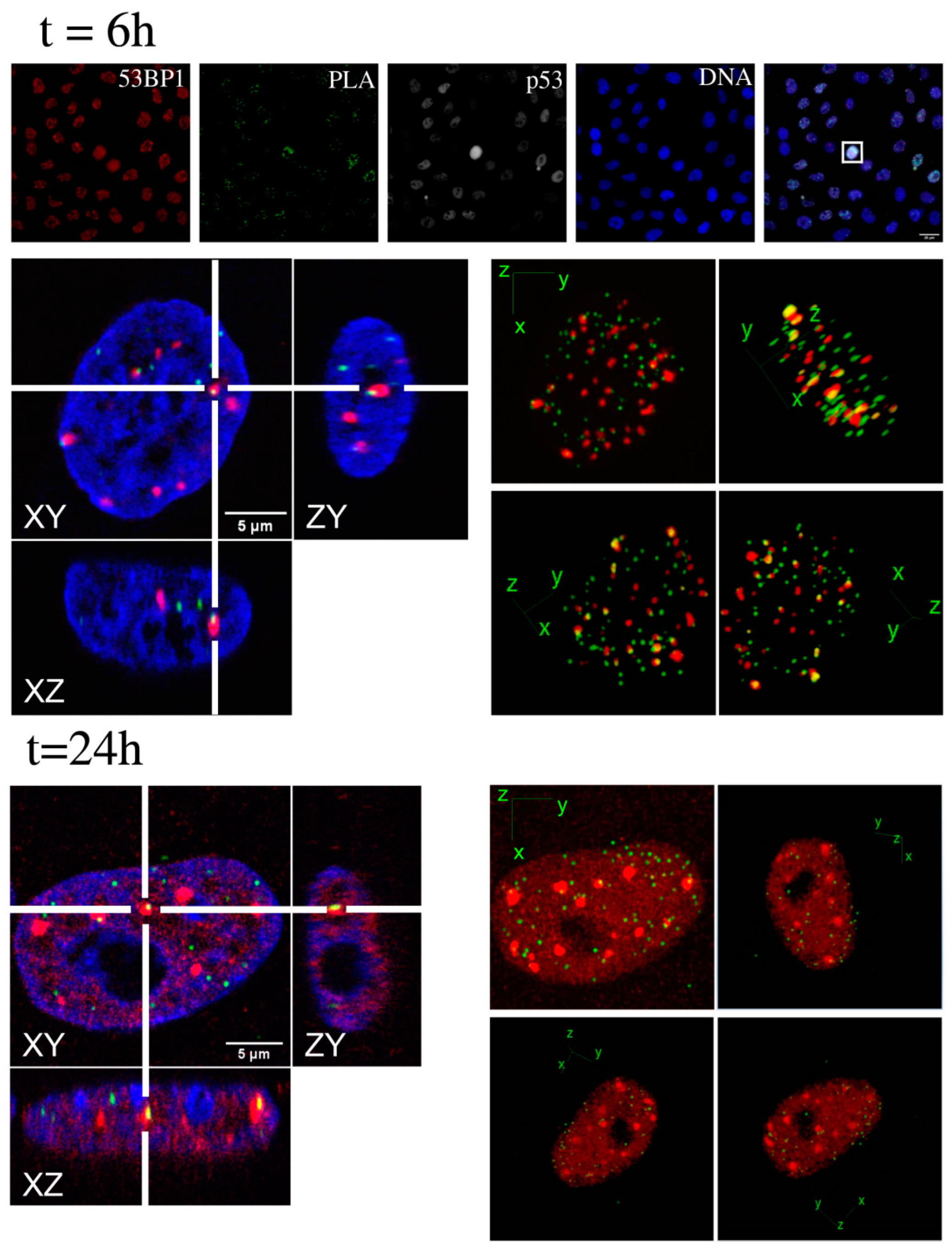
2.2. Kinetic High-Resolution Analysis of the Interactions among DDR and Checkpoint Molecular Networks
3. Material and Methods
3.1. Cell Culture
3.2. EdU Staining and Immunofluorescence of MCF10A Cells
3.3. In-Situ Proximity Ligation Analysis (PLA)
3.4. Automated Microscopy and Image Acquisition
3.5. Image Analysis (A.M.I.CO Analysis Package)
3.6. Pulse and Chase EdU Assay
3.7. dStorm Imaging
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kieffer, S.R.; Lowndes, N.F. Immediate-Early, Early, and Late Responses to DNA Double Stranded Breaks. Front. Genet. 2022, 13, 793884. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. Seminars in Cancer Biology DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 2016, 37–38, 51–64. [Google Scholar] [CrossRef]
- Sperka, T.; Wang, J.; Rudolph, K.L. DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell Biol. 2012, 13, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Mallette, F.A.; Ferbeyre, G. The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell Cycle 2007, 6, 1831–1836. [Google Scholar] [CrossRef]
- Elbakry, A.; Löbrich, M. Homologous Recombination Subpathways: A Tangle to Resolve. Front. Genet. 2021, 12, 723847. [Google Scholar] [CrossRef]
- Chen, Z.; Tyler, J.K. The Chromatin Landscape Channels DNA Double-Strand Breaks to Distinct Repair Pathways. Front. Cell Dev. Biol. 2022, 10, 909696. [Google Scholar] [CrossRef]
- Zimmermann, M.; de Lange, T. 53BP1: Pro choice in DNA repair. Trends Cell Biol. 2014, 24, 108–117. [Google Scholar] [CrossRef]
- Iwabuchi, K.; Basu, B.P.; Kysela, B.; Kurihara, T.; Shibata, M.; Guan, D.; Cao, Y.; Hamada, T.; Imamura, K.; Jeggo, P.A.; et al. Potential role for 53BP1 in DNA end-joining repair through direct interaction with DNA. J. Biol. Chem. 2003, 278, 36487–36495. [Google Scholar] [CrossRef] [Green Version]
- Mirman, Z.; de Lange, T. 53BP1: A DSB escort. Genes Dev. 2020, 34, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Iwabuchi, K.; Bartel, P.L.; Li, B.; Marraccino, R.; Fields, S. Two cellular proteins that bind to wild-type but not mutant p53. Proc. Natl. Acad. Sci. USA 1994, 91, 6098–6102. [Google Scholar] [CrossRef] [PubMed]
- Iwabuchi, K.; Li, B.; Massa, H.F.; Trask, B.J.; Date, T.; Fields, S. Stimulation of p53-mediated transcriptional activation by the p53-binding proteins, 53BP1 and 53BP2. J. Biol. Chem. 1998, 273, 26061–26068. [Google Scholar] [CrossRef] [PubMed]
- Cuella-Martin, R.; Oliveira, C.; Lockstone, H.E.; Snellenberg, S.; Grolmusova, N.; Chapman Correspondence, J.R.; Chapman, J.R. 53BP1 Integrates DNA Repair and p53-Dependent Cell Fate Decisions via Distinct Mechanisms. Mol. Cell 2016, 64, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Ghodke, I.; Remisova, M.; Furst, A.; Kilic, S.; Reina-San-Martin, B.; Poetsch, A.R.; Altmeyer, M.; Soutoglou, E. AHNAK controls 53BP1-mediated p53 response by restraining 53BP1 oligomerization and phase separation. Mol. Cell 2021, 81, 2596–2610.e7. [Google Scholar] [CrossRef]
- Rizzotto, D.; Englmaier, L.; Villunger, A. At a crossroads to cancer: How p53-induced cell fate decisions secure genome integrity. Int. J. Mol. Sci. 2021, 22, 10883. [Google Scholar] [CrossRef]
- Parnandi, N.; Rendo, V.; Cui, G.; Botuyan, M.V.; Remisova, M.; Nguyen, H.; Drané, P.; Beroukhim, R.; Altmeyer, M.; Mer, G.; et al. TIRR inhibits the 53BP1-p53 complex to alter cell-fate programs. Mol. Cell 2021, 81, 2583–2595.e6. [Google Scholar] [CrossRef]
- Hell, S.W. Nanoscopy with Focused Light (Nobel Lecture). Angew. Chemie Int. Ed. 2015, 54, 8054–8066. [Google Scholar] [CrossRef]
- Huang, B.; Jones, S.A.; Brandenburg, B.; Zhuang, X. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nat. Methods 2008, 5, 1047–1052. [Google Scholar] [CrossRef]
- Sahl, S.J.; Hell, S.W.; Jakobs, S. Fluorescence nanoscopy in cell biology. Nat. Rev. Mol. Cell Biol. 2017, 18, 685–701. [Google Scholar] [CrossRef]
- Situ, I.; Imaging, S.; Dna, G.; Beliveau, B.J.; Boettiger, A.N.; Nir, G.; Bintu, B.; Yin, P.; Zhuang, X.; Wu, C. Super-Resolution Microscopy; Springer: Berlin/Heidelberg, Germany, 2017; Volume 1663, pp. 231–252. [Google Scholar] [CrossRef]
- Furia, L.; Pelicci, P.G.; Faretta, M. A computational platform for robotized fluorescence microscopy (I): High-content image-based cell-cycle analysis. Cytometry. A 2013, 83, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Furia, L.; Pelicci, P.; Faretta, M. Confocal microscopy for high-resolution and high-content analysis of the cell cycle. Curr. Protoc. Cytom. 2014, 2014, 7.42.1–7.42.14. [Google Scholar] [CrossRef] [PubMed]
- Furia, L.; Pelicci, P.; Faretta, M. High-resolution cytometry for high-content cell cycle analysis. Curr. Protoc. Cytom. 2014, 2014, 7.41.1–7.41.15. [Google Scholar] [CrossRef]
- Furia, L.; Pelicci, P.G.; Faretta, M. A computational platform for robotized fluorescence microscopy (II): DNA damage, replication, checkpoint activation, and cell cycle progression by high-content high-resolution multiparameter image-cytometry. Cytometry. A 2013, 83, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Leuchowius, K.; Weibrecht, I.; Ola, S. In Situ Proximity Ligation Assay for Microscopy and Flow Cytometry. Curr. Protoc. Cytom. 2011, 56, 9–36. [Google Scholar] [CrossRef]
- Söderberg, O.; Gullberg, M.; Jarvius, M.; Ridderstråle, K.; Leuchowius, K.J.; Jarvius, J.; Wester, K.; Hydbring, P.; Bahram, F.; Larsson, L.G.; et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 2006, 3, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Hong, R.; Roberts, E.; Bieniarz, C. In Situ Detection of Protein Complexes and Modi fi cations by Chemical Ligation Proximity Assay. Bioconjug. Chem. 2016, 27, 1690–1696. [Google Scholar] [CrossRef]
- Bergamaschi, D.; Ronzoni, S.; Taverna, S.; Faretta, M.; De Feudis, P.; Faircloth, G.; Jimeno, J.; Erba, E.; D’Incalci, M. Cell cycle perturbations and apoptosis induced by isohomohalichondrin B (IHB), a natural marine compound. Br. J. Cancer 1999, 79, 267–277. [Google Scholar] [CrossRef]
- Dion, V.; Kalck, V.; Horigome, C.; Towbin, B.D.; Gasser, S.M. Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat. Cell Biol. 2012, 14, 502–509. [Google Scholar] [CrossRef]
- Schrank, B.; Gautier, J. Assembling nuclear domains: Lessons from DNA repair. J. Cell Biol. 2019, 218, 2444–2455. [Google Scholar] [CrossRef]
- Kruhlak, M.J.; Celeste, A.; Dellaire, G.; Fernandez-Capetillo, O.; Müller, W.G.; McNally, J.G.; Bazett-Jones, D.P.; Nussenzweig, A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 2006, 172, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Carr, S.M.; Munro, S.; Zalmas, L.-P.; Fedorov, O.; Johansson, C.; Krojer, T.; Sagum, C.A.; Bedford, M.T.; Oppermann, U.; La Thangue, N.B. Lysine methylation-dependent binding of 53BP1 to the pRb tumor suppressor. Proc. Natl. Acad. Sci. USA 2014, 111, 11341–11346. [Google Scholar] [CrossRef] [PubMed]
- Ticli, G.; Cazzalini, O.; Stivala, L.A.; Prosperi, E. Revisiting the Function of p21CDKN1A in DNA Repair: The Influence of Protein Interactions and Stability. Int. J. Mol. Sci. 2022, 23, 7058. [Google Scholar] [CrossRef] [PubMed]
- Pelicci, S.; Furia, L.; Scanarini, M.; Pelicci, P.G.; Lanzan, L.; Faretta, M. Novel Tools to Measure Single Molecules Colocalization in Fluorescence Nanoscopy by Image Cross Correlation Spectroscopy. Nanomaterials 2022, 12, 686. [Google Scholar] [CrossRef] [PubMed]
- van de Kamp, G.; Heemskerk, T.; Kanaar, R.; Essers, J. DNA Double Strand Break Repair Pathways in Response to Different Types of Ionizing Radiation. Front. Genet. 2021, 12, 738230. [Google Scholar] [CrossRef]
- Matejka, N.; Reindl, J. Influence of α-Particle Radiation on Intercellular Communication Networks of Tunneling Nanotubes in U87 Glioblastoma Cells. Front. Oncol. 2020, 10, 1691. [Google Scholar] [CrossRef]
- Klammer, H.; Mladenov, E.; Li, F.; Iliakis, G. Bystander effects as manifestation of intercellular communication of DNA damage and of the cellular oxidative status. Cancer Lett. 2015, 356, 58–71. [Google Scholar] [CrossRef]
- Berzsenyi, I.; Pantazi, V.; Borsos, B.N.; Pankotai, T. Systematic overview on the most widespread techniques for inducing and visualizing the DNA double-strand breaks. Mutat. Res. Rev. Mutat. Res. 2021, 788, 108397. [Google Scholar] [CrossRef]
- Yan, W.X.; Mirzazadeh, R.; Garnerone, S.; Scott, D.; Schneider, M.W.; Kallas, T.; Custodio, J.; Wernersson, E.; Li, Y.; Gao, L.; et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 2017, 8, 15058. [Google Scholar] [CrossRef] [Green Version]
- Dellino, G.I.; Palluzzi, F.; Chiariello, A.M.; Piccioni, R.; Bianco, S.; Furia, L.; De Conti, G.; Bouwman, B.A.M.; Melloni, G.; Guido, D.; et al. Release of paused RNA polymerase II at specific loci favors DNA double-strand-break formation and promotes cancer translocations. Nat. Genet. 2019, 51, 1011–1023. [Google Scholar] [CrossRef]
- Crosetto, N.; Mitra, A.; Silva, M.J.; Bienko, M.; Dojer, N.; Wang, Q.; Karaca, E.; Chiarle, R.; Skrzypczak, M.; Ginalski, K.; et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 2013, 10, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Ho, T.L.F.; Hariharan, A.; Goh, H.C.; Wong, Y.L.; Verkaik, N.S.; Lee, M.Y.; Tam, W.L.; van Gent, D.C.; Venkitaraman, A.R.; et al. Rapid recruitment of p53 to DNA damage sites directs DNA repair choice and integrity. Proc. Natl. Acad. Sci. USA 2022, 119, e2113233119. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furia, L.; Pelicci, S.; Scanarini, M.; Pelicci, P.G.; Faretta, M. From Double-Strand Break Recognition to Cell-Cycle Checkpoint Activation: High Content and Resolution Image Cytometry Unmasks 53BP1 Multiple Roles in DNA Damage Response and p53 Action. Int. J. Mol. Sci. 2022, 23, 10193. https://doi.org/10.3390/ijms231710193
Furia L, Pelicci S, Scanarini M, Pelicci PG, Faretta M. From Double-Strand Break Recognition to Cell-Cycle Checkpoint Activation: High Content and Resolution Image Cytometry Unmasks 53BP1 Multiple Roles in DNA Damage Response and p53 Action. International Journal of Molecular Sciences. 2022; 23(17):10193. https://doi.org/10.3390/ijms231710193
Chicago/Turabian StyleFuria, Laura, Simone Pelicci, Mirco Scanarini, Pier Giuseppe Pelicci, and Mario Faretta. 2022. "From Double-Strand Break Recognition to Cell-Cycle Checkpoint Activation: High Content and Resolution Image Cytometry Unmasks 53BP1 Multiple Roles in DNA Damage Response and p53 Action" International Journal of Molecular Sciences 23, no. 17: 10193. https://doi.org/10.3390/ijms231710193
APA StyleFuria, L., Pelicci, S., Scanarini, M., Pelicci, P. G., & Faretta, M. (2022). From Double-Strand Break Recognition to Cell-Cycle Checkpoint Activation: High Content and Resolution Image Cytometry Unmasks 53BP1 Multiple Roles in DNA Damage Response and p53 Action. International Journal of Molecular Sciences, 23(17), 10193. https://doi.org/10.3390/ijms231710193