
Distress-Mediated Remodeling of Cardiac Connexin-43 in a Novel Cell Model for Arrhythmogenic Heart Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
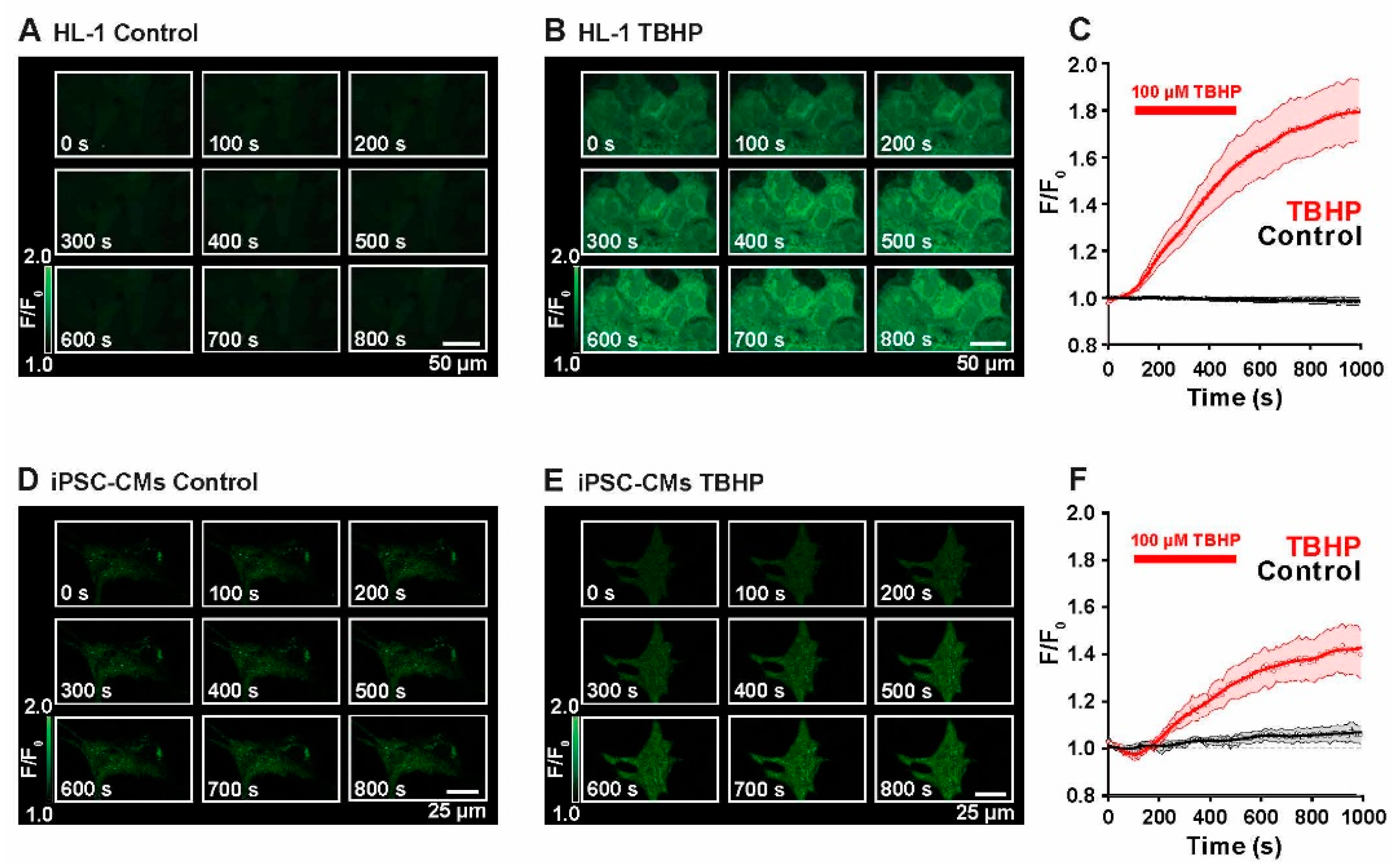
2.1. Live Monitoring of ROS Production
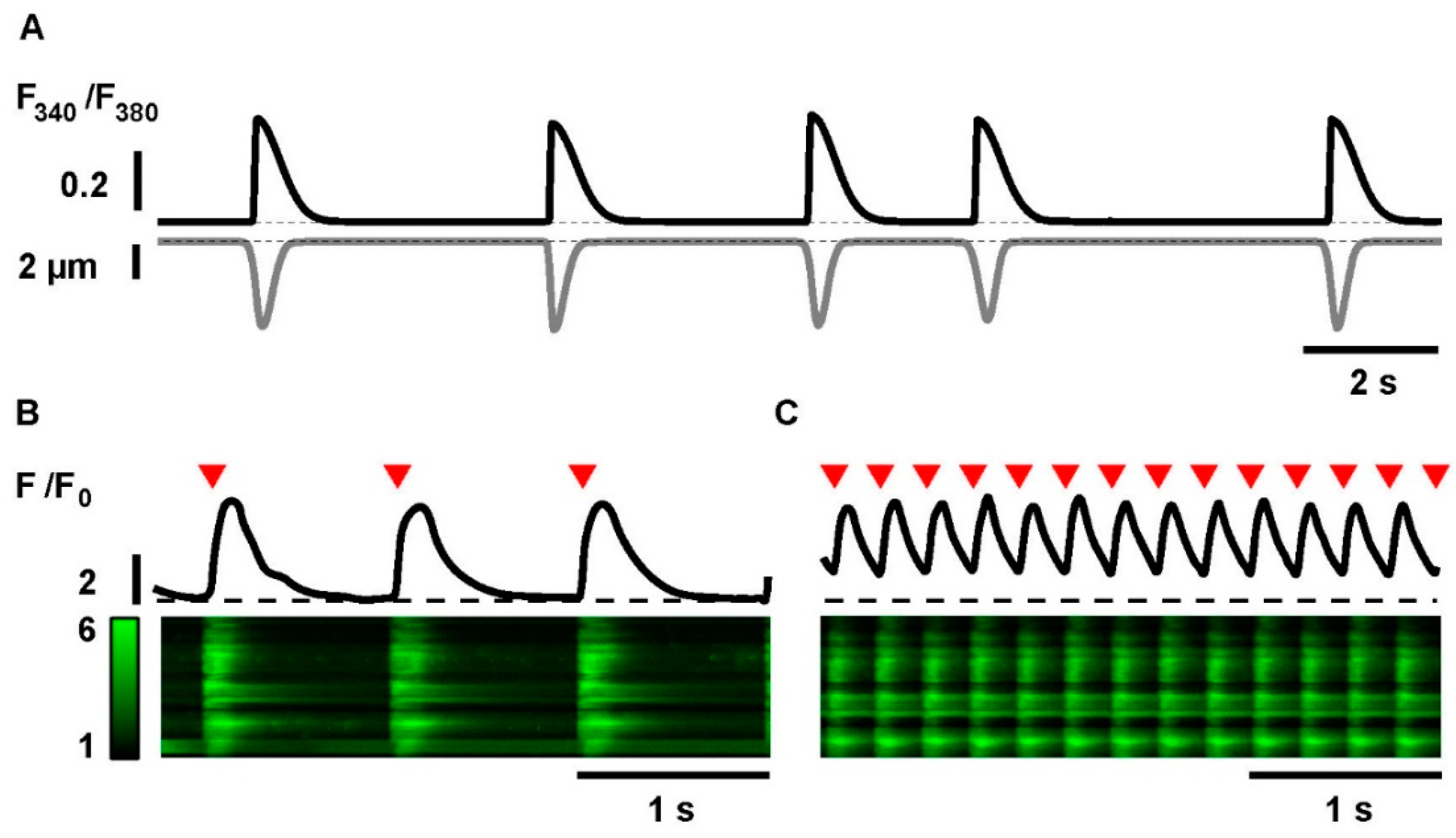
2.2. Electrical Stimulation and Tachypacing of iPSC-CMs
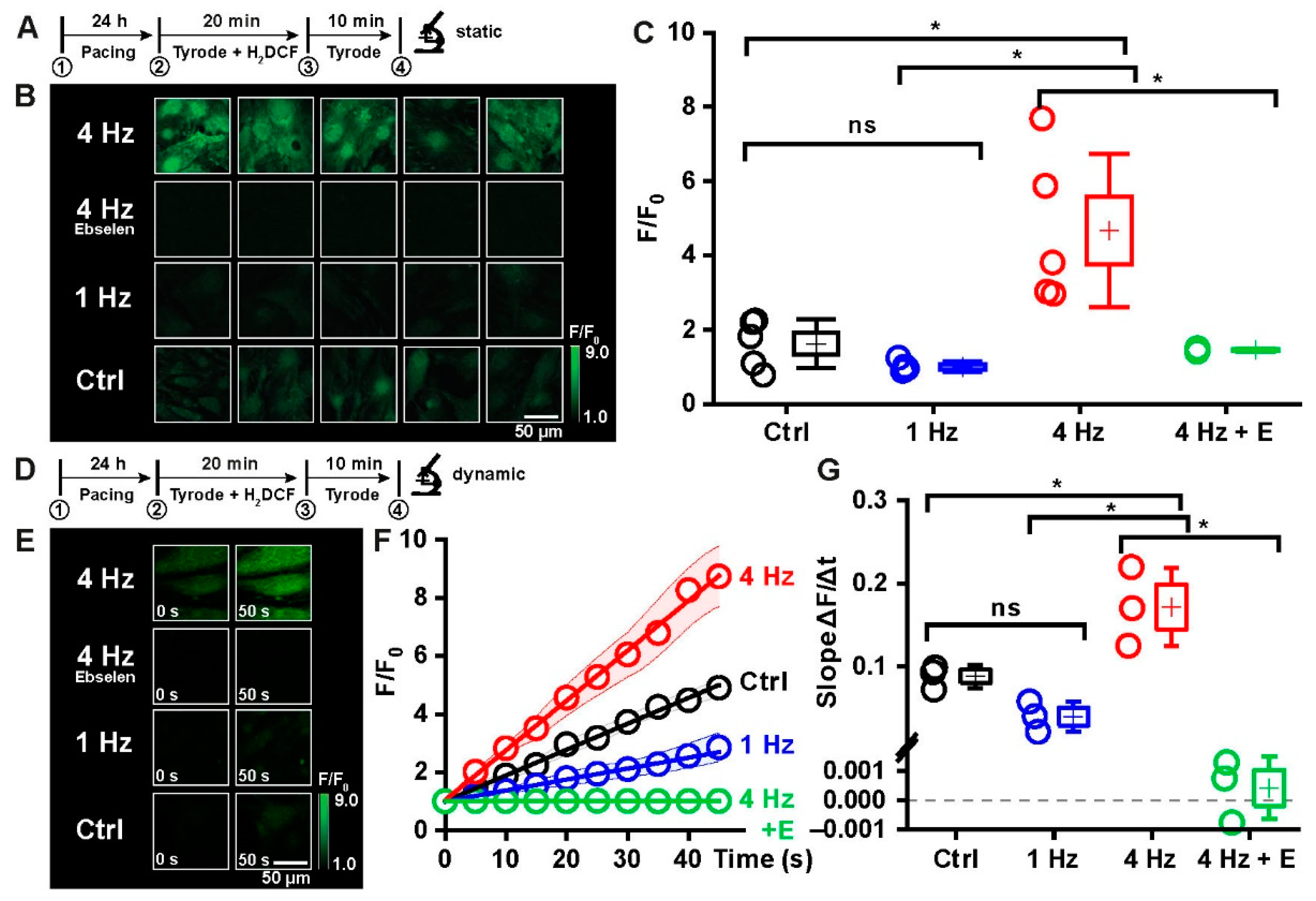
2.3. Tachypacing as a Model to Induce Oxidative Distress in iPSC-CMs
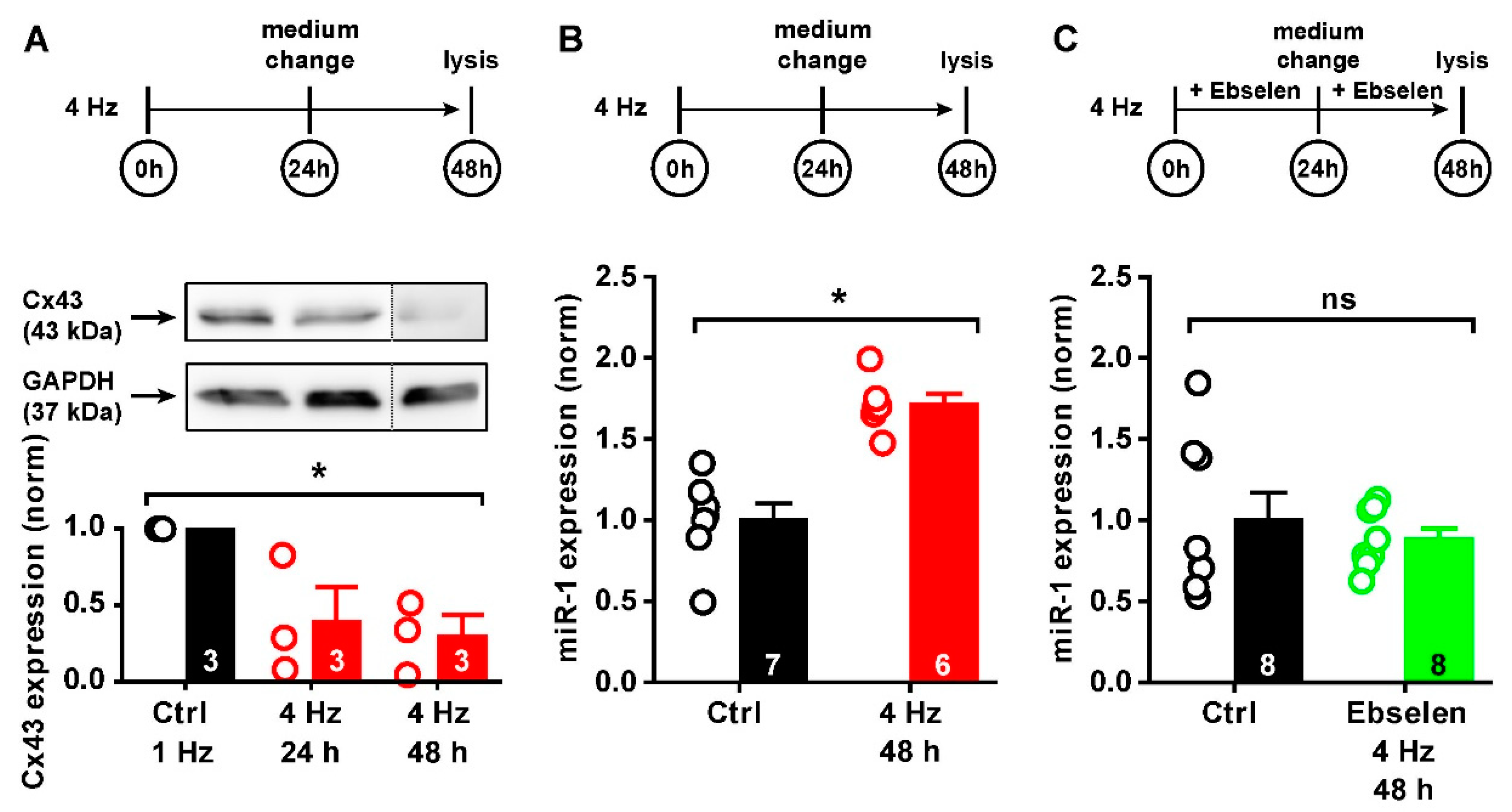
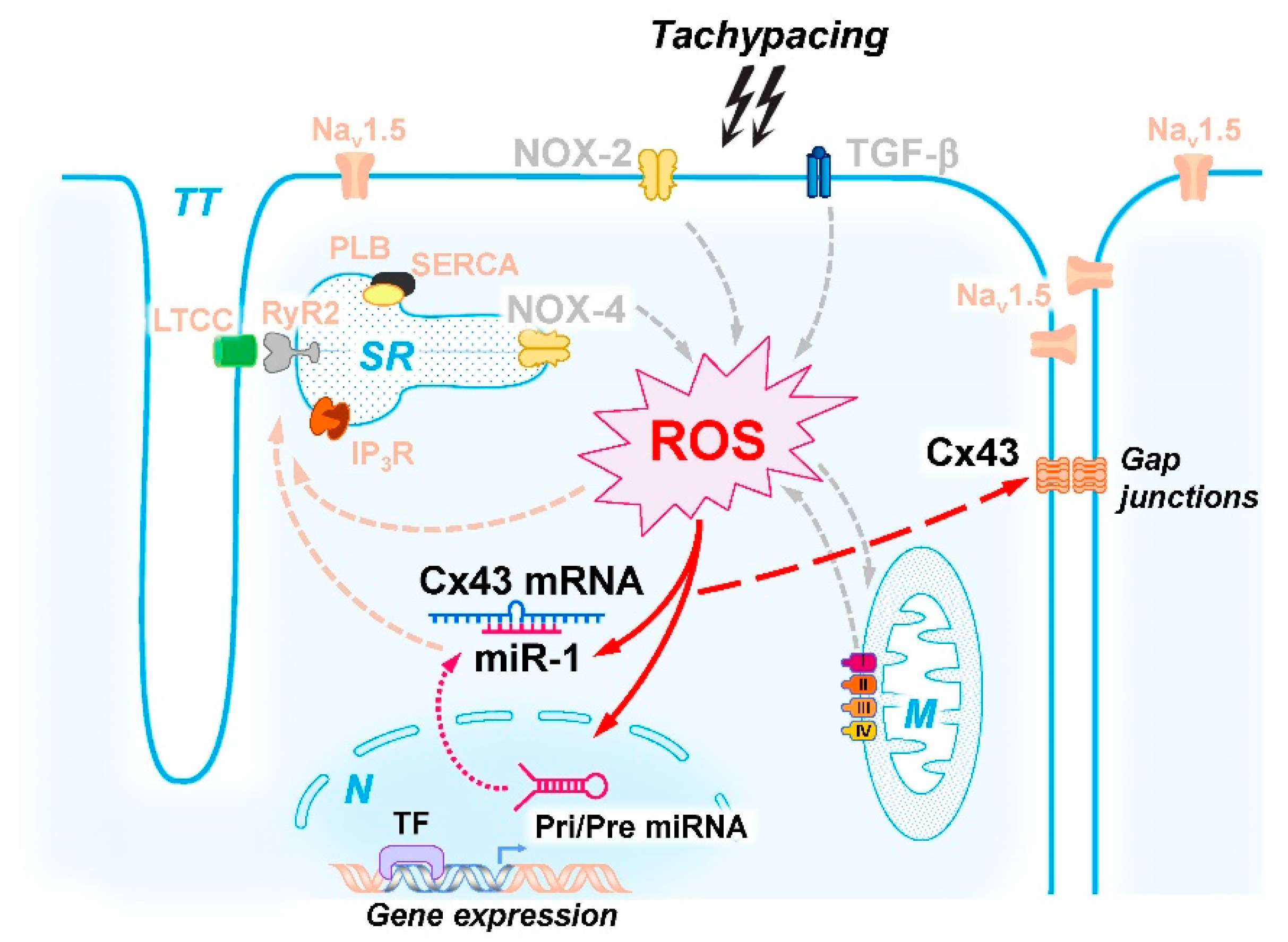
2.4. ROS-Mediated Cx43 Reduction via miR-1
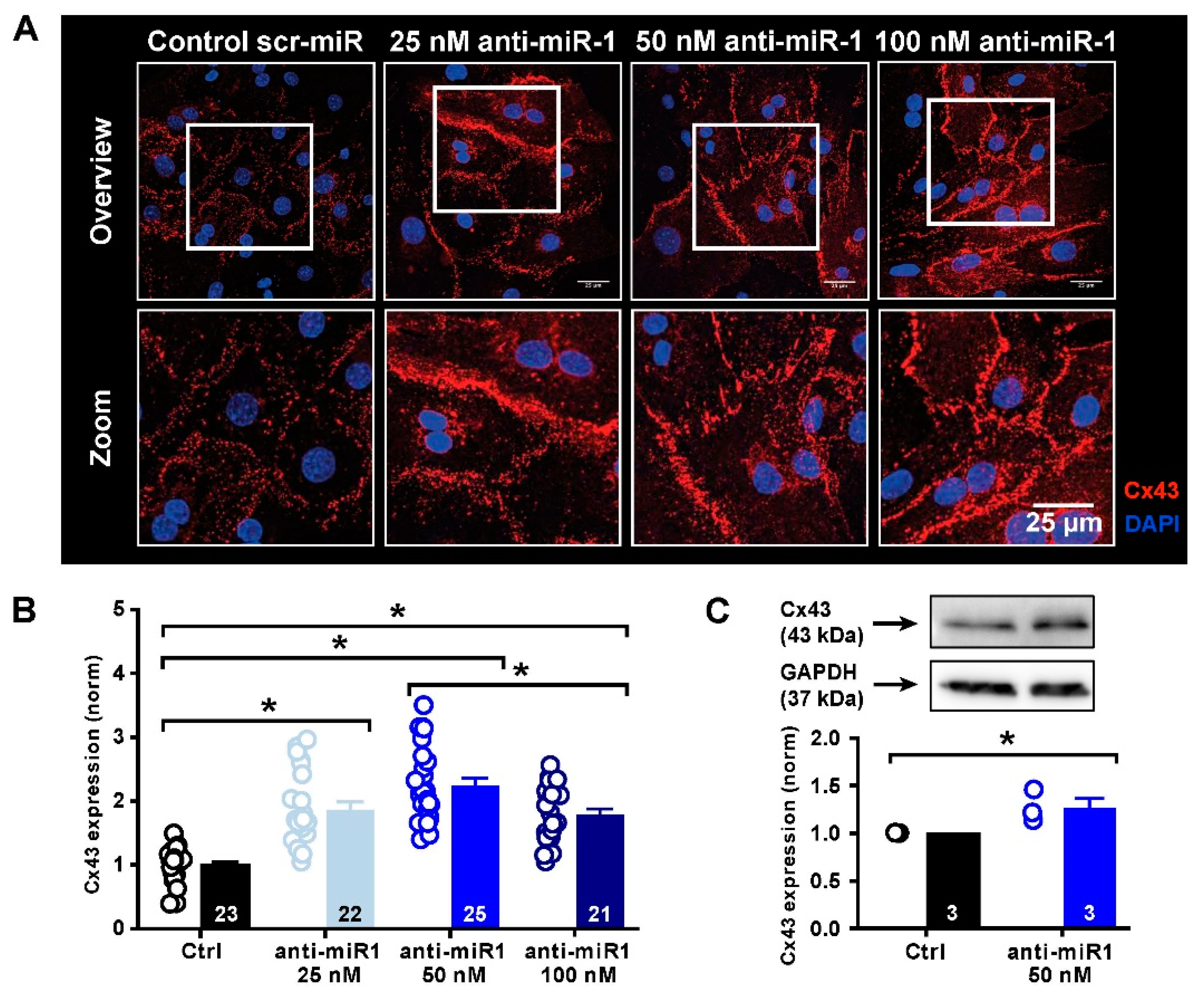
2.5. miR-1 Control of Cx43 Expression
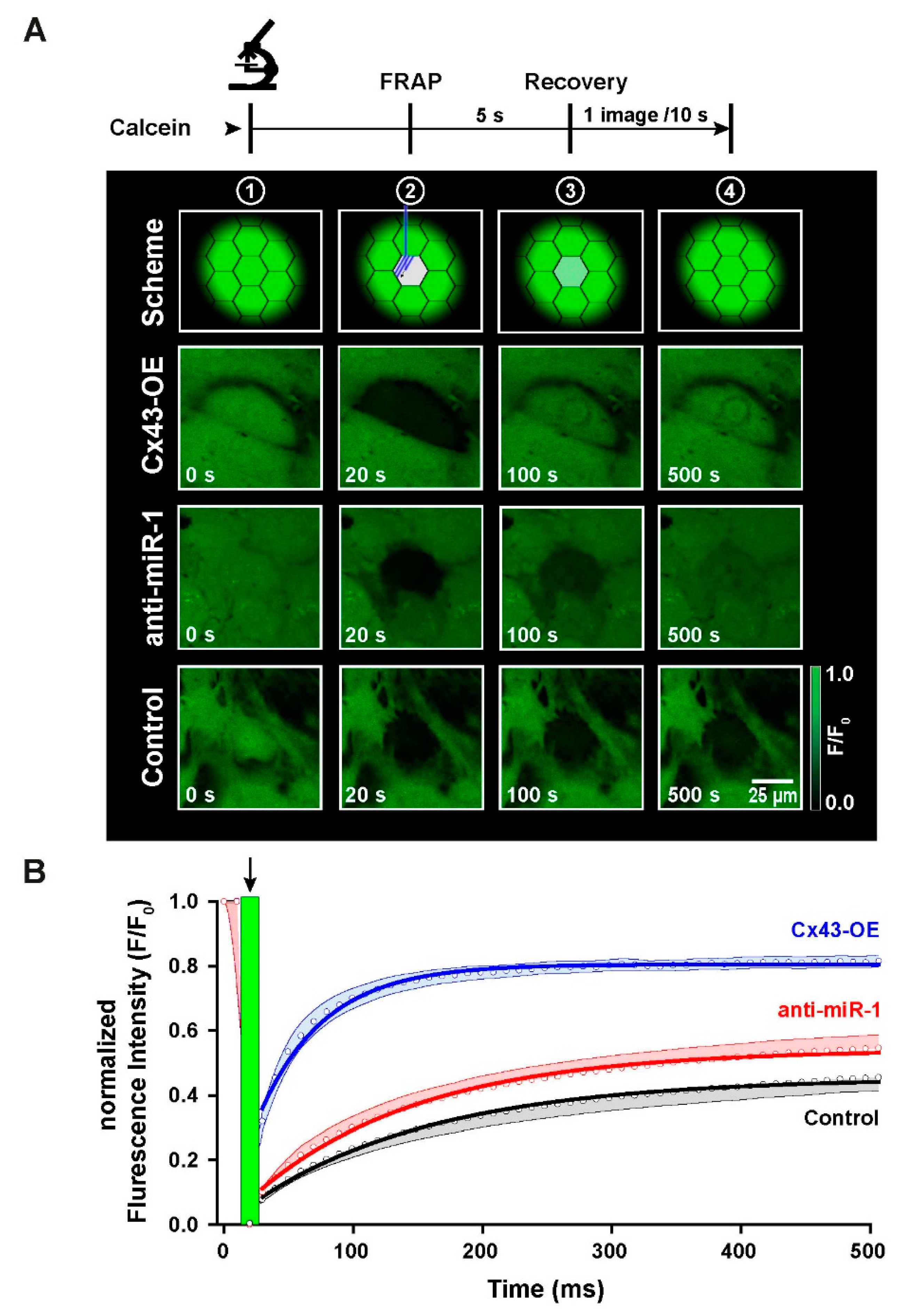
2.6. Functional Characterization of Intercellular Coupling by Modulation of Cx43 Expression in iPSC-CMs
3. Discussion
3.1. Experimental Models to Induce Cell Stress in iPSC-CMs
3.2. Molecular Mechanisms of Tachypacing-Induced Cell Stress
3.3. Molecular Control of Cx43 Protein Expression during Stress
3.4. Implications for iPSC-CM Properties and Clinical Perspective
3.5. Limitations of the Study
4. Materials and Methods
Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Strait, J.B.; Lakatta, E.G. Aging-Associated Cardiovascular Changes and Their Relationship to Heart Failure. Heart Fail. Clin. 2012, 8, 143–164. [Google Scholar] [CrossRef] [PubMed]
- Tribulova, N.; Egan Benova, T.; Szeiffova Bacova, B.; Viczenczova, C.; Barancik, M. New aspects of pathogenesis of atrial fibrillation: Remodeling of intercalated discs. J. Physiol. Pharmacol. 2015, 66, 625–634. [Google Scholar] [PubMed]
- Steenman, M.; Lande, G. Cardiac aging and heart disease in humans. Biophys. Rev. 2017, 9, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Gude, N.A.; Broughton, K.M.; Firouzi, F.; Sussman, M.A. Cardiac ageing: Extrinsic and intrinsic factors in cellular renewal and senescence. Nat. Rev. Cardiol. 2018, 15, 523–542. [Google Scholar] [CrossRef]
- Husti, Z.; Varró, A.; Baczkó, I. Arrhythmogenic remodeling in the failing heart. Cells 2021, 10, 3203. [Google Scholar] [CrossRef] [PubMed]
- Andelova, K.; Bacova, B.S.; Sykora, M.; Hlivak, P.; Barancik, M.; Tribulova, N. Mechanisms Underlying Antiarrhythmic Properties of Cardioprotective Agents Impacting Inflammation and Oxidative Stress. Int. J. Mol. Sci. 2022, 23, 1416. [Google Scholar] [CrossRef]
- Dhein, S.; Salameh, A. Remodeling of cardiac gap junctional cell–cell coupling. Cells 2021, 10, 2422. [Google Scholar] [CrossRef]
- Nielsen, M.S.; Axelsen, L.N.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N.H. Gap junctions. Compr. Physiol. 2012, 2, 1981–2035. [Google Scholar] [CrossRef]
- Stroemlund, L.W.; Jensen, C.F.; Qvortrup, K.; Delmar, M.; Nielsen, M.S. Gap junctions—Guards of excitability. Biochem. Soc. Trans. 2015, 43, 508–512. [Google Scholar] [CrossRef]
- Spach, M.S.; Heidlage, J.F. The stochastic nature of cardiac propagation at a microscopic level: Electrical description of myocardial architecture and its application to conduction. Circ. Res. 1995, 76, 366–380. [Google Scholar] [CrossRef]
- Hesketh, G.G.; Shah, M.H.; Halperin, V.L.; Cooke, C.A.; Akar, F.G.; Yen, T.E.; Kass, D.A.; MacHamer, C.E.; Van Eyk, J.E.; Tomaselli, G.F. Ultrastructure and regulation of lateralized connexin43 in the failing heart. Circ. Res. 2010, 106, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O. Posttranscriptional regulation of connexin-43 expression. Arch. Biochem. Biophys. 2012, 524, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Van Wagoner, D.R. Oxidative stress and inflammation in atrial fibrillation: Role in pathogenesis and potential as a therapeutic target. J. Cardiovasc. Pharmacol. 2008, 52, 306–313. [Google Scholar] [CrossRef]
- Sies, H. Findings in redox biology: From H2O2 to oxidative stress. J. Biol. Chem. 2020, 295, 13458–13473. [Google Scholar] [CrossRef]
- Marcu, I.C.; Illaste, A.; Heuking, P.; Jaconi, M.E.; Ullrich, N.D. Functional characterization and comparison of intercellular communication in stem cell-derived cardiomyocytes. Stem Cells 2015, 33, 2208–2218. [Google Scholar] [CrossRef] [PubMed]
- Kucera, J.P.; Prudat, Y.; Marcu, I.C.; Azzarito, M.; Ullrich, N.D. Slow conduction in mixed cultured strands of primary ventricular cells and stem cell-derived cardiomyocytes. Front. Cell Dev. Biol. 2015, 3, 58. [Google Scholar] [CrossRef]
- Sottas, V.; Wahl, C.M.; Trache, M.C.; Bartolf-Kopp, M.; Cambridge, S.; Hecker, M.; Ullrich, N.D. Improving electrical properties of iPSC-cardiomyocytes by enhancing Cx43 expression. J. Mol. Cell. Cardiol. 2018, 120, 31–41. [Google Scholar] [CrossRef]
- Shi, Y.; Ducharme, A.; Li, D.; Gaspo, R.; Nattel, S.; Tardif, J.C. Remodeling of atrial dimensions and emptying function in canine models of atrial fibrillation. Cardiovasc. Res. 2001, 52, 217–225. [Google Scholar] [CrossRef]
- Citerni, C.; Kirchhoff, J.; Olsen, L.H.; Sattler, S.M.; Gentilini, F.; Forni, M.; Zannoni, A.; Grunnet, M.; Edvardsson, N.; Bentzen, B.H.; et al. Characterization of Atrial and Ventricular Structural Remodeling in a Porcine Model of Atrial Fibrillation Induced by Atrial Tachypacing. Front. Vet. Sci. 2020, 7, 179. [Google Scholar] [CrossRef]
- Fenner, M.F.; Carstensen, H.; Dalgas Nissen, S.; Melis Hesselkilde, E.; Scott Lunddahl, C.; Adler Hess Jensen, M.; Loft-Andersen, A.V.; Sattler, S.M.; Platonov, P.; El-Haou, S.; et al. Effect of selective IK, ACh inhibition by XAF-1407 in an equine model of tachypacing-induced persistent atrial fibrillation. Br. J. Pharmacol. 2020, 177, 3778–3794. [Google Scholar] [CrossRef]
- Yeh, Y.H.; Kuo, C.T.; Chan, T.H.; Chang, G.J.; Qi, X.Y.; Tsai, F.; Nattel, S.; Chen, W.J. Transforming growth factor-β and oxidative stress mediate tachycardia-induced cellular remodelling in cultured atrial-derived myocytes. Cardiovasc. Res. 2011, 91, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Townley-Tilson, W.H.D.; Callis, T.E.; Wang, D. MicroRNAs 1, 133, and 206: Critical factors of skeletal and cardiac muscle development, function, and disease. Int. J. Biochem. Cell Biol. 2010, 42, 1252–1255. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Liang, H.; Wang, H.; Chen, G.; Jiang, H.; Wu, Q.; Liu, T.; Liu, Q.; Yu, T.; Gu, Y.; et al. Over-expression of microRNA-1 causes arrhythmia by disturbing intracellular trafficking system. Sci. Rep. 2017, 7, 46259. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, L.; Zhang, Y.; Liang, H.; Li, X.; Cai, R.; Wang, L.; Du, W.; Zhang, R.; Li, J.; et al. Overexpression of microRNA-1 causes atrioventricular block in rodents. Int. J. Biol. Sci. 2013, 9, 445–462. [Google Scholar] [CrossRef]
- Liu, Q.; Zhao, X.; Peng, R.; Wang, M.; Zhao, W.; Gui, Y.J.; Liao, C.X.; Xu, D.Y. Soluble epoxide hydrolase inhibitors might prevent ischemic arrhythmias via microRNA-1 repression in primary neonatal mouse ventricular myocytes. Mol. Biosyst. 2017, 13, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Jiang, L.; Zhang, H.; Shimoda, L.A.; Deberardinis, R.J.; Semenza, G.L. Analysis of hypoxia-induced metabolic reprogramming. Methods Enzymol. 2014, 542, 425–455. [Google Scholar]
- Puranam, K.L.; Laird, D.W.; Revel, J.P. Trapping an intermediate form of connexin43 in the Golgi. Exp. Cell Res. 1993, 206, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Li, D. Ionic remodeling in the heart: Pathophysiological significance and new therapeutic opportunities for atrial fibrillation. Circ. Res. 2000, 87, 440–447. [Google Scholar] [CrossRef]
- Hanna, N.; Cardin, S.; Leung, T.K.; Nattel, S. Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing-induced congestive heart failure. Cardiovasc. Res. 2004, 63, 236–244. [Google Scholar] [CrossRef]
- Akar, F.G.; Spragg, D.D.; Tunin, R.S.; Kass, D.A.; Tomaselli, G.F. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ. Res. 2004, 95, 717–725. [Google Scholar] [CrossRef]
- Akar, F.G.; Nass, R.D.; Hahn, S.; Cingolani, E.; Shah, M.; Hesketh, G.G.; DiSilvestre, D.; Tunin, R.S.; Kass, D.A.; Tomaselli, G.F. Dynamic changes in conduction velocity and gap junction properties during development of pacing-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1223–H1230. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Gaspo, R.; Leblanc, N.; Nattel, S. Cellular mechanisms of atrial contractile dysfunction caused by sustained atrial tachycardia. Circulation 1998, 98, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Meraviglia, V.; Azzimato, V.; Colussi, C.; Florio, M.C.; Binda, A.; Panariti, A.; Qanud, K.; Suffredini, S.; Gennaccaro, L.; Miragoli, M.; et al. Acetylation mediates Cx43 reduction caused by electrical stimulation. J. Mol. Cell. Cardiol. 2015, 87, 54–64. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Silbernagel, N.; Körner, A.; Balitzki, J.; Jaggy, M.; Bertels, S.; Richter, B.; Hippler, M.; Hellwig, A.; Hecker, M.; Bastmeyer, M.; et al. Shaping the heart: Structural and functional maturation of iPSC-cardiomyocytes in 3D-micro-scaffolds. Biomaterials 2020, 227, 119551. [Google Scholar] [CrossRef] [PubMed]
- Körner, A.; Mosqueira, M.; Hecker, M.; Ullrich, N.D. Substrate Stiffness Influences Structural and Functional Remodeling in Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Front. Physiol. 2021, 12, 1306. [Google Scholar] [CrossRef]
- Daoud, E.G.; Knight, B.P.; Weiss, R.; Bahu, M.; Paladino, W.; Goyal, R.; Man, K.C.; Strickberger, S.A.; Morady, F. Effect of verapamil and procainamide on atrial fibrillation-induced electrical remodeling in humans. Circulation 1997, 96, 1542–1550. [Google Scholar] [CrossRef]
- Yu, W.C.; Chen, S.A.; Lee, S.H.; Tai, C.T.; Feng, A.N.; Kuo, B.I.T.; Ding, Y.A.; Chang, M.S. Tachycardia-induced change of atrial refractory period in humans: Rate dependency and effects of antiarrhythmic drugs. Circulation 1998, 97, 2331–2337. [Google Scholar] [CrossRef]
- Reilly, S.N.; Jayaram, R.; Nahar, K.; Antoniades, C.; Verheule, S.; Channon, K.M.; Alp, N.J.; Schotten, U.; Casadei, B. Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: Implications for the antiarrhythmic effect of statins. Circulation 2011, 124, 1107–1117. [Google Scholar] [CrossRef]
- Müller, A.; Cadenas, E.; Graf, P.; Sies, H. A novel biologically active seleno-organic compound-1. Glutathione peroxidase-like activity in vitro and antioxidant capacity of PZ 51 (Ebselen). Biochem. Pharmacol. 1984, 33, 3235–3239. [Google Scholar] [CrossRef]
- Santi, C.; Scimmi, C.; Sancineto, L. Ebselen and analogues: Pharmacological properties and synthetic strategies for their preparation. Molecules 2021, 26, 4230. [Google Scholar] [CrossRef]
- Neuman, R.B.; Bloom, H.L.; Shukrullah, I.; Darrow, L.A.; Kleinbaum, D.; Jones, D.P.; Dudley, S.C. Oxidative Stress Markers Are Associated with Persistent Atrial Fibrillation. Clin. Chem. 2007, 53, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Saraf, A.; Rampoldi, A.; Chao, M.; Li, D.; Armand, L.; Hwang, H.; Liu, R.; Jha, R.; Fu, H.; Maxwell, J.T.; et al. Functional and molecular effects of TNF-α on human iPSC-derived cardiomyocytes. Stem Cell Res. 2021, 52, 102218. [Google Scholar] [CrossRef]
- Weissman, D.; Maack, C. Redox signaling in heart failure and therapeutic implications. Free Radic. Biol. Med. 2021, 171, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Tribulova, N.; Knezl, V.; Szeiffova Bacova, B.; Egan Benova, T.; Viczenczova, C.; Gonçalvesova, E.; Slezak, J. Disordered myocardial Ca2+ homeostasis results in substructural alterations that may promote occurrence of malignant arrhythmias. Physiol. Res. 2016, 65, S139–S148. [Google Scholar] [CrossRef] [PubMed]
- Andelova, K.; Benova, T.E.; Bacova, B.S.; Sykora, M.; Prado, N.J.; Diez, E.R.; Hlivak, P.; Tribulova, N. Cardiac connexin-43 hemichannels and pannexin1 channels: Provocative antiarrhythmic targets. Int. J. Mol. Sci. 2021, 22, 260. [Google Scholar] [CrossRef]
- Climent, M.; Viggiani, G.; Chen, Y.W.; Coulis, G.; Castaldi, A. Microrna and ros crosstalk in cardiac and pulmonary diseases. Int. J. Mol. Sci. 2020, 21, 4370. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction). J. Mol. Cell. Cardiol. 2016, 94, 107–121. [Google Scholar] [CrossRef]
- Santulli, G.; Iaccarino, G.; De Luca, N.; Trimarco, B.; Condorelli, G. Atrial fibrillation and microRNAs. Front. Physiol. 2014, 5, 15. [Google Scholar] [CrossRef]
- Girmatsion, Z.; Biliczki, P.; Bonauer, A.; Wimmer-Greinecker, G.; Scherer, M.; Moritz, A.; Bukowska, A.; Goette, A.; Nattel, S.; Hohnloser, S.H.; et al. Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm 2009, 6, 1802–1809. [Google Scholar] [CrossRef]
- Stein, C.A.; Hansen, J.B.; Lai, J.; Wu, S.J.; Voskresenskiy, A.; Høg, A.; Worm, J.; Hedtjärn, M.; Souleimanian, N.; Miller, P.; et al. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 2009, 38, e3. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Yang, T.; Shen, L.; Xu, D. Soluble Epoxide Hydrolase Inhibitors Regulate Ischemic Arrhythmia by Targeting MicroRNA-1. Front. Physiol. 2021, 12, 717119. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, Y.; Shan, H.; Pan, Z.; Li, X.; Li, B.; Xu, C.; Zhang, B.; Zhang, F.; Dong, D.; et al. MicroRNA-1 downregulation by propranolol in a rat model of myocardial infarction: A new mechanism for ischaemic cardioprotection. Cardiovasc. Res. 2009, 84, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar] [CrossRef]
- Smyth, J.W.; Hong, T.T.; Gao, D.; Vogan, J.M.; Jensen, B.C.; Fong, T.S.; Simpson, P.C.; Stainier, D.Y.R.; Chi, N.C.; Shaw, R.M. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J. Clin. Investig. 2010, 120, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Laird, D.W.; Castillo, M.; Kasprzak, L. Gap junction turnover, intracellular trafficking, and phosphorylation of connexin43 in brefeldin A-treated rat mammary tumor cells. J. Cell Biol. 1995, 131, 1193–1203. [Google Scholar] [CrossRef]
- Qin, H.; Shao, Q.; Igdoura, S.A.; Alaoui-Jamali, M.A.; Laird, D.W. Lysosomal and proteasomal degradation play distinct roles in the life cycle of Cx43 in gap junctional intercellular communication-deficient and -competent breast tumor cells. J. Biol. Chem. 2003, 278, 30005–30014. [Google Scholar] [CrossRef]
- Thomas, T.; Jordan, K.; Simek, J.; Shao, Q.; Jedeszko, C.; Walton, P.; Laird, D.W. Mechanism of Cx43 and Cx26 transport to the plasma membrane and gap junction regeneration. J. Cell Sci. 2005, 118, 4451–4462. [Google Scholar] [CrossRef]
- Li, N.; Long, B.; Han, W.; Yuan, S.; Wang, K. MicroRNAs: Important regulators of stem cells. Stem Cell Res. Ther. 2017, 8, 110. [Google Scholar] [CrossRef]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Biliczki, P.; Boon, R.A.; Girmatsion, Z.; Bukowska, A.; Ördög, B.; Kaess, B.M.; Hohnloser, S.H.; Goette, A.; Varró, A.; Moritz, A.; et al. Age-related regulation and region-specific distribution of ion channel subunits promoting atrial fibrillation in human left and right atria. Europace 2019, 21, 1261–1269. [Google Scholar] [CrossRef]
- Topkara, V.K.; Mann, D.L. Role of microRNAs in cardiac remodeling and heart failure. Cardiovasc. Drugs Ther. 2011, 25, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.B.; Sanderson, J.E.; Izzat, M.B.; Yu, C.M. Micro-RNA and mRNA myocardial tissue expression in biopsy specimen from patients with heart failure. Int. J. Cardiol. 2015, 199, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Glukhov, A.V.; Fedorov, V.V.; Kalish, P.W.; Ravikumar, V.K.; Lou, Q.; Janks, D.; Schuessler, R.B.; Moazami, N.; Efimov, I.R. Conduction remodeling in human end-stage nonischemic left ventricular cardiomyopathy. Circulation 2012, 125, 1835–1847. [Google Scholar] [CrossRef] [PubMed]
- Claycomb, W.C.; Lanson, N.A.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Wiedmann, F.; Zhou, X.B.; Heijman, J.; Voigt, N.; Ratte, A.; Lang, S.; Kallenberger, S.M.; Campana, C.; Weymann, A.; et al. Inverse remodelling of K2P 3.1 K+ channel expression and action potential duration in left ventricular dysfunction and atrial fibrillation: Implications for patient-specific antiarrhythmic drug therapy. Eur. Heart J. 2017, 38, 1764–1774. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.A.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J.; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wahl, C.-M.; Schmidt, C.; Hecker, M.; Ullrich, N.D. Distress-Mediated Remodeling of Cardiac Connexin-43 in a Novel Cell Model for Arrhythmogenic Heart Diseases. Int. J. Mol. Sci. 2022, 23, 10174. https://doi.org/10.3390/ijms231710174
Wahl C-M, Schmidt C, Hecker M, Ullrich ND. Distress-Mediated Remodeling of Cardiac Connexin-43 in a Novel Cell Model for Arrhythmogenic Heart Diseases. International Journal of Molecular Sciences. 2022; 23(17):10174. https://doi.org/10.3390/ijms231710174
Chicago/Turabian StyleWahl, Carl-Mattheis, Constanze Schmidt, Markus Hecker, and Nina D. Ullrich. 2022. "Distress-Mediated Remodeling of Cardiac Connexin-43 in a Novel Cell Model for Arrhythmogenic Heart Diseases" International Journal of Molecular Sciences 23, no. 17: 10174. https://doi.org/10.3390/ijms231710174
APA StyleWahl, C.-M., Schmidt, C., Hecker, M., & Ullrich, N. D. (2022). Distress-Mediated Remodeling of Cardiac Connexin-43 in a Novel Cell Model for Arrhythmogenic Heart Diseases. International Journal of Molecular Sciences, 23(17), 10174. https://doi.org/10.3390/ijms231710174