
Transcriptome-Wide Detection of Intron/Exon Definition in the Endogenous Pre-mRNA Transcripts of Mammalian Cells and Its Regulation by Depolarization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
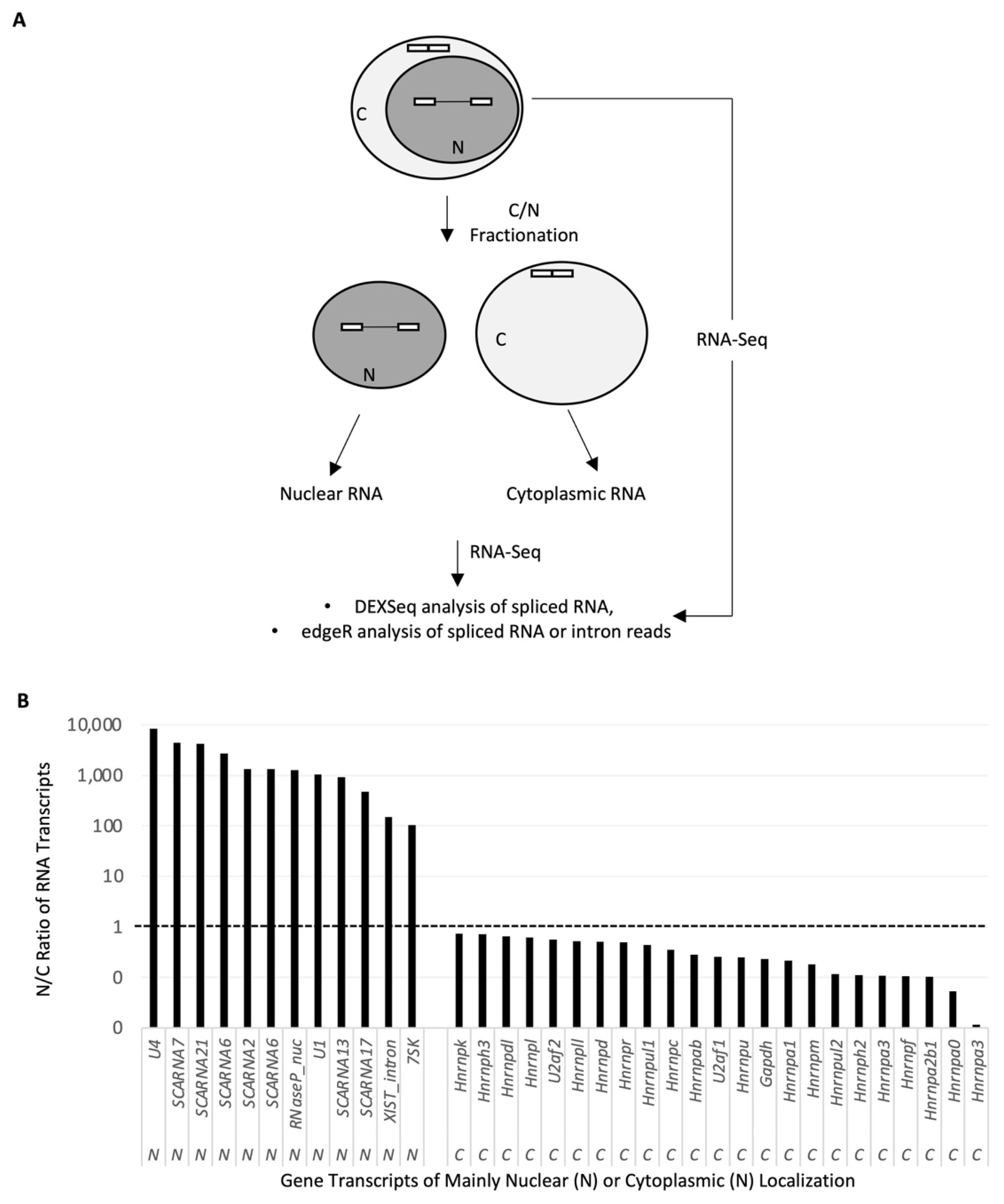
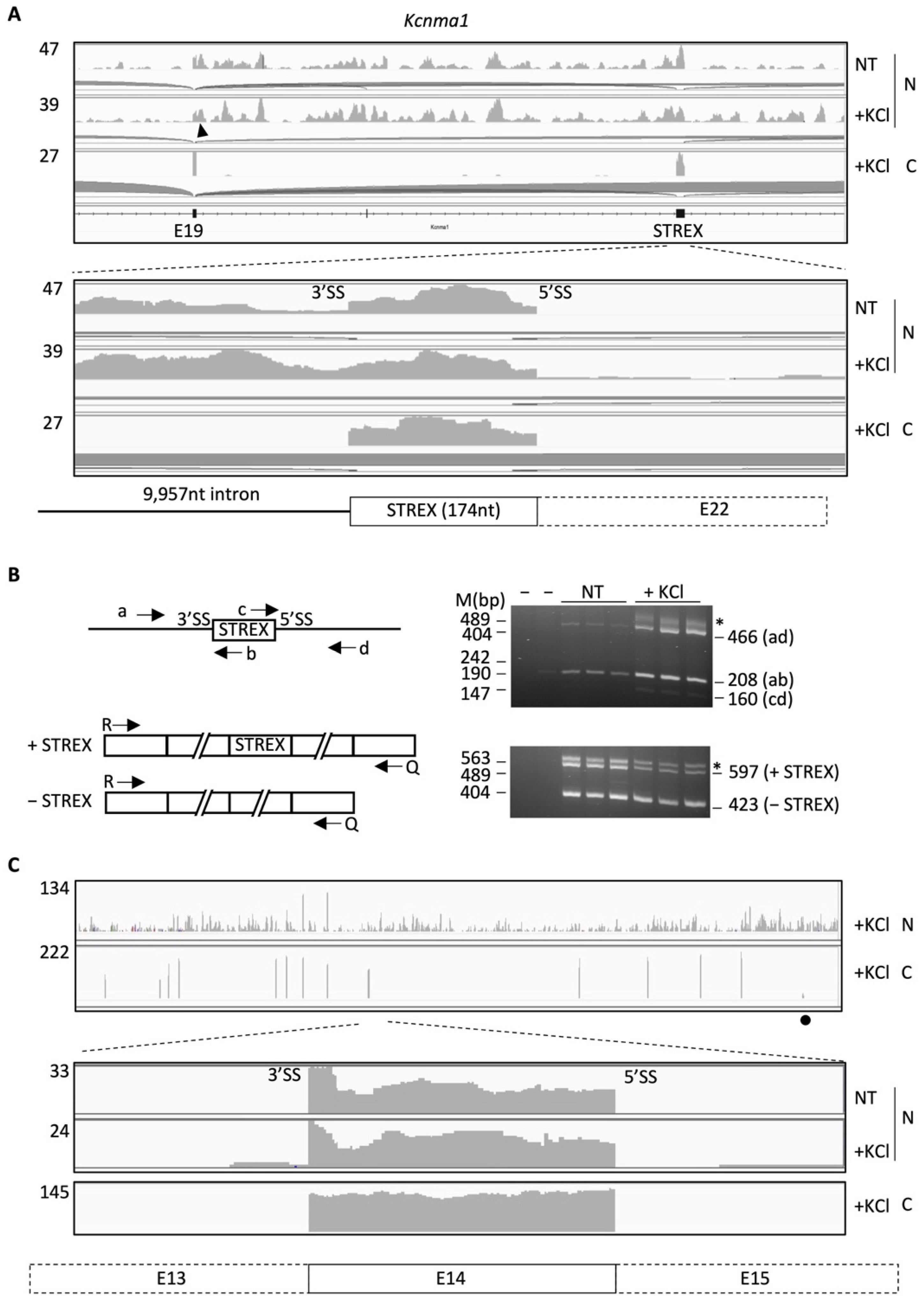
2.1. Nuclear RNA Sequencing Reveals Accumulation of Splice Site-Specific Reads at the Intron Start or End of Alternative Exons
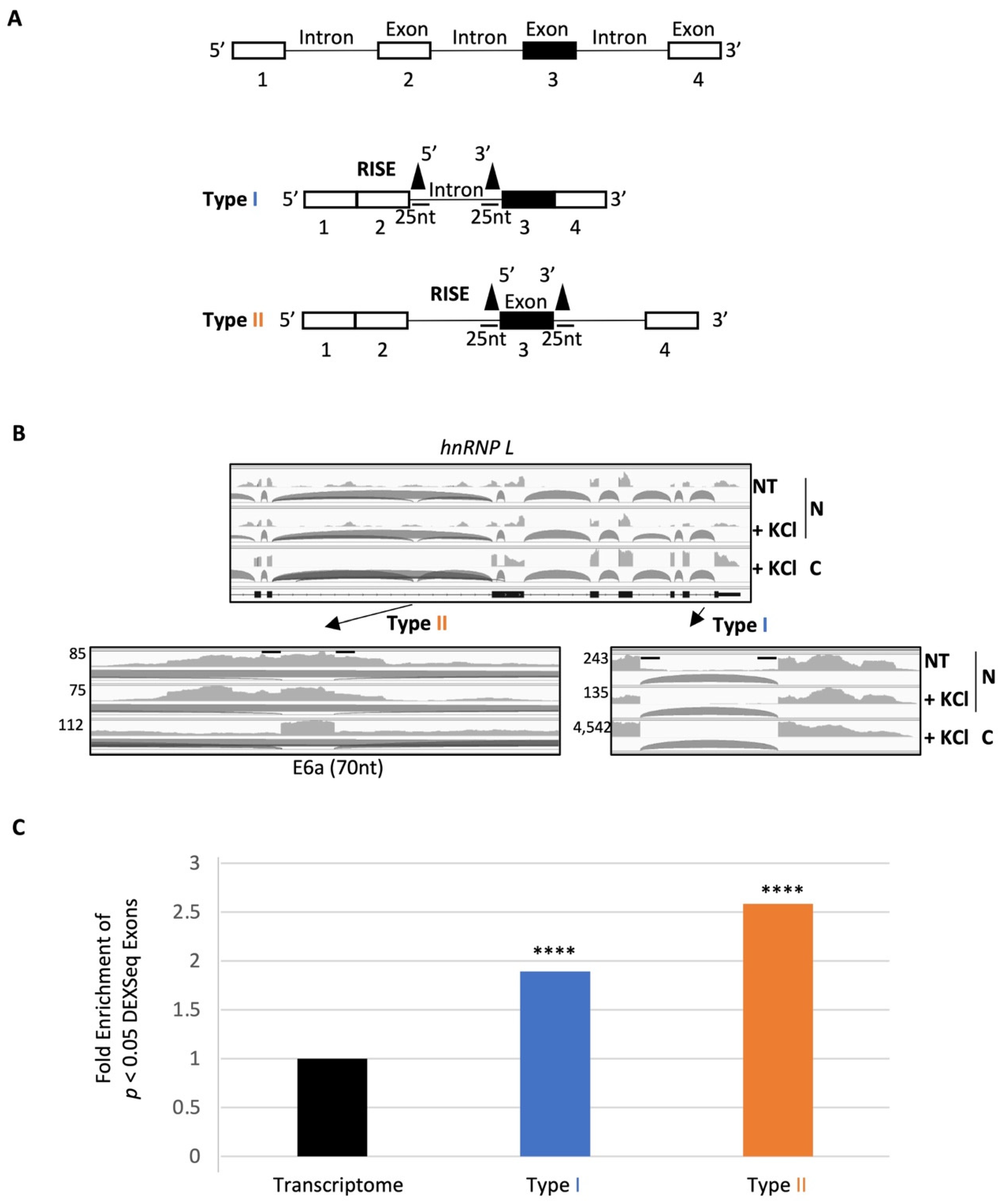
2.2. A Group of RISE Are Associated with Depolarization-Regulated Alternative Exons
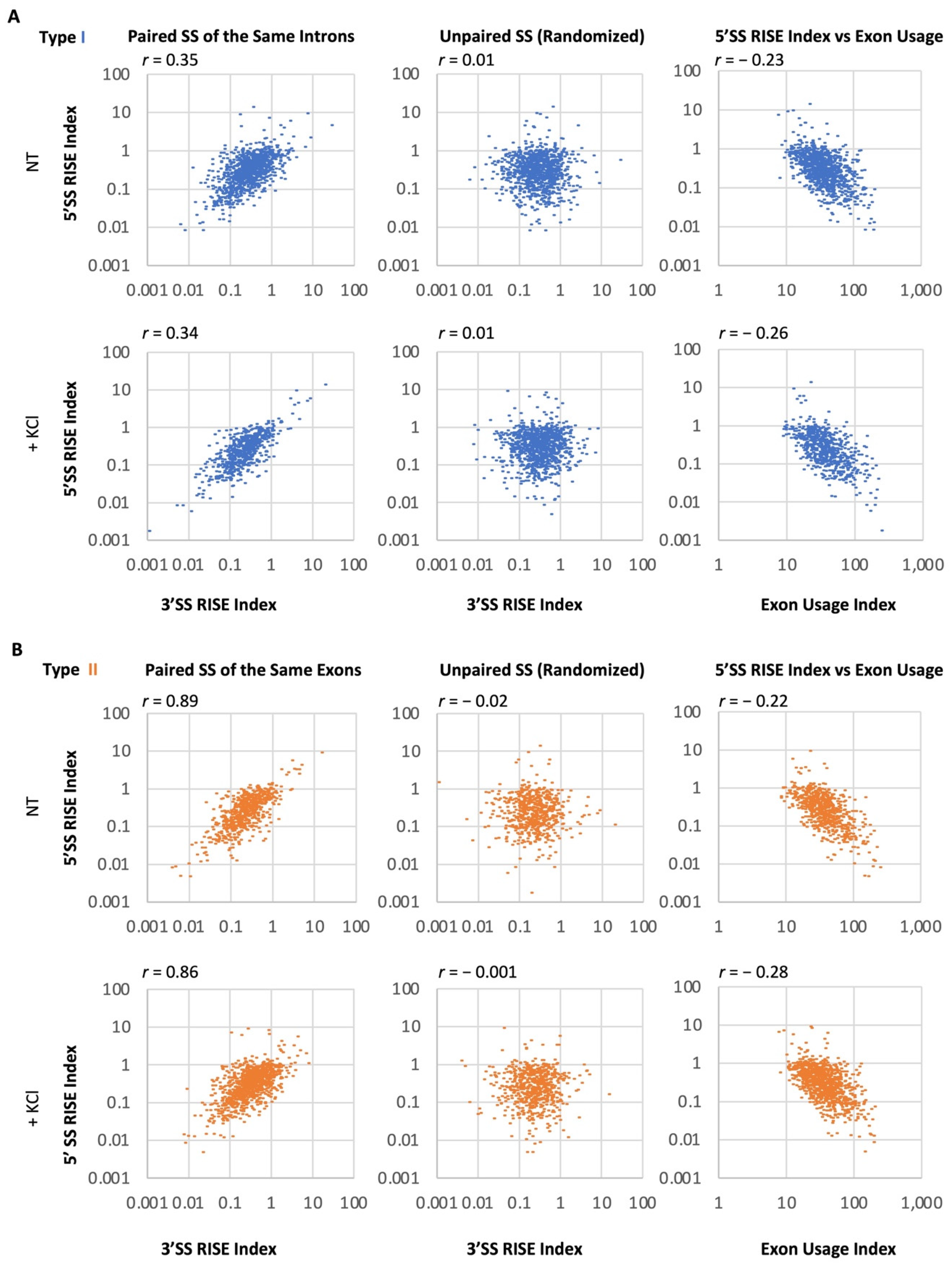
2.3. Correlation of RISE Abundance between Splice Sites across Introns or Exons in the Transcriptome
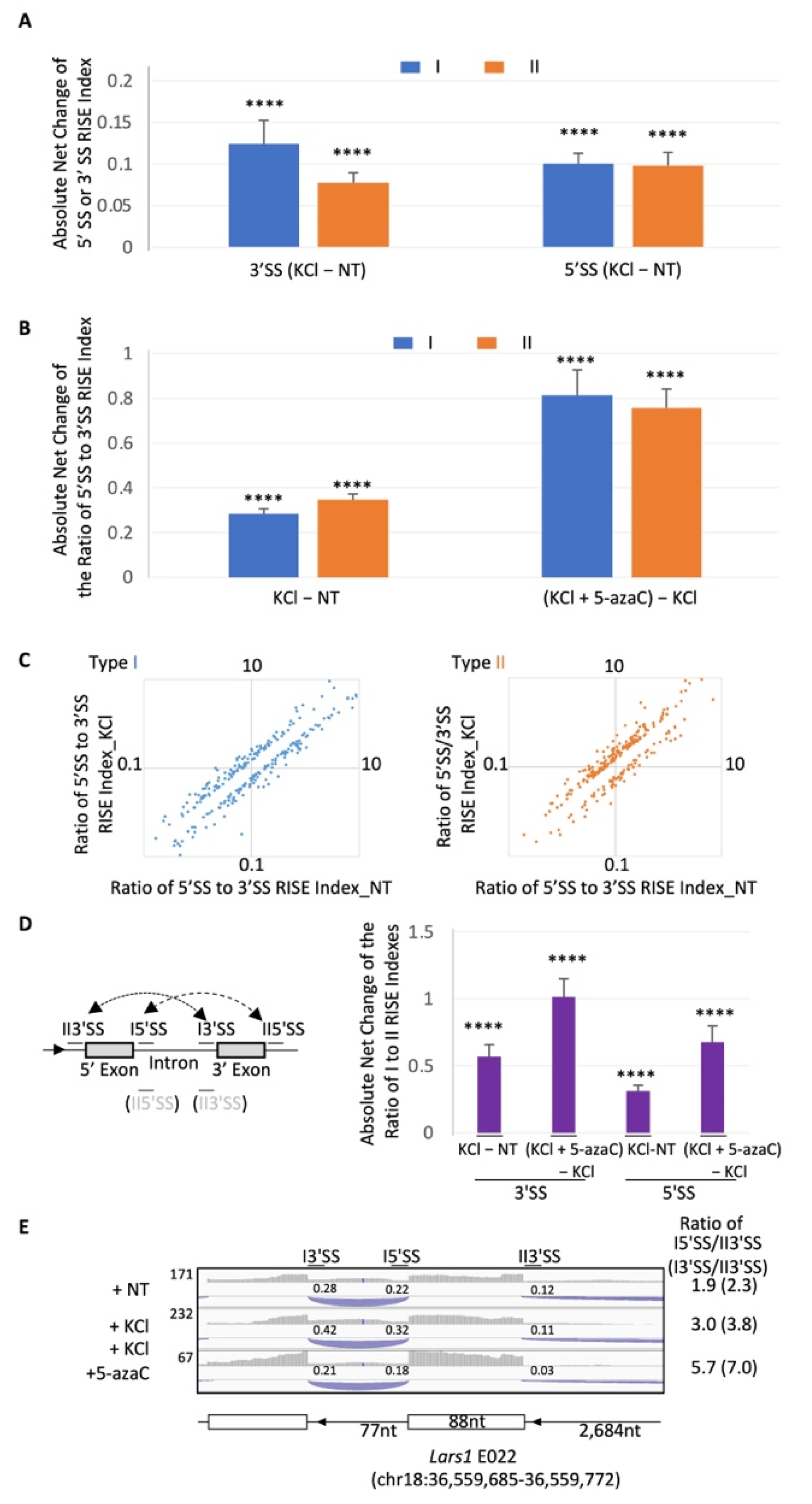
2.4. Global Modulation of the RISE Indexes and Their 5′SS/3′SS Index Ratios by Depolarization
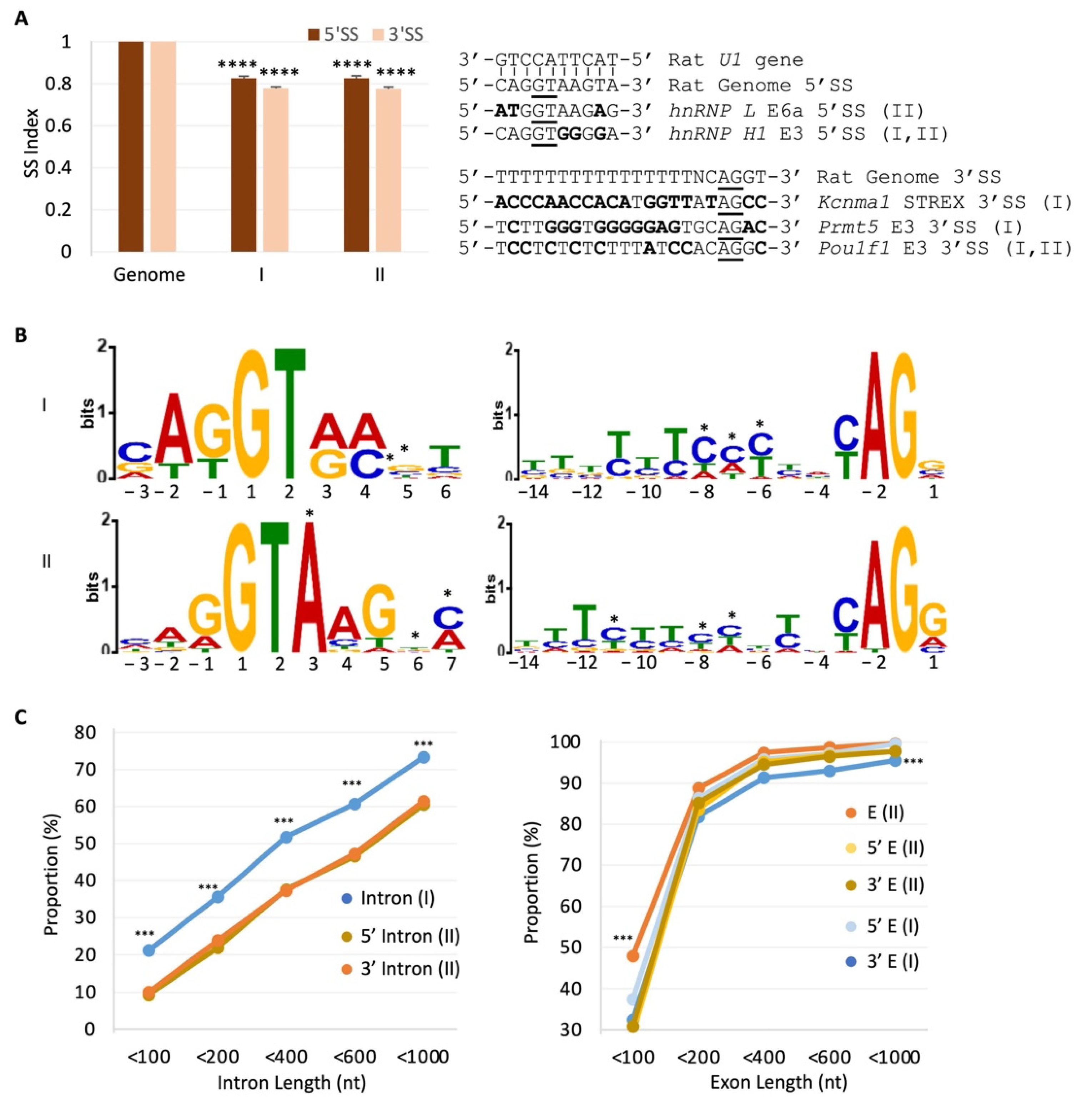
2.5. RISE Are Associated with Weak Splice Sites and Short Introns or Exons
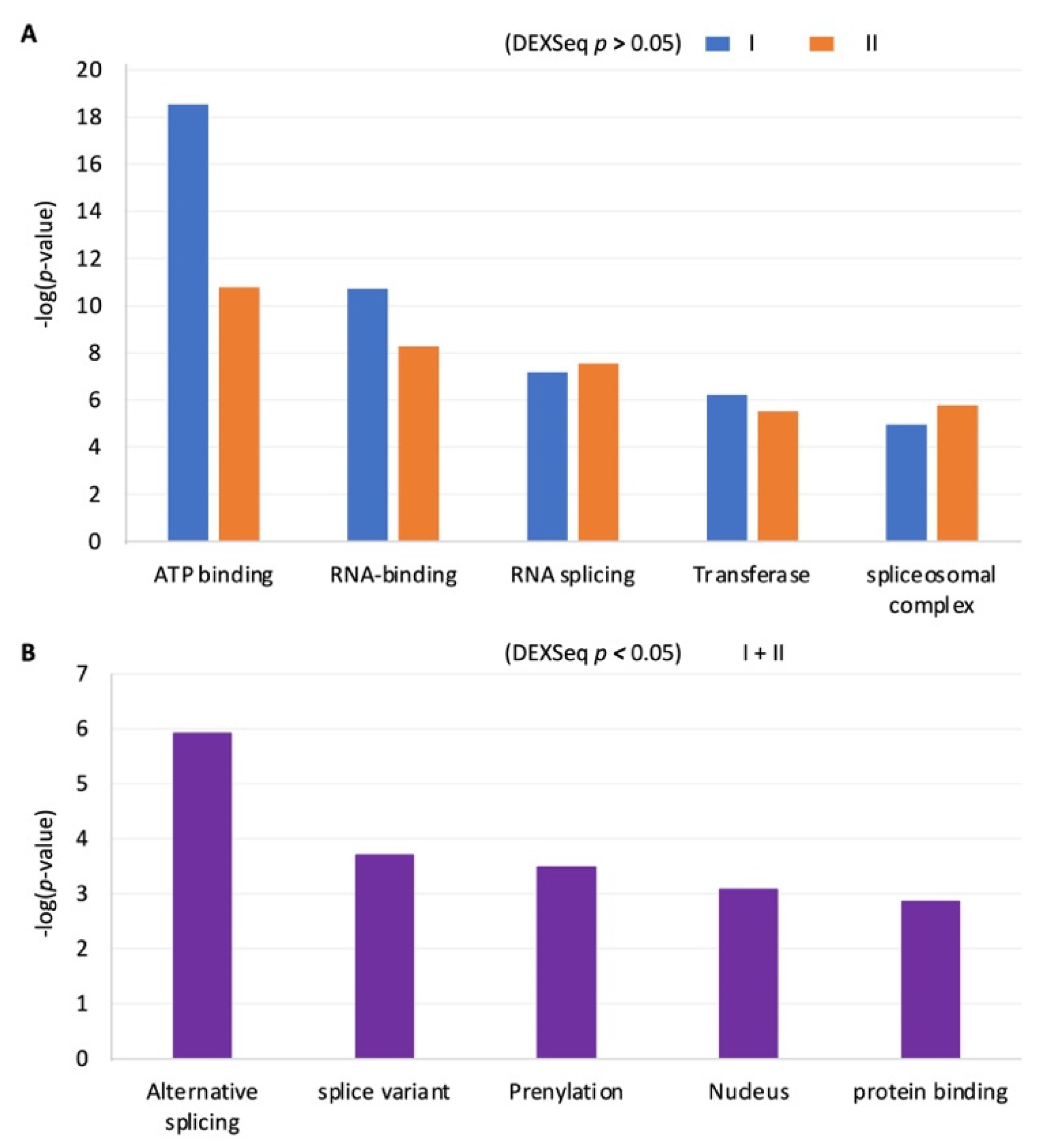
2.6. The RISE Are Highly Associated with Genes in Basic Cellular Processes or in Hormone Production
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robberson, B.L.; Cote, G.J.; Berget, S.M. Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol. Cell. Biol. 1990, 10, 84–94. [Google Scholar] [PubMed]
- Berget, S.M. Exon recognition in vertebrate splicing. J. Biol. Chem. 1995, 270, 2411–2414. [Google Scholar] [CrossRef] [PubMed]
- Talerico, M.; Berget, S.M. Intron definition in splicing of small Drosophila introns. Mol. Cell. Biol. 1994, 14, 3434–3445. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, J.; Xiao, J.; Wang, L.; Zhong, J.; Yin, H.; Wu, S.; Zhang, Z.; Yu, J. Systematic analysis of intron size and abundance parameters in diverse lineages. Sci. China Life Sci. 2013, 56, 968–974. [Google Scholar] [CrossRef]
- Wu, J.Y.; Maniatis, T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell 1993, 75, 1061–1070. [Google Scholar] [CrossRef]
- Martinez-Contreras, R.; Fisette, J.F.; Nasim, F.U.; Madden, R.; Cordeau, M.; Chabot, B. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol. 2006, 4, e21. [Google Scholar] [CrossRef]
- Sharma, S.; Wongpalee, S.P.; Vashisht, A.; Wohlschlegel, J.A.; Black, D.L. Stem-loop 4 of U1 snRNA is essential for splicing and interacts with the U2 snRNP-specific SF3A1 protein during spliceosome assembly. Genes Dev. 2014, 28, 2518–2531. [Google Scholar] [CrossRef]
- Khodor, Y.L.; Menet, J.S.; Tolan, M.; Rosbash, M. Cotranscriptional splicing efficiency differs dramatically between Drosophila and mouse. RNA 2012, 18, 2174–2186. [Google Scholar] [CrossRef]
- Pandya-Jones, A.; Black, D.L. Co-transcriptional splicing of constitutive and alternative exons. RNA 2009, 15, 1896–1908. [Google Scholar] [CrossRef]
- Khodor, Y.L.; Rodriguez, J.; Abruzzi, K.C.; Tang, C.H.; Marr, M.T., 2nd; Rosbash, M. Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev. 2011, 25, 2502–2512. [Google Scholar] [CrossRef]
- Boutz, P.L.; Bhutkar, A.; Sharp, P.A. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev. 2015, 29, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Cao, W.; Liu, L.; Das, U.; Wu, Y.; Liu, G.; Sohail, M.; Chen, Y.; Xie, J. Multilevel Differential Control of Hormone Gene Expression Programs by hnRNP L and LL in Pituitary Cells. Mol. Cell. Biol. 2018, 38, e00651-17. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.Y.; Black, D.L. A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature 2001, 410, 936–939. [Google Scholar] [PubMed]
- Yu, J.; Hai, Y.; Liu, G.; Fang, T.; Kung, S.K.P.; Xie, J. The Heterogeneous Nuclear Ribonucleoprotein L Is an Essential Component in the Ca2+/Calmodulin-dependent Protein Kinase IV-regulated Alternative Splicing through Cytidine-Adenosine Repeats. J. Biol. Chem. 2009, 284, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Razanau, A.; Hai, Y.; Yu, J.; Sohail, M.; Lobo, V.G.; Chu, J.; Kung, S.K.P.; Xie, J. A Conserved Serine of Heterogeneous Nuclear Ribonucleoprotein L (hnRNP L) Mediates Depolarization-regulated Alternative Splicing of Potassium Channels. J. Biol. Chem. 2012, 287, 22709–22716. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; McCobb, D. Control of alternative splicing of potassium channels by stress hormones. Science 1998, 280, 443–446. [Google Scholar] [CrossRef]
- Li, B.; Suutari, B.S.; Sun, S.D.; Luo, Z.; Wei, C.; Chenouard, N.; Mandelberg, N.J.; Zhang, G.; Wamsley, B.; Tian, G.; et al. Neuronal Inactivity Co-opts LTP Machinery to Drive Potassium Channel Splicing and Homeostatic Spike Widening. Cell 2020, 181, 1547–1565.e15. [Google Scholar] [CrossRef]
- Razanau, A.; Xie, J. Emerging mechanisms and consequences of calcium regulation of alternative splicing in neurons and endocrine cells. Cell. Mol. Life Sci. 2013, 70, 4527–4536. [Google Scholar] [CrossRef]
- Iijima, T.; Wu, K.; Witte, H.; Hanno-Iijima, Y.; Glatter, T.; Richard, S.; Scheiffele, P. SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell 2011, 147, 1601–1614. [Google Scholar] [CrossRef]
- Penn, A.C.; Balik, A.; Wozny, C.; Cais, O.; Greger, I.H. Activity-mediated AMPA receptor remodeling, driven by alternative splicing in the ligand-binding domain. Neuron 2012, 76, 503–510. [Google Scholar] [CrossRef]
- Shaw, D.J.; Eggleton, P.; Young, P.J. Joining the dots: Production, processing and targeting of U snRNP to nuclear bodies. Biochim. Biophys. Acta 2008, 1783, 2137–2144. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Darzacq, X.; Bertrand, E.; Jady, B.E.; Verheggen, C.; Kiss, T. A common sequence motif determines the Cajal body-specific localization of box H/ACA scaRNAs. EMBO J. 2003, 22, 4283–4293. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.R.; Cao, L.G.; Taneja, K.; Singer, R.H.; Wang, Y.L.; Pederson, T. Nuclear domains of the RNA subunit of RNase P. J. Cell Sci. 1997, 110 Pt 7, 829–837. [Google Scholar] [CrossRef]
- Clemson, C.M.; McNeil, J.A.; Willard, H.F.; Lawrence, J.B. XIST RNA paints the inactive X chromosome at interphase: Evidence for a novel RNA involved in nuclear/chromosome structure. J. Cell Biol. 1996, 132, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Prasanth, K.V.; Camiolo, M.; Chan, G.; Tripathi, V.; Denis, L.; Nakamura, T.; Hubner, M.R.; Spector, D.L. Nuclear organization and dynamics of 7SK RNA in regulating gene expression. Mol. Biol. Cell 2010, 21, 4184–4196. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar]
- Liu, G.; Lei, L.; Yu, J.; Kung, S.; Xie, J. Refinement of the spectra of exon usage by combined effects of extracellular stimulus and intracellular factors. Biochim. Biophys. Acta 2014, 1839, 537–545. [Google Scholar] [CrossRef]
- Xie, J.Y.; Jan, C.; Stoilov, P.; Park, J.; Black, D.L. A consensus CaMK IV-responsive RNA sequence mediates regulation of alternative exons in neurons. RNA 2005, 11, 1825–1834. [Google Scholar]
- Sohail, M.; Cao, W.; Mahmood, N.; Myschyshyn, M.; Hong, S.P.; Xie, J. Evolutionarily emerged G tracts between the polypyrimidine tract and 3 ‘ AG are splicing silencers enriched in genes involved in cancer. Bmc Genom. 2014, 15, 1143. [Google Scholar] [CrossRef]
- Sohail, M.; Xie, J. Evolutionary emergence of a novel splice variant with an opposite effect on the cell cycle. Mol. Cell. Biol. 2015, 35, 2203–2214. [Google Scholar]
- Rossbach, O.; Hung, L.H.; Schreiner, S.; Grishina, I.; Heiner, M.; Hui, J.; Bindereif, A. Auto- and cross-regulation of the hnRNP L proteins by alternative splicing. Mol. Cell. Biol. 2009, 29, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.; Willson, V.L. One-Sample T-Test. In Basic and Advanced Statistical Tests; SensePublishers: Rotterdam, The Netherlands, 2017. [Google Scholar]
- Cheng, T.L.; Chen, J.; Wan, H.; Tang, B.; Tian, W.; Liao, L.; Qiu, Z. Regulation of mRNA splicing by MeCP2 via epigenetic modifications in the brain. Sci. Rep. 2017, 7, 42790. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013, 23, 1256–1269. [Google Scholar] [CrossRef]
- Young, J.I.; Hong, E.P.; Castle, J.C.; Crespo-Barreto, J.; Bowman, A.B.; Rose, M.F.; Kang, D.; Richman, R.; Johnson, J.M.; Berget, S.; et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 2005, 102, 17551–17558. [Google Scholar] [CrossRef]
- Lopez Soto, E.J.; Lipscombe, D. Cell-specific exon methylation and CTCF binding in neurons regulate calcium ion channel splicing and function. eLife 2020, 9, e54879. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kohlstaedt, L.A.; Damianov, A.; Rio, D.C.; Black, D.L. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat. Struct. Mol. Biol. 2008, 15, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Das, U.; Wang, B.; Xie, J.Y. The matrices and constraints of GT/AG splice sites of more than 1000 species/lineages. Gene 2018, 660, 92–101. [Google Scholar] [CrossRef]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef]
- Zhu, X.; Gleiberman, A.S.; Rosenfeld, M.G. Molecular physiology of pituitary development: Signaling and transcriptional networks. Physiol. Rev. 2007, 87, 933–963. [Google Scholar] [CrossRef]
- Mangalam, H.J.; Albert, V.R.; Ingraham, H.A.; Kapiloff, M.; Wilson, L.; Nelson, C.; Elsholtz, H.; Rosenfeld, M.G. A pituitary POU domain protein, Pit-1, activates both growth hormone and prolactin promoters transcriptionally. Genes Dev. 1989, 3, 946–958. [Google Scholar] [CrossRef]
- Li, X.; Zhou, L.; Peng, G.; Liao, M.; Zhang, L.; Hu, H.; Long, L.; Tang, X.; Qu, H.; Shao, J.; et al. Pituitary P62 deficiency leads to female infertility by impairing luteinizing hormone production. Exp. Mol. Med. 2021, 53, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Takeuchi, S.; Iwama, S.; Kiyota, A.; Yasuda, Y.; Iwata, N.; Enomoto, A.; Arima, H.; Sugimura, Y. Cullin-associated NEDD8-dissociated protein 1, a novel interactor of rabphilin-3A, deubiquitylates rabphilin-3A and regulates arginine vasopressin secretion in PC12 cells. Endocr. J. 2018, 65, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.R.; Emmett, W.; Blazquez, L.; Faro, A.; Haberman, N.; Briese, M.; Trabzuni, D.; Ryten, M.; Weale, M.E.; Hardy, J.; et al. Recursive splicing in long vertebrate genes. Nature 2015, 521, 371–375. [Google Scholar] [CrossRef]
- Hatton, A.R.; Subramaniam, V.; Lopez, A.J. Generation of alternative Ultrabithorax isoforms and stepwise removal of a large intron by resplicing at exon-exon junctions. Mol. Cell 1998, 2, 787–796. [Google Scholar] [CrossRef]
- Burnette, J.M.; Miyamoto-Sato, E.; Schaub, M.A.; Conklin, J.; Lopez, A.J. Subdivision of large introns in Drosophila by recursive splicing at nonexonic elements. Genetics 2005, 170, 661–674. [Google Scholar] [CrossRef]
- Niwa, M.; Berget, S.M. Polyadenylation precedes splicing in vitro. Gene Expr. 1991, 1, 5–14. [Google Scholar]
- Niwa, M.; Berget, S.M. Mutation of the AAUAAA polyadenylation signal depresses in vitro splicing of proximal but not distal introns. Genes Dev. 1991, 5, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.L.; Yang, X.J.; Liang, G.N.; Wang, K. Isoform switching and exon skipping induced by the DNA methylation inhibitor 5-Aza-2 ‘-deoxycytidine. Sci. Rep. 2016, 6, 24545. [Google Scholar] [CrossRef]
- Gelfman, S.; Cohen, N.; Yearim, A.; Ast, G. DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon-intron structure. Genome Res. 2013, 23, 789–799. [Google Scholar] [CrossRef]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011, 479, 74–U99. [Google Scholar] [CrossRef]
- Veloso, A.; Kirkconnell, K.S.; Magnuson, B.; Biewen, B.; Paulsen, M.T.; Wilson, T.E.; Ljungman, M. Rate of elongation by RNA polymerase II is associated with specific gene features and epigenetic modifications. Genome Res. 2014, 24, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Liu, G.; Sun, Y.; Xie, J. Relocalization of the polypyrimidine tract-binding protein during PKA-induced neurite growth. Biochim. Biophys. Acta 2007, 1773, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.E.; Katolik, A.; Roberts, K.M.; Taylor, A.B.; Holloway, S.P.; Schuermann, J.P.; Montemayor, E.J.; Stevens, S.W.; Fitzpatrick, P.F.; Damha, M.J.; et al. Metal dependence and branched RNA cocrystal structures of the RNA lariat debranching enzyme Dbr1. Proc. Natl. Acad. Sci. USA 2016, 113, 14727–14732. [Google Scholar] [CrossRef]
- Arenas, J.; Hurwitz, J. Purification of a RNA debranching activity from HeLa cells. J. Biol. Chem. 1987, 262, 4274–4279. [Google Scholar] [PubMed]
- Sogo, J.M.; Ness, P.J.; Widmer, R.M.; Parish, R.W.; Koller, T. Psoralen-crosslinking of DNA as a probe for the structure of active nucleolar chromatin. J. Mol. Biol. 1984, 178, 897–919. [Google Scholar] [CrossRef]
- Niwa, M.; Rose, S.D.; Berget, S.M. In vitro polyadenylation is stimulated by the presence of an upstream intron. Genes Dev. 1990, 4, 1552–1559. [Google Scholar] [CrossRef]
- Wan, Y.; Anastasakis, D.G.; Rodriguez, J.; Palangat, M.; Gudla, P.; Zaki, G.; Tandon, M.; Pegoraro, G.; Chow, C.C.; Hafner, M.; et al. Dynamic imaging of nascent RNA reveals general principles of transcription dynamics and stochastic splice site selection. Cell 2021, 184, 2878–2895.e2820. [Google Scholar] [CrossRef]
- Anders, S.; Reyes, A.; Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Das, U.; Ogunsola, S.; Xie, J. Transcriptome-Wide Detection of Intron/Exon Definition in the Endogenous Pre-mRNA Transcripts of Mammalian Cells and Its Regulation by Depolarization. Int. J. Mol. Sci. 2022, 23, 10157. https://doi.org/10.3390/ijms231710157
Liu L, Das U, Ogunsola S, Xie J. Transcriptome-Wide Detection of Intron/Exon Definition in the Endogenous Pre-mRNA Transcripts of Mammalian Cells and Its Regulation by Depolarization. International Journal of Molecular Sciences. 2022; 23(17):10157. https://doi.org/10.3390/ijms231710157
Chicago/Turabian StyleLiu, Ling, Urmi Das, Samuel Ogunsola, and Jiuyong Xie. 2022. "Transcriptome-Wide Detection of Intron/Exon Definition in the Endogenous Pre-mRNA Transcripts of Mammalian Cells and Its Regulation by Depolarization" International Journal of Molecular Sciences 23, no. 17: 10157. https://doi.org/10.3390/ijms231710157
APA StyleLiu, L., Das, U., Ogunsola, S., & Xie, J. (2022). Transcriptome-Wide Detection of Intron/Exon Definition in the Endogenous Pre-mRNA Transcripts of Mammalian Cells and Its Regulation by Depolarization. International Journal of Molecular Sciences, 23(17), 10157. https://doi.org/10.3390/ijms231710157