
Sodium-Calcium Exchanger 2: A Pivotal Role in Oxaliplatin Induced Peripheral Neurotoxicity and Axonal Damage?

 , , ,
, , ,  ,
,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
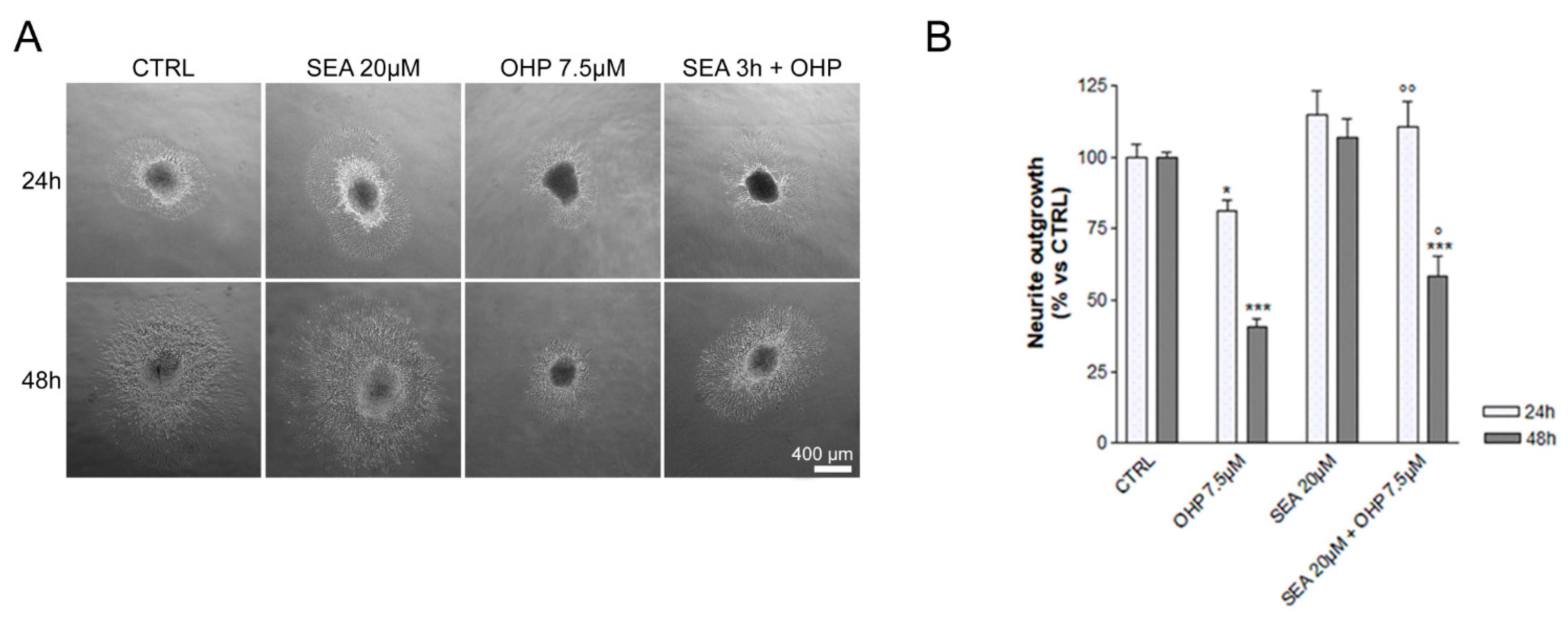
2.1. In Vitro Observations
2.2. In Vivo Observations
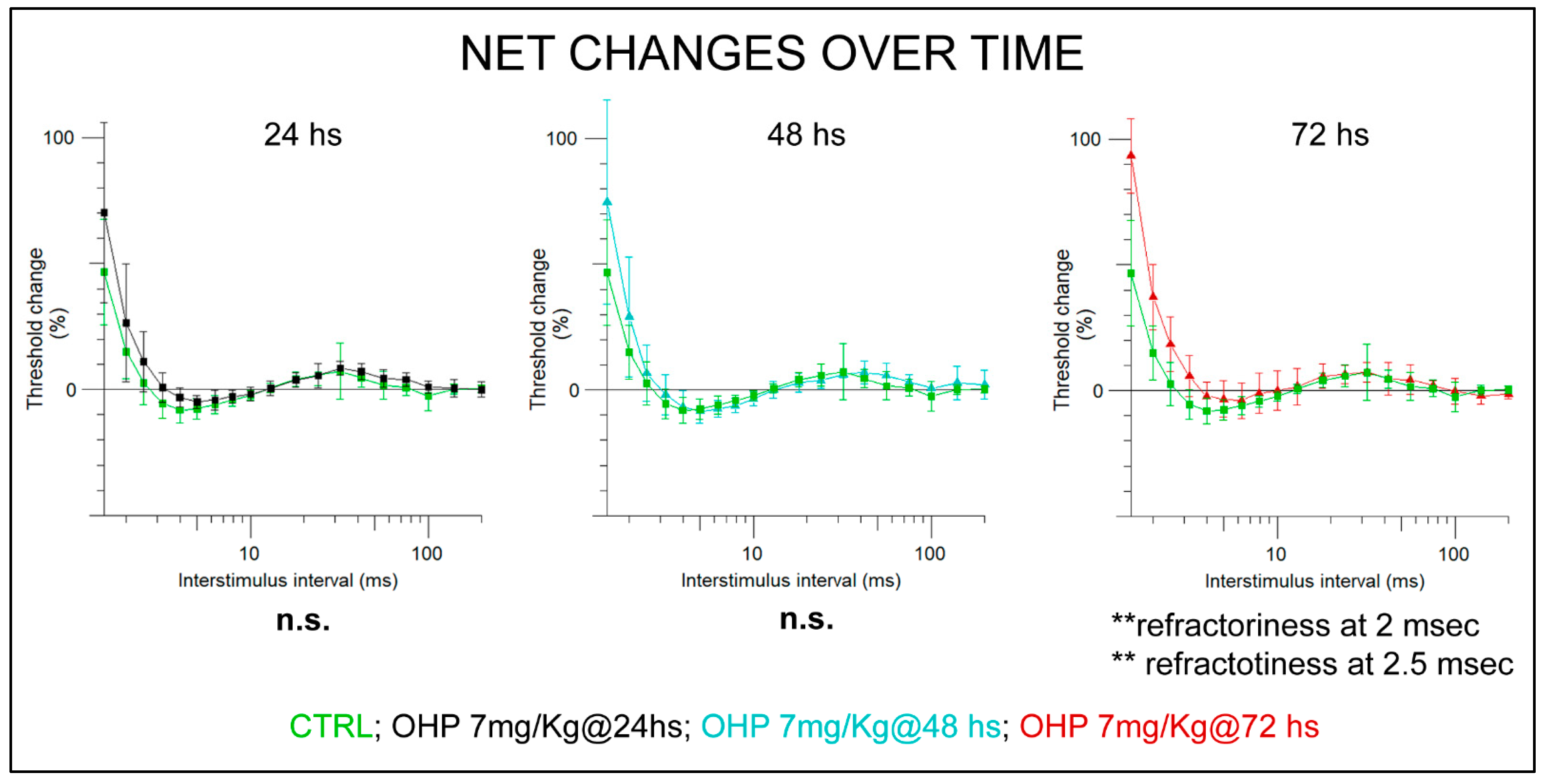
2.2.1. NET after the 1st Administration
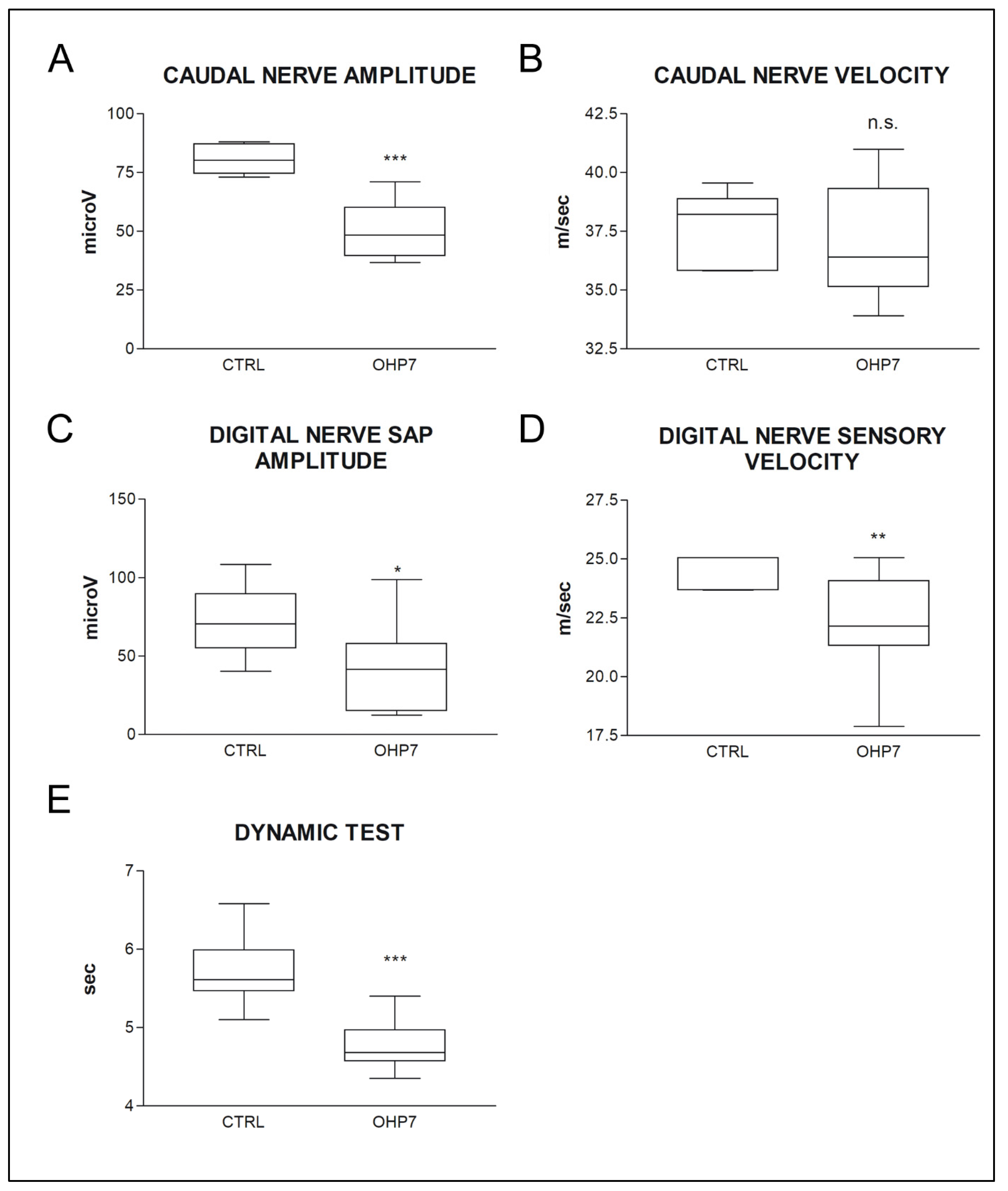
2.2.2. NCS and Behavioral Tests at the End of Treatment
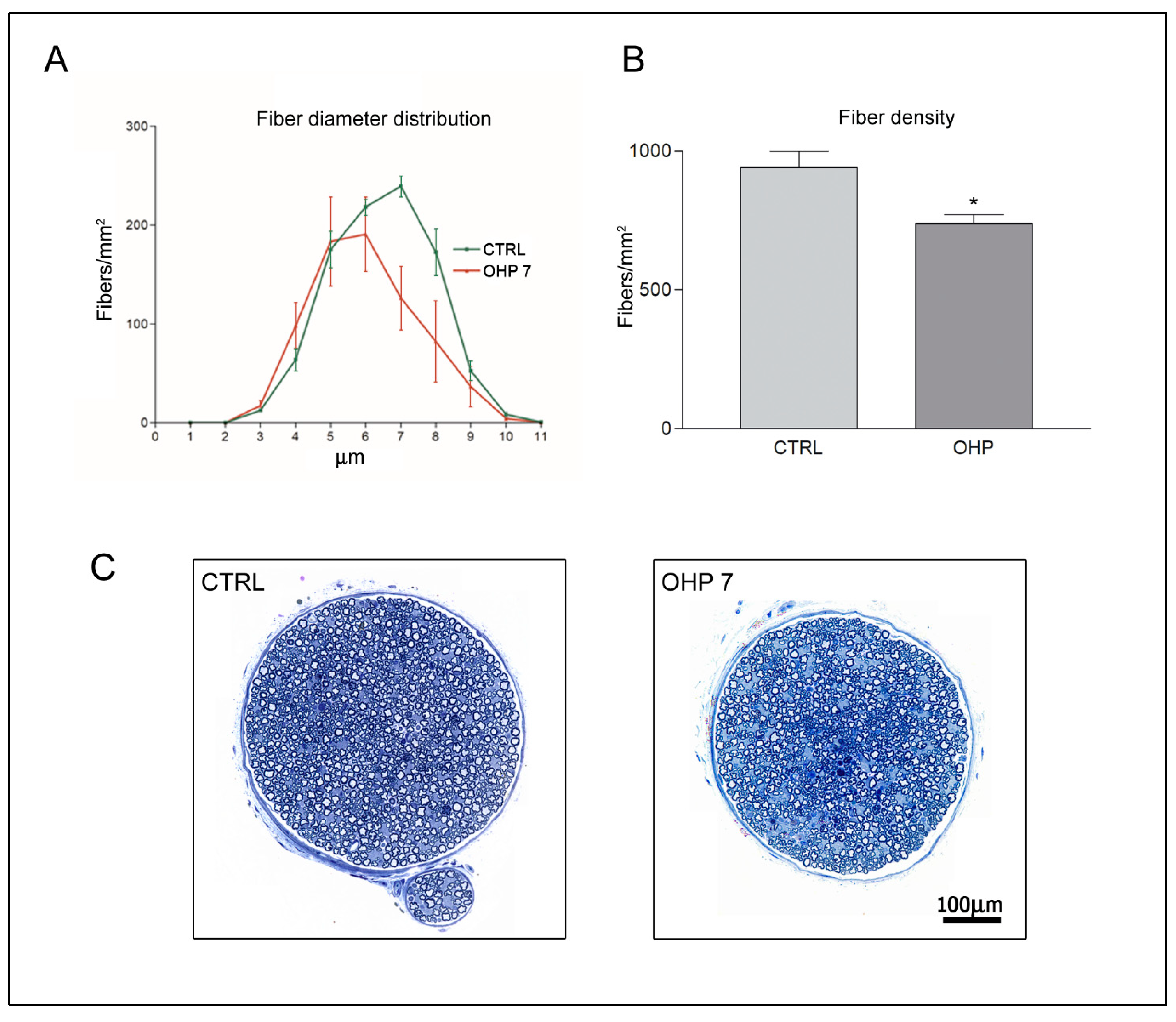
2.2.3. Caudal Nerve Morphology and Morphometry
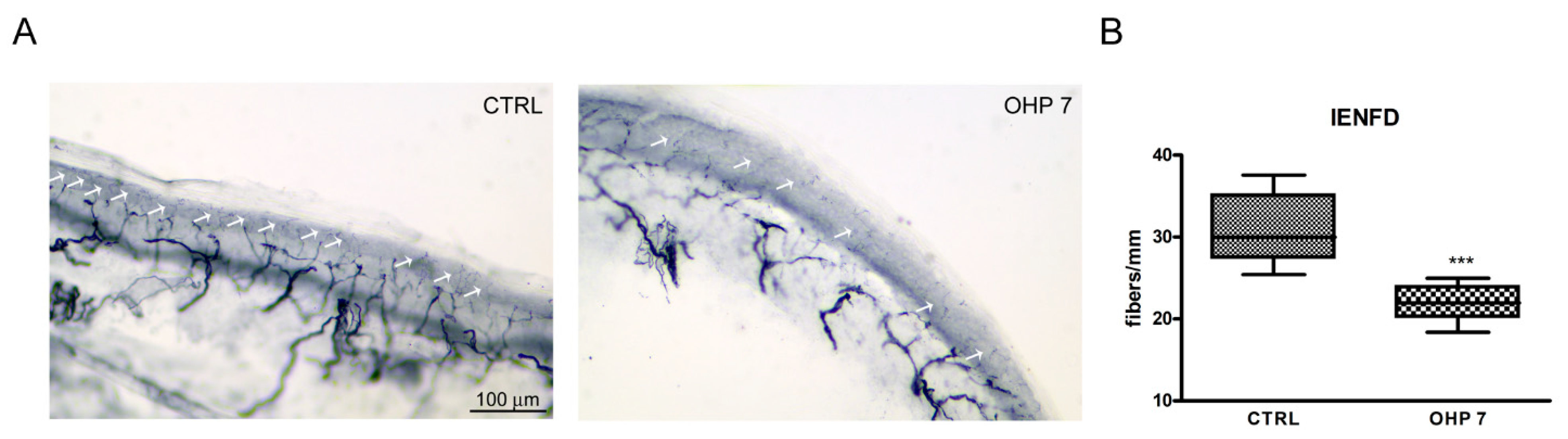
2.2.4. Intraepidermal Nerve Fiber Density (IENFD)
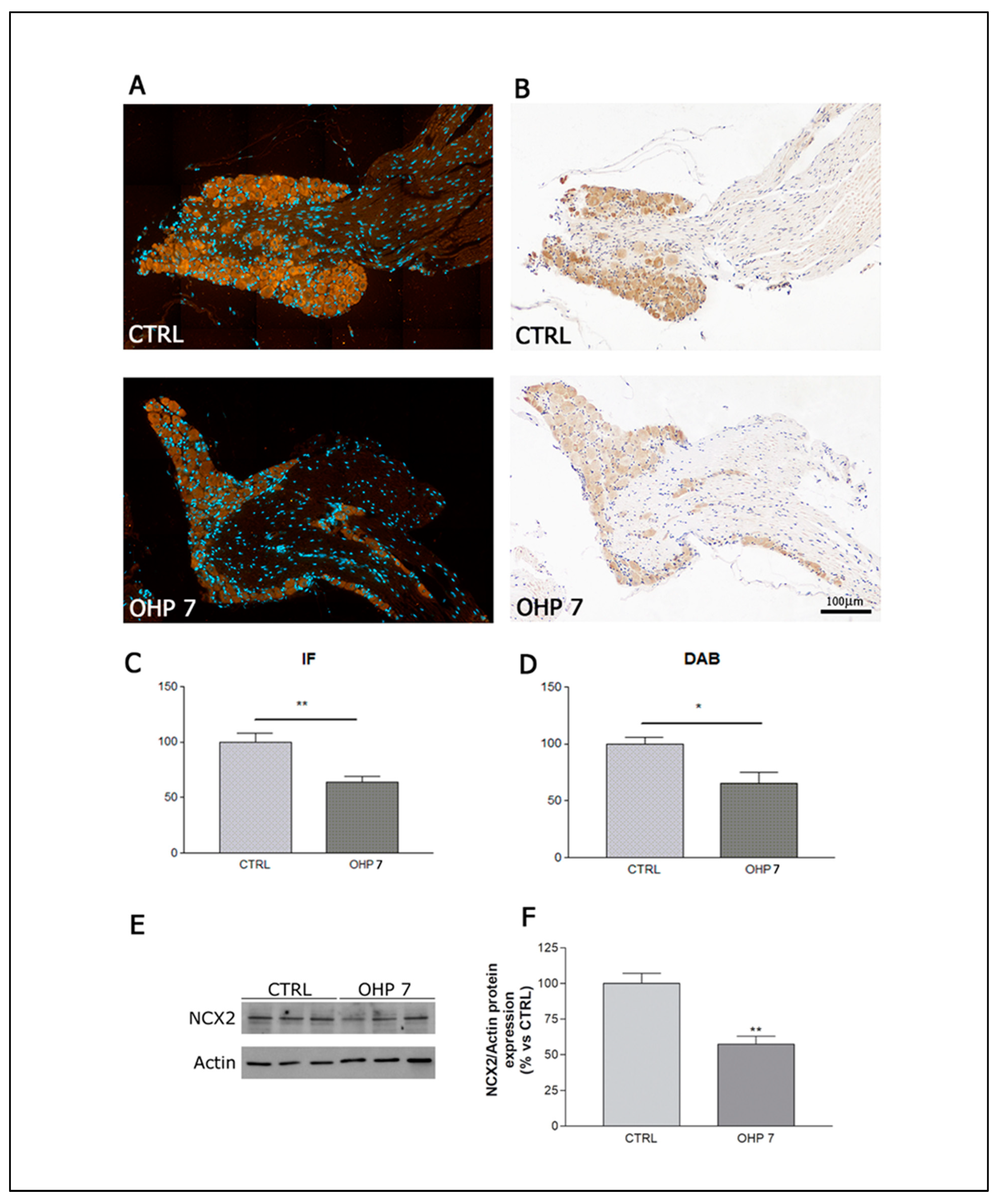
2.2.5. Immunohistochemistry (IHC), Immunofluorescence (IF), and Western Blotting (WB) for NCX2
3. Discussion
4. Materials and Methods
4.1. In Vitro Experiments
4.1.1. Drugs
4.1.2. Dorsal Root Ganglia (DRG) Explants
4.2. In Vivo Observations
4.2.1. Animal and Housing
4.2.2. Study Design
4.2.3. Drug Administration
4.2.4. Dynamic Test
4.2.5. Caudal Nerve Morphology and Morphometry
4.2.6. IENFD
4.2.7. Immunohistochemistry for NCX2
4.2.8. Immunofluorescence for NCX2
4.2.9. Western Blotting for NCX2
4.2.10. NCS
4.2.11. NET
4.2.12. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alberti, P. Platinum-drugs induced peripheral neurotoxicity: Clinical course and preclinical evidence. Expert. Opin. Drug Metab. Toxicol. 2019, 15, 487–497. [Google Scholar] [CrossRef]
- Briani, C.; Argyriou, A.A.; Izquierdo, C.; Velasco, R.; Campagnolo, M.; Alberti, P.; Frigeni, B.; Cacciavillani, M.; Bergamo, F.; Cortinovis, D.; et al. Long-term course of oxaliplatin-induced polyneuropathy: A prospective 2-year follow-up study. J. Peripher. Nerv. Syst. 2014, 19, 299–306. [Google Scholar] [CrossRef]
- Cavaletti, G.; Alberti, P.; Argyriou, A.A.; Lustberg, M.; Staff, N.P.; Tamburin, S.; Toxic Neuropathy Consortium of the Peripheral Nerve Society. Chemotherapy-induced peripheral neurotoxicity: A multifaceted, still unsolved issue. J. Peripher. Nerv. Syst. 2019, 24, S6–S12. [Google Scholar] [CrossRef]
- Alberti, P.; Cavaletti, G.; Cornblath, D.R. Toxic neuropathies: Chemotherapy-induced peripheral neurotoxicity. Curr. Opin. Neurol. 2019, 35, 676–683. [Google Scholar] [CrossRef]
- Loprinzi, C.L.; Lacchetti, C.; Bleeker, J.; Cavaletti, G.; Chauhan, C.; Hertz, D.L.; Kelley, M.R.; Lavino, A.; Lustberg, M.B.; Paice, J.A.; et al. Prevention and Management of Chemotherapy-Induced Peripheral Neuropathy in Survivors of Adult Cancers: ASCO Guideline Update. J. Clin. Oncol. 2020, 28, 3325–3348. [Google Scholar] [CrossRef]
- Alberti, P.; Lehmann, H.C. Chemotherapy induced peripheral neurotoxicity: Six essential articles for effective future research. Exp. Neurol. 2021, 337, 113555. [Google Scholar] [CrossRef]
- Alberti, P.; Cavaletti, G. Management of side effects in the personalized medicine era: Chemotherapy-induced peripheral neuropathy. Methods Mol. Biol. 2014, 1175, 301–322. [Google Scholar] [CrossRef]
- Velasco, R.; Alberti, P.; Bruna, J.; Psimaras, D.; Argyriou, A.A. Bortezomib and other proteosome inhibitors-induced peripheral neurotoxicity: From pathogenesis to treatment. J. Peripher. Nerv. Syst. 2019, 24, S52–S62. [Google Scholar] [CrossRef]
- Tamburin, S.; Park, S.B.; Alberti, P.; Demichelis, C.; Schenone, A.; Argyriou, A.A. Taxane and epothilone-induced peripheral neurotoxicity: From pathogenesis to treatment. J. Peripher. Nerv. Syst. 2019, 24, S40–S51. [Google Scholar] [CrossRef]
- Islam, B.; Lustberg, M.; Staff, N.P.; Kolb, N.; Alberti, P.; Argyriou, A.A. Vinca alkaloids, thalidomide and eribulin-induced peripheral neurotoxicity: From pathogenesis to treatment. J. Peripher. Nerv. Syst. 2019, 24, S63–S73. [Google Scholar] [CrossRef]
- Staff, N.P.; Cavaletti, G.; Islam, B.; Lustberg, M.; Psimaras, D.; Tamburin, S. Platinum-induced peripheral neurotoxicity: From pathogenesis to treatment. J. Peripher. Nerv. Syst. 2019, 24, S26–S39. [Google Scholar] [CrossRef]
- Lucchetta, M.; Lonardi, S.; Bergamo, F.; Alberti, P.; Velasco, R.; Argyriou, A.A.; Briani, C.; Bruna, J.; Cazzaniga, M.; Cortinovis, D.; et al. Incidence of atypical acute nerve hyperexcitability symptoms in oxaliplatin-treated patients with colorectal cancer. Cancer Chemother. Pharmacol. 2012, 70, 899–902. [Google Scholar] [CrossRef]
- Argyriou, A.A.; Velasco, R.; Briani, C.; Cavaletti, G.; Bruna, J.; Alberti, P.; Cacciavillani, M.; Lonardi, S.; Santos, C.; Cortinovis, D.; et al. Peripheral neurotoxicity of oxaliplatin in combination with 5-fluorouracil (FOLFOX) or capecitabine (XELOX): A prospective evaluation of 150 colorectal cancer patients. Ann. Oncol. 2012, 23, 3116–3122. [Google Scholar] [CrossRef]
- Grolleau, F.; Gamelin, L.; Boisdron-Celle, M.; Lapied, B.; Pelhate, M.; Gamelin, E. A possible explanation for a neurotoxic effect of the anticancer agent oxaliplatin on neuronal voltage-gated sodium channels. J. Neurophysiol. 2001, 85, 2293–2297. [Google Scholar] [CrossRef]
- Park, S.B.; Lin, C.S.; Krishnan, A.V.; Goldstein, D.; Friedlander, M.L.; Kiernan, M.C. Oxaliplatin-induced neurotoxicity: Changes in axonal excitability precede development of neuropathy. Brain 2009, 132, 2712–2723. [Google Scholar] [CrossRef]
- Wu, S.N.; Chen, B.S.; Wu, Y.H.; Peng, H.; Chen, L.T. The mechanism of the actions of oxaliplatin on ion currents and action potentials in differentiated NG108-15 neuronal cells. Neurotoxicology 2009, 30, 677–685. [Google Scholar] [CrossRef]
- Dimitrov, A.G.; Dimitrova, N.A. A possible link of oxaliplatin-induced neuropathy with potassium channel deficit. Muscle Nerve 2012, 45, 403–411. [Google Scholar] [CrossRef]
- Makker, P.G.S.; White, D.; Lees, J.G.; Parmar, J.; Goldstein, D.; Park, S.B.; Howells, J.; Moalem-Taylor, G. Acute changes in nerve excitability following oxaliplatin treatment in mice. J. Neurophysiol. 2020, 124, 232–244. [Google Scholar] [CrossRef]
- Adelsberger, H.; Quasthoff, S.; Grosskreutz, J.; Lepier, A.; Eckel, F.; Lersch, C. The chemotherapeutic oxaliplatin alters voltage-gated Na+ channel kinetics on rat sensory neurons. Eur. J. Pharmacol. 2000, 406, 25–32. [Google Scholar] [CrossRef]
- Velasco, R.; Bruna, J.; Briani, C.; Argyriou, A.A.; Cavaletti, G.; Alberti, P.; Frigeni, B.; Cacciavillani, M.; Lonardi, S.; Cortinovis, D.; et al. Early predictors of oxaliplatin-induced cumulative neuropathy in colorectal cancer patients. J. Neurol. Neurosurg. Psychiatry 2014, 85, 392–398. [Google Scholar] [CrossRef]
- Argyriou, A.A.; Cavaletti, G.; Antonacopoulou, A.; Genazzani, A.A.; Briani, C.; Bruna, J.; Terrazzino, S.; Velasco, R.; Alberti, P.; Campagnolo, M.; et al. Voltage-gated sodium channel polymorphisms play a pivotal role in the development of oxaliplatin-induced peripheral neurotoxicity: Results from a prospective multicenter study. Cancer 2013, 119, 3570–3577. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Antonacopoulou, A.G.; Alberti, P.; Briani, C.; Bruna, J.; Velasco, R.; Anastopoulou, G.G.; Park, S.B.; Cavaletti, G.; Kalofonos, H.P. Liability of the voltage-gated potassium channel KCNN3 repeat polymorphism to acute oxaliplatin-induced peripheral neurotoxicity. J. Peripher. Nerv. Syst. 2019, 24, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.V.; Goldstein, D.; Friedlander, M.; Kiernan, M.C. Oxaliplatin and axonal Na+ channel function in vivo. Clin. Cancer Res. 2006, 12, 4481–4484. [Google Scholar] [CrossRef]
- Heide, R.; Bostock, H.; Ventzel, L.; Grafe, P.; Bergmans, J.; Fuglsang-Frederiksen, A.; Finnerup, N.B.; Tankisi, H. Axonal excitability changes and acute symptoms of oxaliplatin treatment: In vivo evidence for slowed sodium channel inactivation. Clin. Neurophysiol. 2018, 129, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Alberti, P.; Canta, A.; Chiorazzi, A.; Fumagalli, G.; Meregalli, C.; Monza, L.; Pozzi, E.; Ballarini, E.; Rodriguez-Menendez, V.; Oggioni, N.; et al. Topiramate prevents oxaliplatin-related axonal hyperexcitability and oxaliplatin induced peripheral neurotoxicity. Neuropharmacology 2020, 164, 107905. [Google Scholar] [CrossRef]
- Bruna, J.; Alberti, P.; Calls-Cobos, A.; Caillaud, M.; Damaj, M.I.; Navarro, X. Methods for in vivo studies in rodents of chemotherapy induced peripheral neuropathy. Exp. Neurol. 2020, 325, 113154. [Google Scholar] [CrossRef]
- Morris, C.E.; Boucher, P.A.; Joós, B. Left-shifted nav channels in injured bilayer: Primary targets for neuroprotective nav antagonists? Front. Pharmacol. 2012, 3, 19. [Google Scholar] [CrossRef]
- Persson, A.K.; Black, J.A.; Gasser, A.; Cheng, X.; Fischer, T.Z.; Waxman, S.G. Sodium-calcium exchanger and multiple sodium channel isoforms in intra-epidermal nerve terminals. Mol. Pain 2010, 6, 84. [Google Scholar] [CrossRef]
- Cavaletti, G.; Tredici, G.; Petruccioli, M.G.; Dondè, E.; Tredici, P.; Marmiroli, P.; Minoia, C.; Ronchi, A.; Bayssas, M.; Etienne, G.G. Effects of different schedules of oxaliplatin treatment on the peripheral nervous system of the rat. Eur. J. Cancer 2001, 37, 2457–2463. [Google Scholar] [CrossRef]
- Annunziato, L.; Pignataro, G.; Di Renzo, G.F. Pharmacology of brain Na+/Ca2+ exchanger: From molecular biology to therapeutic perspectives. Pharmacol. Rev. 2004, 56, 633–654. [Google Scholar] [CrossRef]
- Annunziato, L.; Pignataro, G.; Boscia, F.; Sirabella, R.; Formisano, L.; Saggese, M.; Cuomo, O.; Gala, R.; Secondo, A.; Viggiano, D.; et al. ncx1, ncx2, and ncx3 gene product expression and function in neuronal anoxia and brain ischemia. Ann. NY Acad. Sci. 2007, 1099, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.P.; Lederer, W.J. Sodium/calcium exchange: Its physiological implications. Physiol. Rev. 1999, 79, 763–854. [Google Scholar] [CrossRef] [PubMed]
- Persson, A.K.; Kim, I.; Zhao, P.; Estacion, M.; Black, J.A.; Waxman, S.G. Sodium channels contribute to degeneration of dorsal root ganglion neurites induced by mitochondrial dysfunction in an in vitro model of axonal injury. J. Neurosci. 2013, 33, 19250–19261. [Google Scholar] [CrossRef]
- Persson, A.K.; Hoeijmakers, J.G.J.; Estacion, M.; Black, J.A.; Waxman, S.G. Sodium Channels, Mitochondria, and Axonal Degeneration in Peripheral Neuropathy. Trends Mol. Med. 2016, 22, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Neher, E.; Sakaba, T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 2008, 59, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Marty, A.; Zimmerberg, J. Diffusion into the patch-clamp recording pipette of a factor necessary for muscarinic current response. Cell Signal. 1989, 1, 259–268. [Google Scholar] [CrossRef]
- Malenka, R.C.; Kauer, J.A.; Zucker, R.S.; Nicoll, R.A. Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science 1988, 242, 81–84. [Google Scholar] [CrossRef]
- Szekely, A.M.; Costa, E.; Grayson, D.R. Transcriptional program coordination by N-methyl-D-aspartate-sensitive glutamate receptor stimulation in primary cultures of cerebellar neurons. Mol. Pharmacol. 1990, 38, 624–633. [Google Scholar]
- McCormack, J.G.; Denton, R.M. Intracellular calcium ions and intramitochondrial Ca2+ in the regulation of energy metabolism in mammalian tissues. Proc. Nutr. Soc. 1990, 49, 57–75. [Google Scholar] [CrossRef]
- Chalfie, M.; Wolinsky, E. The identification and suppression of inherited neurodegeneration in Caenorhabditis elegans. Nature 1990, 345, 410–416. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Tavernarakis, N. Calcium homeostasis in aging neurons. Front. Genet. 2012, 3, 200. [Google Scholar] [CrossRef]
- Stys, P.K.; Waxman, S.G.; Ransom, B.R. Ionic mechanisms of anoxic injury in mammalian CNS white matter: Role of Na+ channels and Na+-Ca2+ exchanger. J. Neurosci. 1992, 12, 430–439. [Google Scholar] [CrossRef]
- Lehning, E.J.; Doshi, R.; Isaksson, N.; Stys, P.K.; LoPachin, R.M. Mechanisms of injury-induced calcium entry into peripheral nerve myelinated axons: Role of reverse sodium-calcium exchange. J. Neurochem. 1996, 66, 493–500. [Google Scholar] [CrossRef]
- Lehning, E.J.; Doshi, R.; Stys, P.K.; LoPachin, R.M. Mechanisms of injury-induced calcium entry into peripheral nerve myelinated axons: In vitro anoxia and ouabain exposure. Brain Res. 1995, 694, 158–166. [Google Scholar] [CrossRef]
- Pannaccione, A.; Piccialli, I.; Secondo, A.; Ciccone, R.; Molinaro, P.; Boscia, F.; Annunziato, L. The Na. Cell Calcium 2020, 87, 102190. [Google Scholar] [CrossRef]
- Anzilotti, S.; Valsecchi, V.; Brancaccio, P.; Guida, N.; Laudati, G.; Tedeschi, V.; Petrozziello, T.; Frecentese, F.; Magli, E.; Hassler, B.; et al. Prolonged NCX activation prevents SOD1 accumulation, reduces neuroinflammation, ameliorates motor behavior and prolongs survival in a ALS mouse model. Neurobiol. Dis. 2021, 159, 105480. [Google Scholar] [CrossRef]
- Scuteri, A.; Ravasi, M.; Monfrini, M.; Milano, A.; D’Amico, G.; Miloso, M.; Tredici, G. Human Mesenchymal Stem Cells Protect Dorsal Root Ganglia from the Neurotoxic Effect of Cisplatin. Anticancer. Res. 2015, 35, 5383–5389. [Google Scholar]
- Amran, M.S.; Hashimoto, K.; Homma, N. Effects of sodium-calcium exchange inhibitors, KB-R7943 and SEA0400, on aconitine-induced arrhythmias in guinea pigs in vivo, in vitro, and in computer simulation studies. J. Pharmacol. Exp. Ther. 2004, 310, 83–89. [Google Scholar] [CrossRef]
- Alberti, P. Chemotherapy-induced peripheral neurotoxicity - outcome measures: The issue. Expert. Opin. Drug. Metab. Toxicol. 2017, 13, 241–243. [Google Scholar] [CrossRef][Green Version]
- Boscia, F.; Gala, R.; Pignataro, G.; de Bartolomeis, A.; Cicale, M.; Ambesi-Impiombato, A.; Di Renzo, G.; Annunziato, L. Permanent focal brain ischemia induces isoform-dependent changes in the pattern of Na+/Ca2+ exchanger gene expression in the ischemic core, periinfarct area, and intact brain regions. J. Cereb. Blood Flow Metab. 2006, 26, 502–517. [Google Scholar] [CrossRef]
- Argyriou, A.A.; Cavaletti, G.; Briani, C.; Velasco, R.; Bruna, J.; Campagnolo, M.; Alberti, P.; Bergamo, F.; Cortinovis, D.; Cazzaniga, M.; et al. Clinical pattern and associations of oxaliplatin acute neurotoxicity: A prospective study in 170 patients with colorectal cancer. Cancer 2013, 119, 438–444. [Google Scholar] [CrossRef]
- Alberti, P.; Rossi, E.; Argyriou, A.A.; Kalofonos, H.P.; Briani, C.; Cacciavillani, M.; Campagnolo, M.; Bruna, J.; Velasco, R.; Cazzaniga, M.E.; et al. Risk stratification of oxaliplatin induced peripheral neurotoxicity applying electrophysiological testing of dorsal sural nerve. Support. Care Cancer 2018, 26, 3143–3151. [Google Scholar] [CrossRef]
- Grisold, W.; Cavaletti, G.; Windebank, A.J. Peripheral neuropathies from chemotherapeutics and targeted agents: Diagnosis, treatment, and prevention. Neuro. Oncol. 2012, 14, iv45–iv54. [Google Scholar] [CrossRef]
- Boyette-Davis, J.; Dougherty, P.M. Protection against oxaliplatin-induced mechanical hyperalgesia and intraepidermal nerve fiber loss by minocycline. Exp. Neurol. 2011, 229, 353–357. [Google Scholar] [CrossRef]
- Koskinen, M.J.; Kautio, A.L.; Haanpää, M.L.; Haapasalo, H.K.; Kellokumpu-Lehtinen, P.L.; Saarto, T.; Hietaharju, A.J. Intraepidermal nerve fibre density in cancer patients receiving adjuvant chemotherapy. Anticancer Res. 2011, 31, 4413–4416. [Google Scholar]
- Brenneman, D.E.; Kinney, W.A.; Ward, S.J. Knockdown siRNA Targeting the Mitochondrial Sodium-Calcium Exchanger-1 Inhibits the Protective Effects of Two Cannabinoids Against Acute Paclitaxel Toxicity. J. Mol. Neurosci. 2019, 68, 603–619. [Google Scholar] [CrossRef]
- Yilmaz, E.; Gold, M.S. Paclitaxel-induced increase in NCX activity in subpopulations of nociceptive afferents: A protective mechanism against chemotherapy-induced peripheral neuropathy? Cell Calcium 2016, 60, 25–31. [Google Scholar] [CrossRef]
- Matsuda, T.; Arakawa, N.; Takuma, K.; Kishida, Y.; Kawasaki, Y.; Sakaue, M.; Takahashi, K.; Takahashi, T.; Suzuki, T.; Ota, T.; et al. SEA0400, a novel and selective inhibitor of the Na+-Ca2+ exchanger, attenuates reperfusion injury in the in vitro and in vivo cerebral ischemic models. J. Pharmacol. Exp. Ther. 2001, 298, 249–256. [Google Scholar]
- Arakawa, N.; Sakaue, M.; Yokoyama, I.; Hashimoto, H.; Koyama, Y.; Baba, A.; Matsuda, T. KB-R7943 inhibits store-operated Ca2+ entry in cultured neurons and astrocytes. Biochem. Biophys. Res. Commun. 2000, 279, 354–357. [Google Scholar] [CrossRef]
- Lee, C.; Hryshko, L.V. SEA0400: A novel sodium-calcium exchange inhibitor with cardioprotective properties. Cardiovasc. Drug. Rev. 2004, 22, 334–347. [Google Scholar] [CrossRef]
- Koyama, Y.; Matsui, S.; Itoh, S.; Osakada, M.; Baba, A.; Matsuda, T. The selective Na+-Ca2+ exchange inhibitor attenuates brain edema after radiofrequency lesion in rats. Eur. J. Pharmacol. 2004, 489, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Shenoda, B. The role of Na+/Ca2+ exchanger subtypes in neuronal ischemic injury. Transl. Stroke. Res. 2015, 6, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Beaugé, L.; DiPolo, R. SEA-0400, a potent inhibitor of the Na+/Ca2+ exchanger, as a tool to study exchanger ionic and metabolic regulation. Am. J. Physiol. Cell Physiol. 2005, 288, C1374–C1380. [Google Scholar] [CrossRef]
- Nashida, T.; Takuma, K.; Fukuda, S.; Kawasaki, T.; Takahashi, T.; Baba, A.; Ago, Y.; Matsuda, T. The specific Na+/Ca2+ exchange inhibitor SEA0400 prevents nitric oxide-induced cytotoxicity in SH-SY5Y cells. Neurochem. Int. 2011, 59, 51–58. [Google Scholar] [CrossRef]
- Calls, A.; Carozzi, V.; Navarro, X.; Monza, L.; Bruna, J. Pathogenesis of platinum-induced peripheral neurotoxicity: Insights from preclinical studies. Exp. Neurol. 2020, 325, 113141. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.A.; Lockwood, G.F.; Greenslade, D.; Brienza, S.; Bayssas, M.; Gamelin, E. Clinical pharmacokinetics of oxaliplatin: A critical review. Clin. Cancer Res. 2000, 6, 1205–1218. [Google Scholar] [PubMed]
- Lersch, C.; Schmelz, R.; Eckel, F.; Erdmann, J.; Mayr, M.; Schulte-Frohlinde, E.; Quasthoff, S.; Grosskreutz, J.; Adelsberger, H. Prevention of oxaliplatin-induced peripheral sensory neuropathy by carbamazepine in patients with advanced colorectal cancer. Clin. Colorectal. Cancer 2002, 2, 54–58. [Google Scholar] [CrossRef]
- Cavaletti, G.; Nicolini, G.; Marmiroli, P. Neurotoxic effects of antineoplastic drugs: The lesson of pre-clinical studies. Front. Biosci. 2008, 13, 3506–3524. [Google Scholar] [CrossRef][Green Version]
- Soliman, D.; Wang, L.; Hamming, K.S.; Yang, W.; Fatehi, M.; Carter, C.C.; Clanachan, A.S.; Light, P.E. Late sodium current inhibition alone with ranolazine is sufficient to reduce ischemia- and cardiac glycoside-induced calcium overload and contractile dysfunction mediated by reverse-mode sodium/calcium exchange. J. Pharmacol. Exp. Ther. 2012, 343, 325–332. [Google Scholar] [CrossRef]
- Catterall, W.A.; Swanson, T.M. Structural Basis for Pharmacology of Voltage-Gated Sodium and Calcium Channels. Mol. Pharmacol. 2015, 88, 141–150. [Google Scholar] [CrossRef]
- de Lera Ruiz, M.; Kraus, R.L. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 2015, 58, 7093–7118. [Google Scholar] [CrossRef] [PubMed]
- Monza, L.; Fumagalli, G.; Chiorazzi, A.; Alberti, P. Addressing the Need of a Translational Approach in Peripheral Neuropathy Research: Morphology Meets Function. Brain Sci. 2021, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Boërio, D.; Greensmith, L.; Bostock, H. Excitability properties of motor axons in the maturing mouse. J. Peripher. Nerv. Syst. 2009, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Bostock, H. Effects of membrane polarization and ischaemia on the excitability properties of human motor axons. Brain 2000, 123 Pt 12, 2542–2551. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ballarini, E.; Malacrida, A.; Rodriguez-Menendez, V.; Pozzi, E.; Canta, A.; Chiorazzi, A.; Monza, L.; Semperboni, S.; Meregalli, C.; Carozzi, V.A.; et al. Sodium-Calcium Exchanger 2: A Pivotal Role in Oxaliplatin Induced Peripheral Neurotoxicity and Axonal Damage? Int. J. Mol. Sci. 2022, 23, 10063. https://doi.org/10.3390/ijms231710063
Ballarini E, Malacrida A, Rodriguez-Menendez V, Pozzi E, Canta A, Chiorazzi A, Monza L, Semperboni S, Meregalli C, Carozzi VA, et al. Sodium-Calcium Exchanger 2: A Pivotal Role in Oxaliplatin Induced Peripheral Neurotoxicity and Axonal Damage? International Journal of Molecular Sciences. 2022; 23(17):10063. https://doi.org/10.3390/ijms231710063
Chicago/Turabian StyleBallarini, Elisa, Alessio Malacrida, Virginia Rodriguez-Menendez, Eleonora Pozzi, Annalisa Canta, Alessia Chiorazzi, Laura Monza, Sara Semperboni, Cristina Meregalli, Valentina Alda Carozzi, and et al. 2022. "Sodium-Calcium Exchanger 2: A Pivotal Role in Oxaliplatin Induced Peripheral Neurotoxicity and Axonal Damage?" International Journal of Molecular Sciences 23, no. 17: 10063. https://doi.org/10.3390/ijms231710063
APA StyleBallarini, E., Malacrida, A., Rodriguez-Menendez, V., Pozzi, E., Canta, A., Chiorazzi, A., Monza, L., Semperboni, S., Meregalli, C., Carozzi, V. A., Hashemi, M., Nicolini, G., Scuteri, A., Housley, S. N., Cavaletti, G., & Alberti, P. (2022). Sodium-Calcium Exchanger 2: A Pivotal Role in Oxaliplatin Induced Peripheral Neurotoxicity and Axonal Damage? International Journal of Molecular Sciences, 23(17), 10063. https://doi.org/10.3390/ijms231710063