
The Mechanism of Energy Coupling in H+/Na+-Pumping Membrane Pyrophosphatase—Possibilities and Probabilities


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
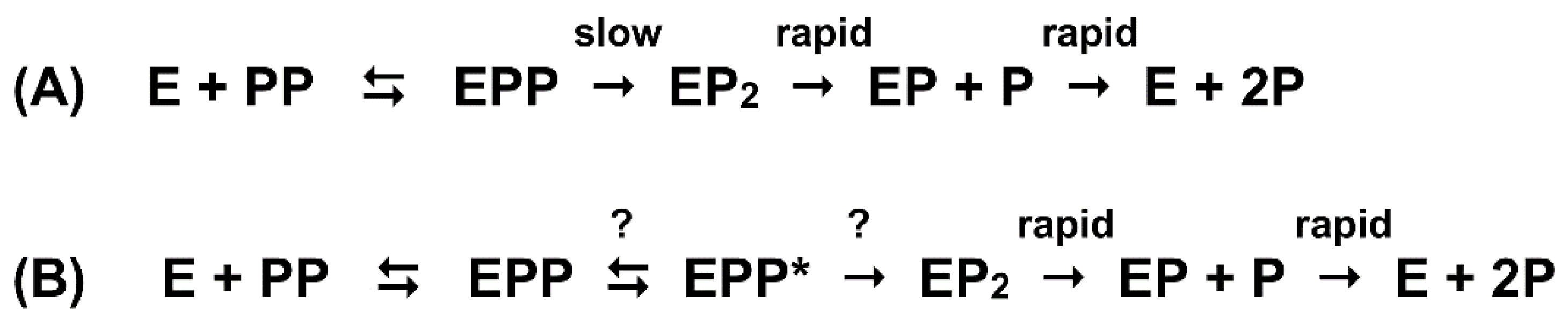
2. The Coupling Step
3. Subunit Cooperation
4. Direct or Indirect Coupling?
5. The Relationship between H+ and Na+ Transport
6. Na+,H+-Pyrophosphatase and Transport Stoichiometry
7. Comparison with Other Na+ Pumps
8. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Baltscheffsky, H.; von Stedingk, L.V.; Heldt, H.W.; Klingenberg, M. Inorganic pyrophosphate: Formation in bacterial photophosphorylation. Science 1966, 153, 1120–1122. [Google Scholar] [CrossRef] [PubMed]
- Baykov, A.A.; Malinen, A.M.; Luoto, H.H.; Lahti, R. Pyrophosphate-fueled Na+ and H+ transport in prokaryotes. Microbiol. Mol. Biol. Rev. 2013, 77, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.Y.; Kellosalo, J.; Sun, Y.J.; Goldman, A. Proton/sodium pumping pyrophosphatases: The last of the primary ion pumps. Curr. Opin. Struct. Biol. 2014, 27, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Pérez-Castiñeira, J.; Baltscheffsky, M.; Baltscheffsky, H. H+-PPases: Yesterday, today and tomorrow. IUBMB Life 2007, 59, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Segami, S.; Asaoka, M.; Kinoshita, S.; Fukuda, M.; Nakanishi, Y.; Maeshima, M. Biochemical, structural and physiological characteristics of vacuolar H+-pyrophosphatase. Plant Cell Physiol. 2018, 59, 1300–1308. [Google Scholar] [CrossRef]
- Gaxiola, R.A.; Fink, G.R.; Hirschi, K.D. Genetic manipulation of vacuolar proton pumps and transporters. Plant Physiol. 2002, 129, 967–973. [Google Scholar] [CrossRef]
- Gutiérrez-Luna, F.M.; Hernandez-Dominguez, E.E.; Gabriela Valencia-Turcotte, L.G.; Rodríguez-Sotres, R. Pyrophosphate and pyrophosphatases in plants, their involvement in stress responses and their possible relationship to secondary metabolism. Plant Sci. 2018, 267, 11–19. [Google Scholar] [CrossRef]
- Belogurov, G.A.; Malinen, A.M.; Turkina, M.V.; Jalonen, U.; Rytkönen, K.; Baykov, A.A.; Lahti, R. Membrane-bound pyrophosphatase of Thermotoga maritima requires sodium for activity. Biochemistry 2005, 44, 2088–2096. [Google Scholar] [CrossRef]
- Malinen, A.M.; Belogurov, G.A.; Baykov, A.A.; Lahti, R. Na+-pyrophosphatase: A novel primary sodium pump. Biochemistry 2007, 46, 8872–8878. [Google Scholar] [CrossRef]
- Luoto, H.; Belogurov, G.A.; Baykov, A.A.; Lahti, R.; Malinen, A.M. Na+-translocating membrane pyrophosphatases are widespread in the microbial world and evolutionarily precede H+-translocating pyrophosphatases. J. Biol. Chem. 2011, 286, 21633–21642. [Google Scholar] [CrossRef]
- Biegel, E.; Müller, V. A Na+-translocating pyrophosphatase in the acetogenic bacterium Acetobacterium woodii. J. Biol. Chem. 2011, 286, 6080–6084. [Google Scholar] [CrossRef] [PubMed]
- Luoto, H.H.; Nordbo, E.; Baykov, A.A.; Lahti, R.; Malinen, A.M. Membrane Na+-pyrophosphatases can transport protons at low sodium concentrations. J. Biol. Chem. 2013, 288, 35489–35499. [Google Scholar] [CrossRef] [PubMed]
- Luoto, H.H.; Baykov, A.A.; Lahti, R.; Malinen, A.M. Membrane-integral pyrophosphatase subfamily capable of translocating both Na+ and H+. Proc. Natl. Acad. Sci. USA 2013, 110, 1255–1260. [Google Scholar] [CrossRef]
- Belogurov, G.A.; Lahti, R. A lysine substitute for K+: A460K mutation eliminates K+ dependence in H+-pyrophosphatase of Carboxydothermus hydrogenoformans. J. Biol. Chem. 2002, 277, 49651–49654. [Google Scholar] [CrossRef] [PubMed]
- Li, K.M.; Wilkinson, C.; Kellosalo, J.; Tsai, J.Y.; Kajander, T.; Jeuken, L.J.C.; Sun, Y.J.; Goldman, A. Membrane pyrophosphatases from Thermotoga maritima and Vigna radiata suggest a conserved coupling mechanism. Nat. Commun. 2016, 7, 13596. [Google Scholar] [CrossRef]
- Baykov, A.A.; Bakuleva, N.P.; Rea, P.A. Steady-state kinetics of substrate hydrolysis by vacuolar H+-pyrophosphatase. A simple three-state model. Eur. J. Biochem. 1993, 217, 755–762. [Google Scholar] [CrossRef]
- Artukka, E.; Luoto, H.H.; Baykov, A.A.; Lahti, R.; Malinen, A.M. Role of the potassium/lysine cationic center in catalysis and functional asymmetry in membrane-bound pyrophosphatases. Biochem. J. 2018, 475, 1141–1158. [Google Scholar] [CrossRef]
- Malinen, A.M.; Baykov, A.A.; Lahti, R. Mutual effects of cationic ligands and substrate on activity of the Na+-transporting pyrophosphatase of Methanosarcina mazei. Biochemistry 2008, 47, 13447–13454. [Google Scholar] [CrossRef]
- Lin, S.M.; Tsai, J.Y.; Hsiao, C.D.; Huang, Y.T.; Chiu, C.L.; Liu, M.H.; Tung, J.Y.; Liu, T.H.; Pan, R.L.; Sun, Y.J. Crystal structure of a membrane-embedded H+-translocating pyrophosphatase. Nature 2012, 484, 399–403. [Google Scholar] [CrossRef]
- Kellosalo, J.; Kajander, T.; Kogan, K.; Pokharel, K.; Goldman, A. The structure and catalytic cycle of a sodium-pumping pyrophosphatase. Science 2012, 337, 473–476. [Google Scholar] [CrossRef]
- Vidilaseris, K.; Kiriazis, A.; Turku, A.; Khattab, A.; Johansson, N.G.; Leino, T.O.; Kiuru, P.S.; Boije af Gennäs, G.; Meri, S.; Yli-Kauhaluoma, J.; et al. Asymmetry in catalysis by Thermotoga maritima membrane bound pyrophosphatase demonstrated by a nonphosphorus allosteric inhibitor. Sci. Adv. 2019, 5, eaav7574. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Malinen, A.M.; Anashkin, V.A.; Orlov, V.N.; Bogachev, A.V.; Lahti, R.; Baykov, A.A. Pre-steady state kinetics and solvent isotope effects support the “billiard-type” transport mechanism in Na+-translocating pyrophosphatase. Prot. Sci. 2022, 31, e4394. [Google Scholar] [CrossRef]
- Baykov, A.A.; Fabrichniy, I.P.; Pohjanjoki, P.; Zyryanov, A.B.; Lahti, R. Fluoride effects along the reaction pathway of pyrophosphatase. Evidence for a second enzyme pyrophosphate intermediate. Biochemistry 2000, 39, 11939–11947. [Google Scholar] [CrossRef]
- Anashkin, V.A.; Malinen, A.M.; Bogachev, A.V.; Baykov, A.A. Catalytic asymmetry in homodimeric H+-pumping membrane pyrophosphatase demonstrated by non-hydrolyzable pyrophosphate analogs. Int. J. Mol. Sci. 2021, 22, 9820. [Google Scholar] [CrossRef]
- Shah, N.R.; Wilkinson, C.; Harborne, S.P.; Turku, A.; Li, K.M.; Sun, Y.J.; Sarah Harris, S.; Goldman, A. Insights into the mechanism of membrane pyrophosphatases by combining experiment and computer simulation. Struct. Dyn. 2017, 4, 032105. [Google Scholar] [CrossRef]
- Baykov, A.A. Energy coupling in cation-pumping pyrophosphatase—Back to Mitchell. Front. Plant Sci. 2020, 11, 107. [Google Scholar] [CrossRef]
- Tsai, J.Y.; Tang, K.Z.; Li, K.M.; Hsu, B.L.; Chiang, Y.W.; Goldman, A.; Sun, Y.-J. Roles of the hydrophobic gate and exit channel in Vigna radiata pyrophosphatase ion translocation. J. Mol. Biol. 2019, 431, 1619–1632. [Google Scholar] [CrossRef]
- Anashkin, V.A.; Baykov, A.A. A lumenal loop associated with catalytic asymmetry in plant vacuolar H+-translocating pyrophosphatase. Int. J. Mol. Sci. 2021, 22, 12902. [Google Scholar] [CrossRef]
- Holmes, A.O.M.; Kalli, A.C.; Goldman, A. The function of membrane integral pyrophosphatases from whole organism to single molecule. Front. Mol. Biosci. 2019, 6, 132. [Google Scholar] [CrossRef]
- Baykov, A.A.; Anashkin, V.A.; Malinen, A.M. Good-practice non-radioactive assays of inorganic pyrophosphatase activities. Molecules 2021, 26, 2356. [Google Scholar] [CrossRef] [PubMed]
- Bisswanger, H. Enzyme Kinetics. Principles and Methods, 2nd ed.; Wiley-VCH Verlag: Weinheim, Germany, 2008; pp. 81–85. [Google Scholar]
- Calisto, F.; Sousa, F.M.; Sena, F.V.; Refojo, P.N.; Pereira, M.M. Mechanisms of energy transduction by charge translocating membrane proteins. Chem. Rev. 2021, 121, 1804–1844. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. A chemiosmotic molecular mechanism for proton translocating adenosine triphosphatase. FEBS Lett. 1974, 43, 189–194. [Google Scholar] [CrossRef]
- Walker, J.E. ATP synthesis by rotary catalysis (Nobel Lecture) Angew. Chem. Int. Ed. 1998, 37, 2309–2319. [Google Scholar] [CrossRef]
- Boyer, P.D. Energy, life, and ATP (Nobel Lecture). Angew. Chem. Int. Ed. 1998, 37, 2297–2307. [Google Scholar] [CrossRef]
- Johannes, E.; Felle, H. Proton gradient across the tonoplast of Riccia fluitans as a result of the joint action of two electroenzymes. Plant Physiol. 1990, 93, 412–417. [Google Scholar] [CrossRef]
- Schmidt, A.L.; Briskin, D.P. Energy transduction in tonoplast vesciles from red beet (Beta vulgaris L.) storage tissue: H+/substrate stoichiometries for the H+-ATPase and H+-PPase. Arch. Biochem. Biophys. 1993, 301, 165–173. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Yabe, I.; Maeshima, M. Patch clamp analysis of a H+ pump heterologously expressed in giant yeast vacuoles. J. Biochem. 2003, 134, 615–623. [Google Scholar] [CrossRef]
- Nordbo, E.; Luoto, H.H.; Baykov, A.A.; Lahti, R.; Malinen, A.M. Two independent evolutionary routes to Na+/H+ cotransport function in membrane pyrophosphatases. Biochem. J. 2016, 473, 3099–3111. [Google Scholar] [CrossRef]
- Wang, Y.; Leigh, R.A.; Kaestner, K.H.; Sze, H. Electrogenic H+-pumping pyrophosphatase in tonoplast vesicles of oat roots. Plant Physiol. 1986, 81, 497–502. [Google Scholar] [CrossRef]
- White, P.J.; Marshall, J.; Smith, J.A. Substrate kinetics of the tonoplast H+-translocating inorganic pyrophosphatase and its activation by free Mg2+. Plant Physiol. 1990, 93, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Weeks, R.; Koren’kov, V.D.; Steele, S.H.; Leigh, R.A. The role of magnesium, pyrophosphate, and their complexes as substrates and activators of the vacuolar H+-pumping inorganic pyrophosphatase. Plant Physiol. 1997, 114, 901–905. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Scholz-Starke, J.; Primo, C.; Yang, J.; Kandel, R.; Gaxiola, R.A.; Hirschi, K.D. The flip side of the Arabidopsis type I proton-pumping pyrophosphatase (AVP1): Using a transmembrane H+ gradient to synthesize pyrophosphate. J. Biol. Chem. 2019, 294, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Primo, C.; Pizzio, G.A.; Yang, J.; Gaxiola, R.A.; Scholz-Starke, J.; Hirschi, K.D. Plant proton pumping pyrophosphatase: The potential for its pyrophosphate synthesis activity to modulate plant growth. Plant Biol. 2019, 21, 989–996. [Google Scholar] [CrossRef]
- Sondergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theor. Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Sosa, A.; Celis, H. H+/PPi stoichiometry of membrane-bound pyrophosphatase of Rhodospirillum rubrum. Arch. Biochem. Biophys. 1995, 316, 421–427. [Google Scholar] [CrossRef]
- Kashket, E.R. Stoichiometry of the H+-ATPase of growing and resting, aerobic Escherichia coli. Biochemistry. 1982, 21, 5534–5538. [Google Scholar] [CrossRef]
- Kashket, E.R. Stoichiometry of the H+-ATPase of Escherichia coli cells during anaerobic growth. FEBS Lett. 1983, 154, 343–346. [Google Scholar] [CrossRef]
- Gober, J.W.; Kashket, E.R. Measurement of the proton motive force in Rhizobium meliloti with the Escherichia coli lacY gene product. J. Bacteriol. 1985, 164, 929–931. [Google Scholar] [CrossRef]
- Heinonen, J.K. Biological Role of Inorganic Pyrophosphate; Kluwer Academic Publishers: London, UK, 2001; pp. 45–48. [Google Scholar]
- Veech, R.L.; Cook, G.A.; King, M.T. Relationship of free cytoplasmic pyrophosphate to liver glucose content and total pyrophosphate to cytoplasmic phosphorylation potential. FEBS Lett. 1980, 117 (Suppl. S1), K65–K72. [Google Scholar] [CrossRef]
- Flodgaard, H.; Fleron, P. Thermodynamic parameters for the hydrolysis of inorganic pyrophosphate at pH 7.4 as a function of [Mg2+], [K+], and ionic strength determined from equilibrium studies of the reaction. J. Biol. Chem. 1974, 249, 3465–3474. [Google Scholar] [CrossRef]
- Junge, W.; Nelson, N. ATP synthase. Annu. Rev. Biochem. 2015, 84, 631–657. [Google Scholar] [CrossRef] [PubMed]
- Breton, S.; Brown, D. Regulation of luminal acidification by the V-ATPase. Physiology 2013, 28, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Palmgren, M.G.; Nissen, P. P-type ATPases. Annu. Rev. Biophys. 2011, 40, 243–266. [Google Scholar] [CrossRef]
- Pedersen, B.P.; Buch-Pedersen, M.J.; Morth, J.P.; Palmgren, M.G.; Nissen, P. Crystal structure of the plasma membrane proton pump. Nature 2007, 450, 1111–1115. [Google Scholar] [CrossRef]
- Kozlova, M.I.; Bushmakin, I.M.; Belyaeva, J.D.; Shalaeva, D.N.; Dibrova, D.V.; Cherepanov, D.A.; Mulkidjanian, A.Y. Expansion of the “Sodium World” through evolutionary time and taxonomic space. Biochemistry 2020, 85, 1518–1542. [Google Scholar] [CrossRef]
- Schlegel, K.; Leone, V.; Faraldo-Gómez, J.D.; Müller, V. Promiscuous archaeal ATP synthase concurrently coupled to Na+ and H+ translocation. Proc. Natl Acad. Sci. USA 2012, 109, 947–952. [Google Scholar] [CrossRef]
- Brandt, K.; Müller, V. Hybrid rotors in F1Fo ATP synthases: Subunit composition, distribution, and physiological significance. J. Biol. Chem. 2015, 396, 1031–1042. [Google Scholar] [CrossRef]
- Leone, V.; Pogoryelov, D.; Meier, T.; Faraldo-Gómez, J.D. On the principle of ion selectivity in Na+/H+-coupled membrane proteins: Experimental and theoretical studies of an ATP synthase rotor. Proc. Natl Acad. Sci. USA 2015, 112, E1057–E1066. [Google Scholar] [CrossRef]
- Skopintsev, P.; Ehrenberg, D.; Weinert, T.; James, D.; Kar, R.K.; Johnson, P.; Ozerov, D.; Furrer, A.; Martiel, I.; Dworkowski, F.; et al. Femtosecond-to-millisecond structural changes in a light-driven sodium pump. Nature 2020, 583, 314–318. [Google Scholar] [CrossRef]
- Bogachev, A.V.; Bertsova, Y.V.; Verkhovskaya, M.L.; Mamedov, M.D.; Skulachev, V.P. Real-time kinetics of electrogenic Na+ transport by rhodopsin from the marine flavobacterium Dokdonia sp. PRO95. Sci Rep. 2016, 6, 21397. [Google Scholar] [CrossRef] [PubMed]
- Bogachev, A.V.; Baykov, A.A.; Bertsova, Y.V.; Mamedov, M.D. Mechanism of ion translocation by Na+-rhodopsin. Biochemistry 2022, 87, 731–741. [Google Scholar] [CrossRef]
- Zhorov, B.S. Possible mechanism of ion selectivity in eukaryotic voltage-gated sodium channels. J. Phys. Chem. B 2021, 125, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Baltscheffsky, H. Energy conversion leading to the origin and early evolution of life: Did inorganic pyrophosphate precede adenosine triphosphate? In Origin and Evolution of Biological Energy Conversion; Baltscheffsky, H., Ed.; VCH: New York, NY, USA, 1996; pp. 1–9. [Google Scholar]
- Luoto, H.H.; Nordbo, E.; Malinen, A.M.; Baykov, A.A.; Lahti, R. Evolutionarily divergent, Na+-regulated H+-transporting membrane-bound pyrophosphatases. Biochem. J. 2015, 467, 281–291. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baykov, A.A.; Anashkin, V.A.; Malinen, A.M.; Bogachev, A.V. The Mechanism of Energy Coupling in H+/Na+-Pumping Membrane Pyrophosphatase—Possibilities and Probabilities. Int. J. Mol. Sci. 2022, 23, 9504. https://doi.org/10.3390/ijms23169504
Baykov AA, Anashkin VA, Malinen AM, Bogachev AV. The Mechanism of Energy Coupling in H+/Na+-Pumping Membrane Pyrophosphatase—Possibilities and Probabilities. International Journal of Molecular Sciences. 2022; 23(16):9504. https://doi.org/10.3390/ijms23169504
Chicago/Turabian StyleBaykov, Alexander A., Viktor A. Anashkin, Anssi M. Malinen, and Alexander V. Bogachev. 2022. "The Mechanism of Energy Coupling in H+/Na+-Pumping Membrane Pyrophosphatase—Possibilities and Probabilities" International Journal of Molecular Sciences 23, no. 16: 9504. https://doi.org/10.3390/ijms23169504
APA StyleBaykov, A. A., Anashkin, V. A., Malinen, A. M., & Bogachev, A. V. (2022). The Mechanism of Energy Coupling in H+/Na+-Pumping Membrane Pyrophosphatase—Possibilities and Probabilities. International Journal of Molecular Sciences, 23(16), 9504. https://doi.org/10.3390/ijms23169504