
Evolution of Graves’ Disease during Immune Reconstitution following Nonmyeloablative Haploidentical Peripheral Blood Stem Cell Transplantation in a Boy Carrying Germline SAMD9L and FLT3 Variants

 and
and
Abstract
1. Introduction
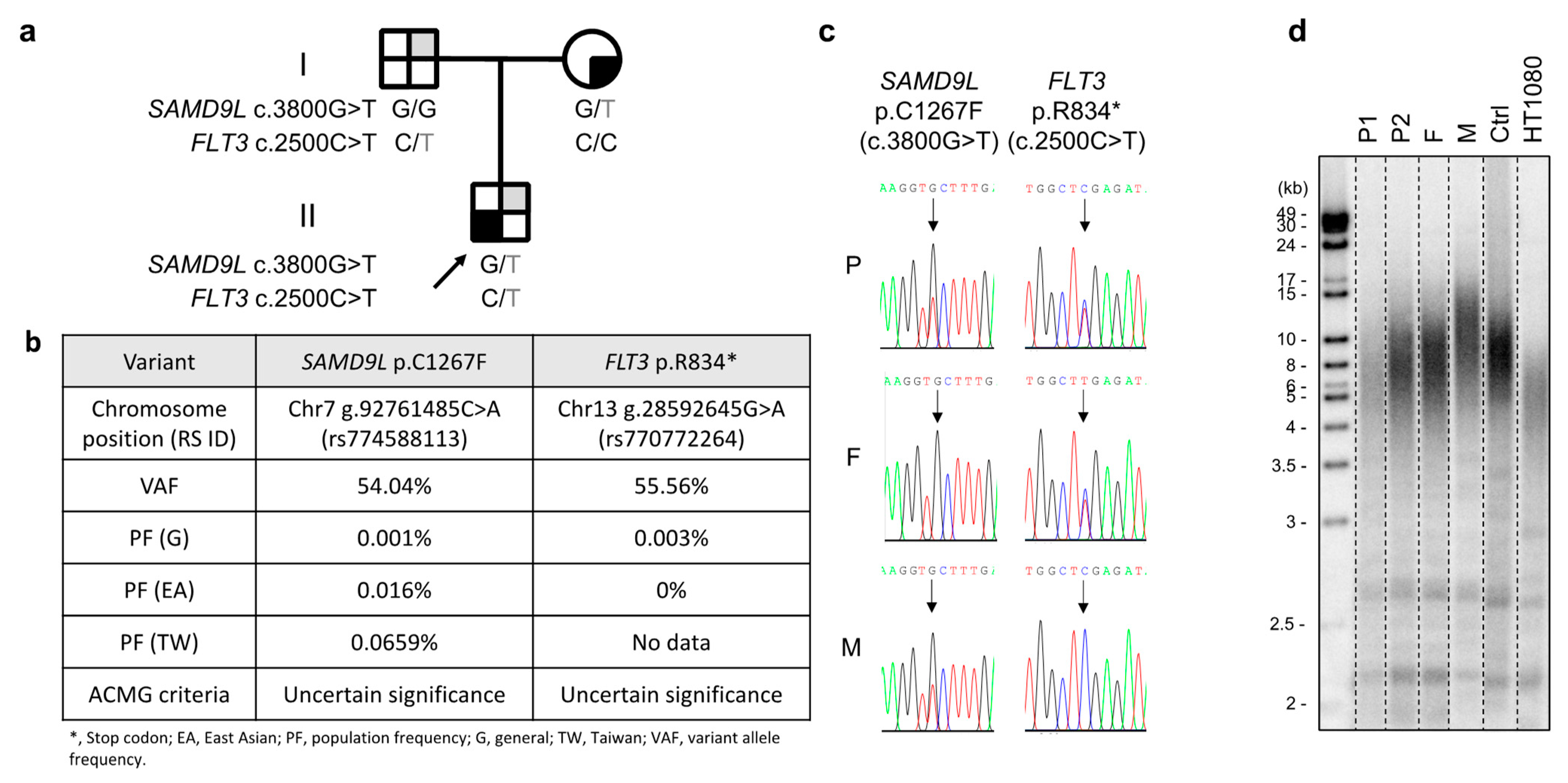
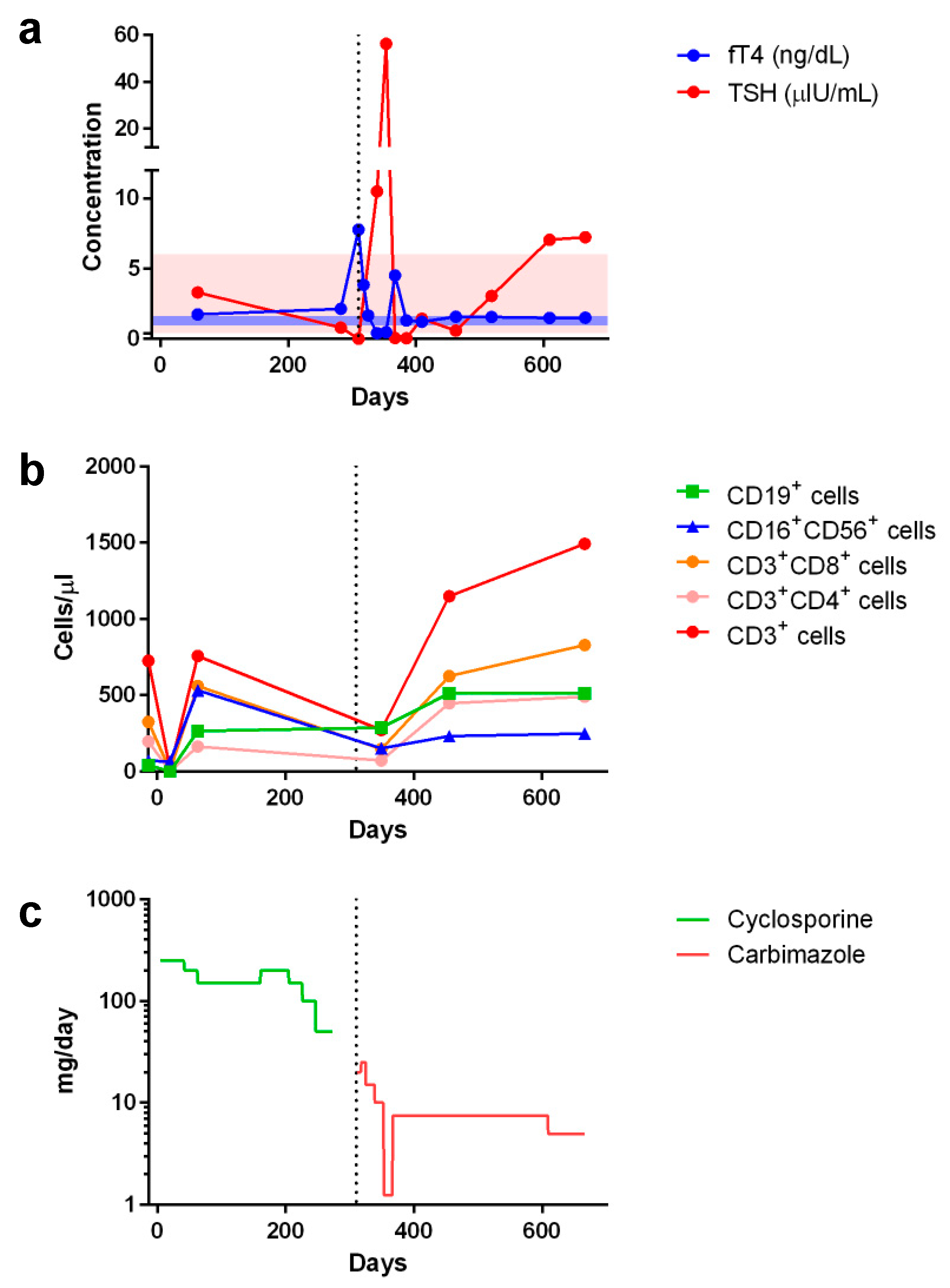
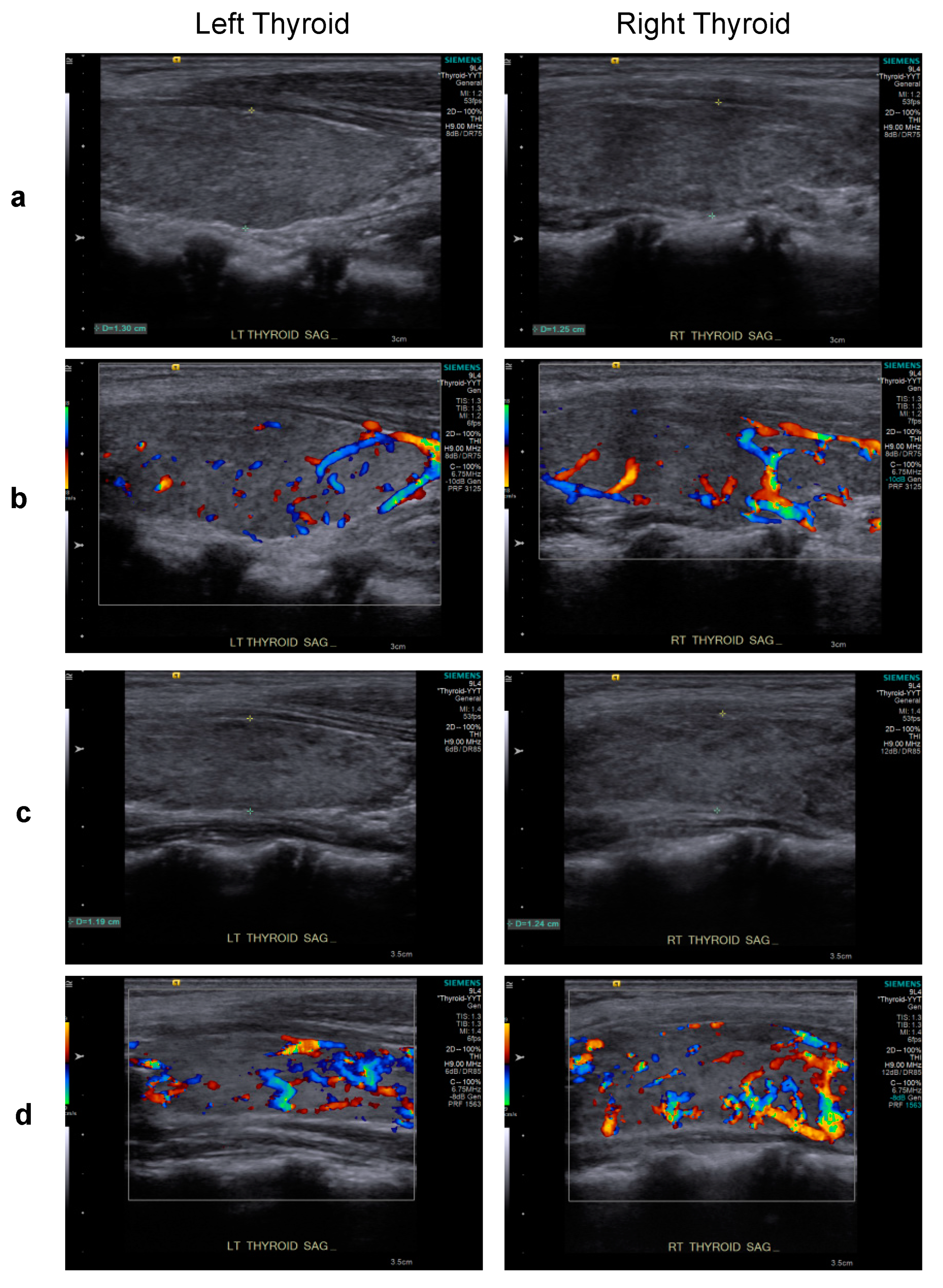
2. Case Report
3. Discussion
4. Materials and Methods
4.1. Identification of Variants by Whole-Exome Sequencing (WES)
4.2. Determination of HLA Haplotypes
4.3. Terminal Restriction Fragment Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- De Leo, S.; Lee, S.Y.; Braverman, L.E. Hyperthyroidism. Lancet 2016, 388, 906–918. [Google Scholar] [CrossRef]
- Smith, T.J.; Hegedus, L. Graves’ Disease. N. Engl. J. Med. 2016, 375, 1552–1565. [Google Scholar] [CrossRef]
- Boguslawska, J.; Godlewska, M.; Gajda, E.; Piekielko-Witkowska, A. Cellular and molecular basis of thyroid autoimmunity. Eur. Thyroid J. 2022, 11, e210024. [Google Scholar] [CrossRef]
- Tomer, Y. Mechanisms of autoimmune thyroid diseases: From genetics to epigenetics. Annu. Rev. Pathol. 2014, 9, 147–156. [Google Scholar] [CrossRef]
- Sarantopoulos, S.; Ritz, J. Aberrant B-cell homeostasis in chronic GVHD. Blood 2015, 125, 1703–1707. [Google Scholar] [CrossRef]
- Drabko, K.; Winnicka, D.; Gaworczyk, A.; Ben-Skowronek, I.; Skomra, D.; Kowalczyk, J.R. Donor origin of Graves disease in a BMT recipient: Evidence from FISH studies of thyroid tissue. Bone Marrow Transpl. 2006, 37, 789–791. [Google Scholar] [CrossRef][Green Version]
- Holland, F.J.; McConnon, J.K.; Volpe, R.; Saunders, E.F. Concordant Graves’ disease after bone marrow transplantation: Implications for pathogenesis. J. Clin. Endocrinol. Metab. 1991, 72, 837–840. [Google Scholar] [CrossRef]
- Ichihashi, T.; Yoshida, H.; Kiyoi, H.; Fukutani, H.; Kubo, K.; Yamauchi, T.; Naoe, T.; Ohno, R. Development of hyperthyroidism in donor and recipient after allogeneic bone marrow transplantation. Bone Marrow Transpl. 1992, 10, 397–398. [Google Scholar]
- Mulligan, S.P.; Joshua, D.E.; Joasoo, A.; Kronenberg, H. Autoimmune hyperthyroidism associated with chronic graft-versus-host disease. Transplantation 1987, 44, 463–464. [Google Scholar]
- Paketci, A.; Demir, K.; Tufekci, O.; Acar, S.; Abaci, A.; Yilmaz, S.; Bober, E. Graves’ disease following allogenic hematopoietic stem cell transplantation for severe aplastic anemia: Case report and literature review. J. Pediatr. Endocrinol. Metab. 2018, 31, 589–593. [Google Scholar] [CrossRef]
- Shimazaki, S.; Kazukawa, I.; Minagawa, M. Autoimmune thyroid disease following hematopoietic stem cell transplantation in childhood cancer survivors. Clin. Pediatr. Endocrinol. 2022, 31, 54–58. [Google Scholar] [CrossRef]
- Saevarsdottir, S.; Olafsdottir, T.A.; Ivarsdottir, E.V.; Halldorsson, G.H.; Gunnarsdottir, K.; Sigurdsson, A.; Johannesson, A.; Sigurdsson, J.K.; Juliusdottir, T.; Lund, S.H.; et al. FLT3 stop mutation increases FLT3 ligand level and risk of autoimmune thyroid disease. Nature 2020, 584, 619–623. [Google Scholar] [CrossRef]
- Chen, P.L.; Fann, C.S.; Chu, C.C.; Chang, C.C.; Chang, S.W.; Hsieh, H.Y.; Lin, M.; Yang, W.S.; Chang, T.C. Comprehensive genotyping in two homogeneous Graves’ disease samples reveals major and novel HLA association alleles. PLoS ONE 2011, 6, e16635. [Google Scholar] [CrossRef]
- Hawkins, B.R.; Ma, J.T.; Lam, K.S.; Wang, C.C.; Yeung, R.T. Association of HLA antigens with thyrotoxic Graves’ disease and periodic paralysis in Hong Kong Chinese. Clin. Endocrinol. 1985, 23, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Vita, R.; Lapa, D.; Trimarchi, F.; Vita, G.; Fallahi, P.; Antonelli, A.; Benvenga, S. Certain HLA alleles are associated with stress-triggered Graves’ disease and influence its course. Endocrine 2017, 55, 93–100. [Google Scholar] [CrossRef]
- Badenhoop, K.; Walfish, P.G.; Rau, H.; Fischer, S.; Nicolay, A.; Bogner, U.; Schleusener, H.; Usadel, K.H. Susceptibility and resistance alleles of human leukocyte antigen (HLA) DQA1 and HLA DQB1 are shared in endocrine autoimmune disease. J. Clin. Endocrinol. Metab. 1995, 80, 2112–2117. [Google Scholar] [CrossRef]
- Ban, Y.; Davies, T.F.; Greenberg, D.A.; Concepcion, E.S.; Osman, R.; Oashi, T.; Tomer, Y. Arginine at position 74 of the HLA-DR beta1 chain is associated with Graves’ disease. Genes Immun. 2004, 5, 203–208. [Google Scholar] [CrossRef]
- Chen, Q.Y.; Huang, W.; She, J.X.; Baxter, F.; Volpe, R.; Maclaren, N.K. HLA-DRB1*08, DRB1*03/DRB3*0101, and DRB3*0202 are susceptibility genes for Graves’ disease in North American Caucasians, whereas DRB1*07 is protective. J. Clin. Endocrinol. Metab. 1999, 84, 3182–3186. [Google Scholar] [CrossRef][Green Version]
- Simmonds, M.J.; Howson, J.M.; Heward, J.M.; Cordell, H.J.; Foxall, H.; Carr-Smith, J.; Gibson, S.M.; Walker, N.; Tomer, Y.; Franklyn, J.A.; et al. Regression mapping of association between the human leukocyte antigen region and Graves disease. Am. J. Hum. Genet. 2005, 76, 157–163. [Google Scholar] [CrossRef]
- Au, W.Y.; Lie, A.K.; Kung, A.W.; Liang, R.; Hawkins, B.R.; Kwong, Y.L. Autoimmune thyroid dysfunction after hematopoietic stem cell transplantation. Bone Marrow Transpl. 2005, 35, 383–388. [Google Scholar] [CrossRef]
- Mangklabruks, A.; Cox, N.; DeGroot, L.J. Genetic factors in autoimmune thyroid disease analyzed by restriction fragment length polymorphisms of candidate genes. J. Clin. Endocrinol. Metab. 1991, 73, 236–244. [Google Scholar] [CrossRef]
- Das, P.K.; Wherrett, D.; Dror, Y. Remission of aplastic anemia induced by treatment for Graves disease in a pediatric patient. Pediatr. Blood Cancer 2007, 49, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Moisidis, A.; Tsiakalos, A.; Alexandraki, K.; Syriou, V.; Kaltsas, G. Antithyroid drug-induced aplastic anemia. Thyroid 2008, 18, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.S.; Kim, H.K.; Han, D.K.; Baek, H.J.; Jang, H.I.; Kim, C.J.; Kook, H. Graves disease following rabbit antithymocyte globulin treatment of severe aplastic anemia in a Korean child. Korean J. Pediatr. 2015, 58, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Goldman, J. Severe aplastic anaemia and Grave’s disease in a paediatric patient. Br. J. Haematol. 2002, 118, 327–329. [Google Scholar] [CrossRef]
- Todd, A.; Todd, J. Graves’ disease following successful treatment of severe aplastic anaemia with antilymphocyte globulin. Clin. Lab. Haematol. 1999, 21, 69–70. [Google Scholar] [CrossRef]
- Zhang, W.; Shao, Z. Grave’s disease following aplastic anemia: Predisposition or coincidence? Indian Pediatr. 2015, 52, 347–348. [Google Scholar]
- Sarnat-Kucharczyk, M.; Swierkot, M.; Handzlik, G.; Kulawik, G.; Jagoda, K.; Grochola-Malecka, I.; Fryzewska, J.; Mrukwa-Kominek, E.; Chudek, J. Antithymocyte globulin as second-line therapy in Graves orbitopathy-preliminary results from a prospective single-center study. Front. Endocrinol. 2022, 13, 871009. [Google Scholar] [CrossRef]
- Sherer, Y.; Shoenfeld, Y. Autoimmune diseases and autoimmunity post-bone marrow transplantation. Bone Marrow Transpl. 1998, 22, 873–881. [Google Scholar] [CrossRef]
- Brasel, K.; Escobar, S.; Anderberg, R.; de Vries, P.; Gruss, H.J.; Lyman, S.D. Expression of the flt3 receptor and its ligand on hematopoietic cells. Leukemia 1995, 9, 1212–1218. [Google Scholar]
- Turner, A.M.; Lin, N.L.; Issarachai, S.; Lyman, S.D.; Broudy, V.C. FLT3 receptor expression on the surface of normal and malignant human hematopoietic cells. Blood 1996, 88, 3383–3390. [Google Scholar] [CrossRef] [PubMed]
- Abu-Duhier, F.M.; Goodeve, A.C.; Wilson, G.A.; Care, R.S.; Peake, I.R.; Reilly, J.T. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br. J. Haematol. 2001, 113, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kiyoi, H.; Nakano, Y.; Suzuki, R.; Kodera, Y.; Miyawaki, S.; Asou, N.; Kuriyama, K.; Yagasaki, F.; Shimazaki, C.; et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001, 97, 2434–2439. [Google Scholar] [CrossRef]
- Sahoo, S.S.; Pastor, V.B.; Goodings, C.; Voss, R.K.; Kozyra, E.J.; Szvetnik, A.; Noellke, P.; Dworzak, M.; Stary, J.; Locatelli, F.; et al. Clinical evolution, genetic landscape and trajectories of clonal hematopoiesis in SAMD9/SAMD9L syndromes. Nat. Med. 2021, 27, 1806–1817. [Google Scholar] [CrossRef]
- Thomas, M.E., 3rd; Abdelhamed, S.; Hiltenbrand, R.; Schwartz, J.R.; Sakurada, S.M.; Walsh, M.; Song, G.; Ma, J.; Pruett-Miller, S.M.; Klco, J.M. Pediatric MDS and bone marrow failure-associated germline mutations in SAMD9 and SAMD9L impair multiple pathways in primary hematopoietic cells. Leukemia 2021, 35, 3232–3244. [Google Scholar] [CrossRef] [PubMed]
- Tesi, B.; Davidsson, J.; Voss, M.; Rahikkala, E.; Holmes, T.D.; Chiang, S.C.C.; Komulainen-Ebrahim, J.; Gorcenco, S.; Nilsson, A.R.; Ripperger, T.; et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood 2017, 129, 2266–2279. [Google Scholar] [CrossRef]
- Ahmed, I.A.; Farooqi, M.S.; Vander Lugt, M.T.; Boklan, J.; Rose, M.; Friehling, E.D.; Triplett, B.; Lieuw, K.; Saldana, B.D.; Smith, C.M.; et al. Outcomes of hematopoietic cell transplantation in patients with germline SAMD9/SAMD9L mutations. Biol Blood Marrow Transpl. 2019, 25, 2186–2196. [Google Scholar] [CrossRef]
- Ka, S.; Lee, S.; Hong, J.; Cho, Y.; Sung, J.; Kim, H.N.; Kim, H.L.; Jung, J. HLAscan: Genotyping of the HLA region using next-generation sequencing data. BMC Bioinform. 2017, 18, 258. [Google Scholar] [CrossRef]
- Chen, L.Y.; Majerska, J.; Lingner, J. Molecular basis of telomere syndrome caused by CTC1 mutations. Genes Dev. 2013, 27, 2099–2108. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Susceptible HLA Haplotypes | Ethnicity [References] | Corresponding HLA Haplotypes of Recipient & Donor | Carrier Status of Susceptible HLA Haplotypes in Recipient and Donor |
|---|---|---|---|
| HLA-DQA1 Arg-52 | Caucasian [16] | HLA-DQA1 Phe-52 & HLA-DQA1 Phe-52 | No and No |
| HLA-DRB1 Arg-74 | Caucasian [17] | HLA-DRB1 Gly-74 & N.D.1 | No and N.D. 1 |
| DRB1*03:01-DQA1*05-DQB1*02 (DR3 haplotype) DRB1*04-DQA1*03-DQB1*03 (DR4 haplotype) | Caucasian [15,21] Caucasian [15] | DRB1*11:01:01/11:01:01-DQA1*05:09/05:09-DQB1*03:01:01:03/03:01:01:03 & DRB1*11:01:01/09:01:02-DQA1*05:05:01:02/03:02-DQB1*03:01:01:01/03:03:02:04 | No and No No and No |
| DPB1*05:01 | Chinese Han [13] | DPB1*02:02/02:01:02 & DPB1*02:02/02:01:02 | No and No |
| DQB1*05:02 DQB1*02 | Chinese Han [13] Caucasian [15] | DQB1*03:01:01:03/03:01:01:03 & DQB1*03:01:01:01/03:03:02:04 | No and No No and No |
| DRB1*15:01 DRB1*16:02 DRB1*09 | Chinese Han [13] Chinese Han [13] Chinese Han [20] | DRB1*11:01:01/11:01:01 & DRB1*11:01:01/09:01:02 | No and No No and No No and Yes |
| B*05 B*46:01 B*08 | Chinese Han [14] Chinese Han [13,14] Caucasian [15] | B*39:01:01:01/51:02:01 & B*46:01:01/51:02:01 | No and No No and Yes No and No |
| C*07 (Cw7) | Caucasian [15] | C*07:02:01:03/15:02:01:02 & C*01:02:30/15:02:01:03 | Yes and No |
| Age (Years) at HSCT/Sex | Disease at HSCT | HSCT Donor/Source | HSCT Regimens * | HSCT-GD Interval | GD Risk Factors | GD Outcome | Reference |
|---|---|---|---|---|---|---|---|
| 8.5/M | SAA refractory | Haplo/PBSC | Flu150/Cy29/TBI4—PTCy/CSA/MMF | 10 months | Described in text | Euthyroid over 1 year under carbimazole | Present case |
| 4/M | SAA at diagnosis | MFD/BMSC | Cy200/rATG—N.A. | 3 years | rATG, DR9 | Under methimazole | [11] |
| 8/F | SAA at diagnosis | MSD/BMSC | Cy200/rATG—CSA/MTX | 3 years | rATG | Euthyroid over 1.5 years after methimazole/thyroidectomy and under L-thyroxine | [10] |
| 14/M | SAA at diagnosis | MSD/BMSC | Cy200/rATG—CSA/MTX | 2 years | rATG, donor GD, HLA-DRB1*0301 (DR3) | Euthyroid over 2 years after lithium carbonicum/thiamazole/thyroidectomy | [6] |
| 17/F | SAA at diagnosis | MSD/BMSC | Cy200/TLI7—CSA | 30 months | Donor GD, HLA-Bw46 | Clinical improvement but persistent T3 elevation over one year after thiamazole | [8] |
| 10/M | SAA at diagnosis | MSD/BMSC | Cy—MTX | 8 years | Donor GD | N.A. | [7] |
| 50/M | SAA at diagnosis | MSD/BMSC | Cy180—MTX | 36 months | GVHD | Euthyroid after propranolol/I131 15 mCi and under thyroxine | [9] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ip, P.P.; Fang, L.-H.; Shen, Y.-L.; Tung, K.-C.; Lai, M.-T.; Juan, L.-Y.; Chen, L.-Y.; Chen, R.-L. Evolution of Graves’ Disease during Immune Reconstitution following Nonmyeloablative Haploidentical Peripheral Blood Stem Cell Transplantation in a Boy Carrying Germline SAMD9L and FLT3 Variants. Int. J. Mol. Sci. 2022, 23, 9494. https://doi.org/10.3390/ijms23169494
Ip PP, Fang L-H, Shen Y-L, Tung K-C, Lai M-T, Juan L-Y, Chen L-Y, Chen R-L. Evolution of Graves’ Disease during Immune Reconstitution following Nonmyeloablative Haploidentical Peripheral Blood Stem Cell Transplantation in a Boy Carrying Germline SAMD9L and FLT3 Variants. International Journal of Molecular Sciences. 2022; 23(16):9494. https://doi.org/10.3390/ijms23169494
Chicago/Turabian StyleIp, Peng Peng, Li-Hua Fang, Yi-Ling Shen, Kuan-Chiun Tung, Ming-Tsong Lai, Li-Ying Juan, Liuh-Yow Chen, and Rong-Long Chen. 2022. "Evolution of Graves’ Disease during Immune Reconstitution following Nonmyeloablative Haploidentical Peripheral Blood Stem Cell Transplantation in a Boy Carrying Germline SAMD9L and FLT3 Variants" International Journal of Molecular Sciences 23, no. 16: 9494. https://doi.org/10.3390/ijms23169494
APA StyleIp, P. P., Fang, L.-H., Shen, Y.-L., Tung, K.-C., Lai, M.-T., Juan, L.-Y., Chen, L.-Y., & Chen, R.-L. (2022). Evolution of Graves’ Disease during Immune Reconstitution following Nonmyeloablative Haploidentical Peripheral Blood Stem Cell Transplantation in a Boy Carrying Germline SAMD9L and FLT3 Variants. International Journal of Molecular Sciences, 23(16), 9494. https://doi.org/10.3390/ijms23169494