
Implications of Polyploidy and Ploidy Alterations in Hepatocytes in Liver Injuries and Cancers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Physiological Polyploidization of Hepatocytes
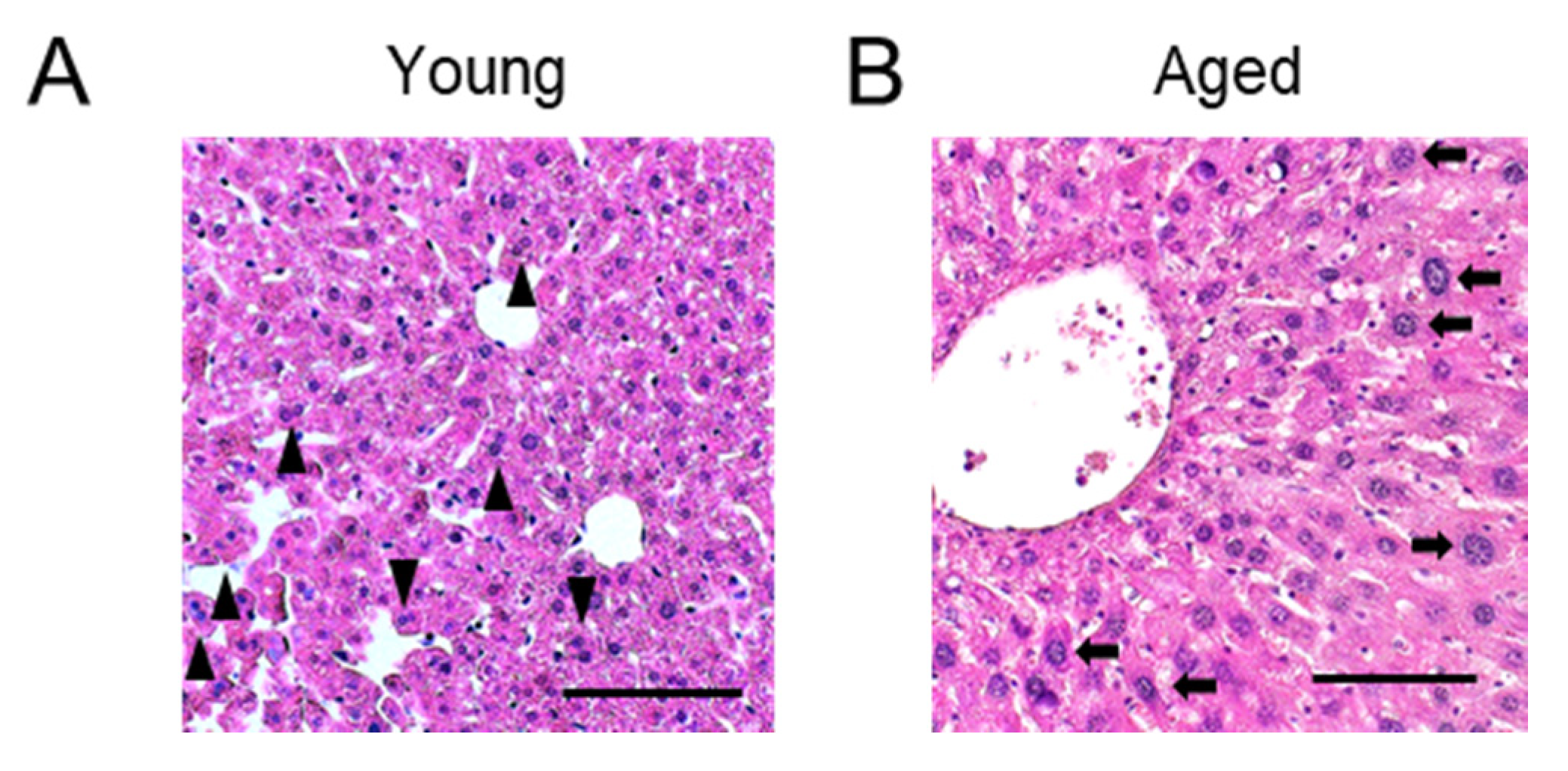
2.1. Overview of Physiological Polyploidization of Hepatocytes
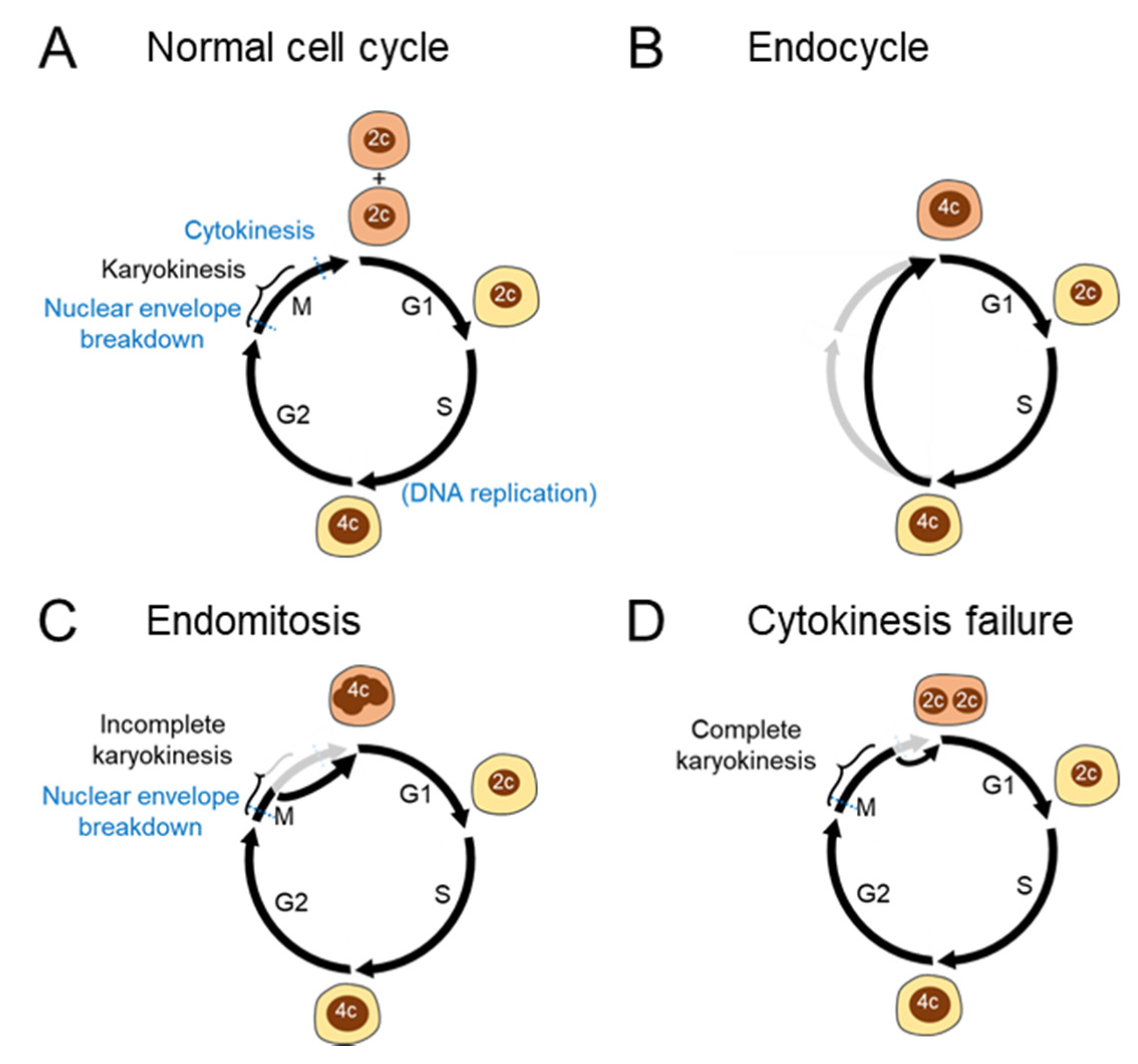
2.2. Mechanisms of Polyploidization
2.3. Molecular Mechanisms Underlying Physiological Polyploidization in Hepatocytes
2.4. Significance of Physiological Polyploidization
3. Polyploid Hepatocytes in Acute and Chronic Liver Damage
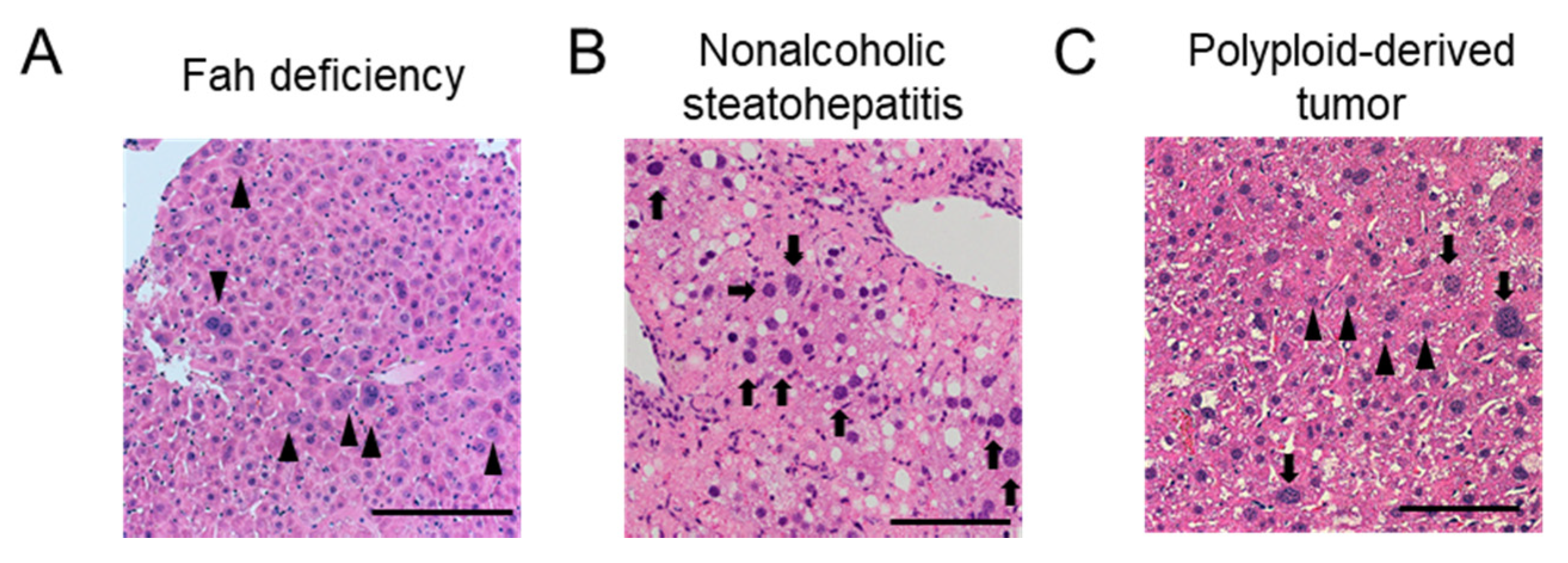
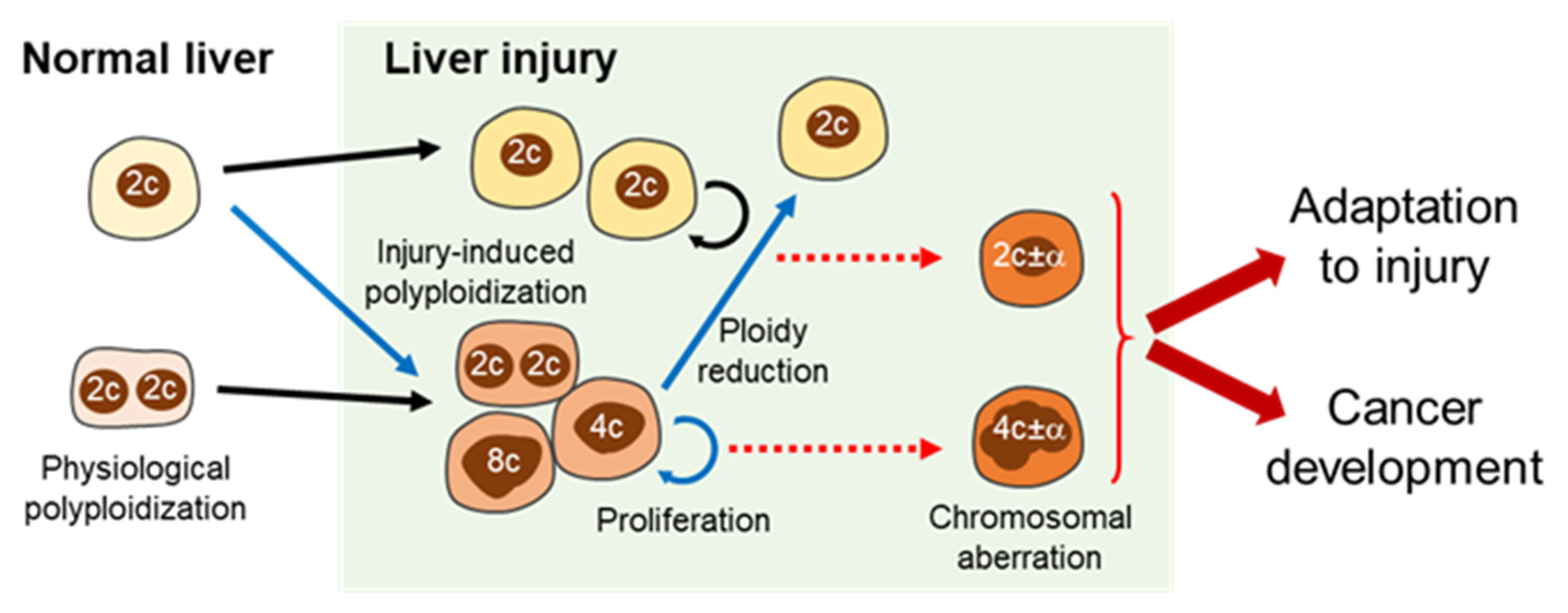
3.1. Ploidy Alterations in Liver Damage
3.2. Liver Regeneration by Polyploid Hepatocytes
3.3. Significance of Ploidy Alterations in Liver Injuries
4. Polyploidy and Ploidy Alterations in the Development of Liver Cancer
4.1. Correlation between Polyploidization and Carcinogenesis
4.2. Impacts of Hepatocyte Ploidy on Liver Cancer Development in Rodent Studies
4.3. Consideration of Carcinogenic Risks Related to Hepatocyte Ploidy
4.4. Polyploidy in Hepatocellular Carcinoma
5. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Carriere, R. The growth of liver parenchymal nuclei and its endocrine regulation. Int. Rev. Cytol. 1969, 25, 201–277. [Google Scholar] [PubMed]
- St Aubin, P.M.; Bucher, N.L. A study of binucleate cell counts in resting and regenerating rat liver employing a mechanical method for the separation of liver cells. Anat. Rec. 1952, 112, 797–809. [Google Scholar] [CrossRef]
- Gjelsvik, K.J.; Besen-McNally, R.; Losick, V.P. Solving the Polyploid Mystery in Health and Disease. Trends Genet. 2019, 35, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, E.; Angelotti, M.L.; Conte, C.; Anders, H.J.; Romagnani, P. Surviving Acute Organ Failure: Cell Polyploidization and Progenitor Proliferation. Trends Mol. Med. 2019, 25, 366–381. [Google Scholar] [CrossRef] [Green Version]
- Donne, R.; Sangouard, F.; Celton-Morizur, S.; Desdouets, C. Hepatocyte Polyploidy: Driver or Gatekeeper of Chronic Liver Diseases. Cancers 2021, 13, 5151. [Google Scholar] [CrossRef]
- Wilkinson, P.D.; Duncan, A.W. Differential Roles for Diploid and Polyploid Hepatocytes in Acute and Chronic Liver Injury. Semin. Liver Dis. 2021, 41, 42–49. [Google Scholar] [CrossRef]
- Sladky, V.C.; Eichin, F.; Reiberger, T.; Villunger, A. Polyploidy control in hepatic health and disease. J. Hepatol. 2021, 75, 1177–1191. [Google Scholar] [CrossRef]
- Wheatley, D.N. Binucleation in mammalian liver. Studies on the control of cytokinesis in vivo. Exp. Cell Res. 1972, 74, 455–465. [Google Scholar] [CrossRef]
- Guidotti, J.E.; Brégerie, O.; Robert, A.; Debey, P.; Brechot, C.; Desdouets, C. Liver cell polyploidization: A pivotal role for binuclear hepatocytes. J. Biol. Chem. 2003, 278, 19095–19101. [Google Scholar] [CrossRef] [Green Version]
- Celton-Morizur, S.; Merlen, G.; Couton, D.; Margall-Ducos, G.; Desdouets, C. The insulin/Akt pathway controls a specific cell division program that leads to generation of binucleated tetraploid liver cells in rodents. J. Clin. Investig. 2009, 119, 1880–1887. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.H.; Delgado, E.R.; Otero, P.A.; Teng, K.Y.; Kutay, H.; Meehan, K.M.; Moroney, J.B.; Monga, J.K.; Hand, N.J.; Friedman, J.R.; et al. MicroRNA-122 regulates polyploidization in the murine liver. Hepatology 2016, 64, 599–615. [Google Scholar] [CrossRef] [Green Version]
- Severin, E.; Willers, R.; Bettecken, T. Flow cytometric analysis of mouse hepatocyte ploidy. II. The development of polyploidy pattern in four mice strains with different life spans. Cell Tissue Res. 1984, 238, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtsev, B.N.; Kudryavtseva, M.V.; Sakuta, G.A.; Stein, G.I. Human hepatocyte polyploidization kinetics in the course of life cycle. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1993, 64, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Hanlon Newell, A.E.; Smith, L.; Wilson, E.M.; Olson, S.B.; Thayer, M.J.; Strom, S.C.; Grompe, M. Frequent aneuploidy among normal human hepatocytes. Gastroenterology 2012, 142, 25–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoda, H.; Bregerie, O.; Vallet, A.; Nalpas, B.; Pivert, G.; Brechot, C.; Desdouets, C. Changes to hepatocyte ploidy and binuclearity profiles during human chronic viral hepatitis. Gut 2005, 54, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Wakefield, L.; Grompe, M. The Significance of Polyploid Hepatocytes During Aging Process. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1347–1349. [Google Scholar] [CrossRef]
- Abmayr, S.M.; Pavlath, G.K. Myoblast fusion: Lessons from flies and mice. Development 2012, 139, 641–656. [Google Scholar] [CrossRef] [Green Version]
- Hernández, J.M.; Podbilewicz, B. The hallmarks of cell-cell fusion. Development 2017, 144, 4481–4495. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.O.; Davidson, J.M.; Duronio, R.J. Endoreplication: Polyploidy with purpose. Genes. Dev. 2009, 23, 2461–2477. [Google Scholar] [CrossRef] [Green Version]
- Øvrebø, J.I.; Edgar, B.A. Polyploidy in tissue homeostasis and regeneration. Development 2018, 145, dev156034. [Google Scholar] [CrossRef] [Green Version]
- Lazzeri, E.; Angelotti, M.L.; Peired, A.; Conte, C.; Marschner, J.A.; Maggi, L.; Mazzinghi, B.; Lombardi, D.; Melica, M.E.; Nardi, S.; et al. Endocycle-related tubular cell hypertrophy and progenitor proliferation recover renal function after acute kidney injury. Nat. Commun. 2018, 9, 1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravid, K.; Lu, J.; Zimmet, J.M.; Jones, M.R. Roads to polyploidy: The megakaryocyte example. J. Cell Physiol. 2002, 190, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, B.; Maddox, A.S. Cytokinesis, ploidy and aneuploidy. J. Pathol. 2012, 226, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Fantone, S.; Tossetta, G.; Graciotti, L.; Galosi, A.B.; Skrami, E.; Marzioni, D.; Morroni, M. Identification of multinucleated cells in human kidney cortex: A way for tissue repairing? J. Anat. 2022, 240, 985–990. [Google Scholar] [CrossRef]
- Margall-Ducos, G.; Celton-Morizur, S.; Couton, D.; Brégerie, O.; Desdouets, C. Liver tetraploidization is controlled by a new process of incomplete cytokinesis. J. Cell Sci. 2007, 120, 3633–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandit, S.K.; Westendorp, B.; Nantasanti, S.; van Liere, E.; Tooten, P.C.; Cornelissen, P.W.; Toussaint, M.J.; Lamers, W.H.; de Bruin, A. E2F8 is essential for polyploidization in mammalian cells. Nat. Cell Biol. 2012, 14, 1181–1191. [Google Scholar] [CrossRef]
- Chao, H.W.; Doi, M.; Fustin, J.M.; Chen, H.; Murase, K.; Maeda, Y.; Hayashi, H.; Tanaka, R.; Sugawa, M.; Mizukuchi, N.; et al. Circadian clock regulates hepatic polyploidy by modulating Mkp1-Erk1/2 signaling pathway. Nat. Commun. 2017, 8, 2238. [Google Scholar] [CrossRef]
- Mayhew, C.N.; Bosco, E.E.; Fox, S.R.; Okaya, T.; Tarapore, P.; Schwemberger, S.J.; Babcock, G.F.; Lentsch, A.B.; Fukasawa, K.; Knudsen, E.S. Liver-specific pRB loss results in ectopic cell cycle entry and aberrant ploidy. Cancer Res. 2005, 65, 4568–4577. [Google Scholar] [CrossRef] [Green Version]
- Sheahan, S.; Bellamy, C.O.; Treanor, L.; Harrison, D.J.; Prost, S. Additive effect of p53, p21 and Rb deletion in triple knockout primary hepatocytes. Oncogene 2004, 23, 1489–1497. [Google Scholar] [CrossRef] [Green Version]
- Pandit, S.K.; Westendorp, B.; de Bruin, A. Physiological significance of polyploidization in mammalian cells. Trends Cell Biol. 2013, 23, 556–566. [Google Scholar] [CrossRef]
- Lu, P.; Prost, S.; Caldwell, H.; Tugwood, J.D.; Betton, G.R.; Harrison, D.J. Microarray analysis of gene expression of mouse hepatocytes of different ploidy. Mamm. Genome. 2007, 18, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Zhang, S.; Zhu, M.; Lu, T.; Chen, K.; Wen, Z.; Wang, S.; Xiao, G.; Luo, D.; Jia, Y.; et al. Mice With Increased Numbers of Polyploid Hepatocytes Maintain Regenerative Capacity But Develop Fewer Hepatocellular Carcinomas Following Chronic Liver Injury. Gastroenterology 2020, 158, 1698–1712.e14. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E. Somatic polyploidy promotes cell function under stress and energy depletion: Evidence from tissue-specific mammal transcriptome. Funct. Integr. Genom. 2010, 10, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Kreutz, C.; MacNelly, S.; Follo, M.; Wäldin, A.; Binninger-Lacour, P.; Timmer, J.; Bartolomé-Rodríguez, M.M. Hepatocyte Ploidy Is a Diversity Factor for Liver Homeostasis. Front. Physiol. 2017, 8, 862. [Google Scholar] [CrossRef] [Green Version]
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. Int. J. Mol. Sci. 2022, 23, 3542. [Google Scholar] [CrossRef]
- Richter, M.L.; Deligiannis, I.K.; Yin, K.; Danese, A.; Lleshi, E.; Coupland, P.; Vallejos, C.A.; Matchett, K.P.; Henderson, N.C.; Colome-Tatche, M.; et al. Single-nucleus RNA-seq2 reveals functional crosstalk between liver zonation and ploidy. Nat. Commun. 2021, 12, 4264. [Google Scholar] [CrossRef]
- Matsumoto, T.; Wakefield, L.; Tarlow, B.D.; Grompe, M. In Vivo Lineage Tracing of Polyploid Hepatocytes Reveals Extensive Proliferation during Liver Regeneration. Cell Stem. Cell 2020, 26, 34–47.e3. [Google Scholar] [CrossRef]
- Tanami, S.; Ben-Moshe, S.; Elkayam, A.; Mayo, A.; Bahar Halpern, K.; Itzkovitz, S. Dynamic zonation of liver polyploidy. Cell Tissue Res. 2017, 368, 405–410. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, K.; Luo, X.; Li, L.; Tu, H.C.; Sehgal, A.; Nguyen, L.H.; Zhang, Y.; Gopal, P.; Tarlow, B.D.; et al. The Polyploid State Plays a Tumor-Suppressive Role in the Liver. Dev. Cell 2018, 44, 447–459.e5. [Google Scholar] [CrossRef] [Green Version]
- Bahar Halpern, K.; Tanami, S.; Landen, S.; Chapal, M.; Szlak, L.; Hutzler, A.; Nizhberg, A.; Itzkovitz, S. Bursty gene expression in the intact mammalian liver. Mol. Cell 2015, 58, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Gentric, G.; Maillet, V.; Paradis, V.; Couton, D.; L’Hermitte, A.; Panasyuk, G.; Fromenty, B.; Celton-Morizur, S.; Desdouets, C. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Madra, S.; Styles, J.; Smith, A.G. Perturbation of hepatocyte nuclear populations induced by iron and polychlorinated biphenyls in C57BL/10ScSn mice during carcinogenesis. Carcinogenesis 1995, 16, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, Y.; Yamada, T.; Moralejo, D.H.; Mochizuki, H.; Sogawa, K.; Matsumoto, K. Increased polyploid incidence is associated with abnormal copper accumulation in the liver of LEC mutant rat. Res. Commun. Mol. Pathol. Pharmacol. 2000, 107, 129–136. [Google Scholar] [PubMed]
- Wilkinson, P.D.; Alencastro, F.; Delgado, E.R.; Leek, M.P.; Weirich, M.P.; Otero, P.A.; Roy, N.; Brown, W.K.; Oertel, M.; Duncan, A.W. Polyploid Hepatocytes Facilitate Adaptation and Regeneration to Chronic Liver Injury. Am. J. Pathol. 2019, 189, 1241–1255. [Google Scholar] [CrossRef] [PubMed]
- Sigal, S.H.; Rajvanshi, P.; Gorla, G.R.; Sokhi, R.P.; Saxena, R.; Gebhard, D.R., Jr.; Reid, L.M.; Gupta, S. Partial hepatectomy-induced polyploidy attenuates hepatocyte replication and activates cell aging events. Am. J. Physiol. 1999, 276, G1260–G1272. [Google Scholar] [CrossRef]
- Matsumoto, T.; Wakefield, L.; Peters, A.; Peto, M.; Spellman, P.; Grompe, M. Proliferative polyploid cells give rise to tumors via ploidy reduction. Nat. Commun. 2021, 12, 646. [Google Scholar] [CrossRef]
- Johmura, Y.; Shimada, M.; Misaki, T.; Naiki-Ito, A.; Miyoshi, H.; Motoyama, N.; Ohtani, N.; Hara, E.; Nakamura, M.; Morita, A.; et al. Necessary and sufficient role for a mitosis skip in senescence induction. Mol. Cell 2014, 55, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Aravinthan, A.D.; Alexander, G.J.M. Senescence in chronic liver disease: Is the future in aging? J. Hepatol. 2016, 65, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Sinha, D.; Duijf, P.H.G.; Khanna, K.K. Mitotic slippage: An old tale with a new twist. Cell Cycle. 2019, 18, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Park, S.Y.; Yong, H.; Famulski, J.K.; Chae, S.; Lee, J.H.; Kang, C.M.; Saya, H.; Chan, G.K.; Cho, H. HBV X protein targets hBubR1, which induces dysregulation of the mitotic checkpoint. Oncogene 2008, 27, 3457–3464. [Google Scholar] [CrossRef] [Green Version]
- Ahodantin, J.; Bou-Nader, M.; Cordier, C.; Mégret, J.; Soussan, P.; Desdouets, C.; Kremsdorf, D. Hepatitis B virus X protein promotes DNA damage propagation through disruption of liver polyploidization and enhances hepatocellular carcinoma initiation. Oncogene 2019, 38, 2645–2657. [Google Scholar] [CrossRef]
- Wang, X.; Willenbring, H.; Akkari, Y.; Torimaru, Y.; Foster, M.; Al-Dhalimy, M.; Lagasse, E.; Finegold, M.; Olson, S.; Grompe, M. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 2003, 422, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Willenbring, H.; Bailey, A.S.; Foster, M.; Akkari, Y.; Dorrell, C.; Olson, S.; Finegold, M.; Fleming, W.H.; Grompe, M. Myelomonocytic cells are sufficient for therapeutic cell fusion in liver. Nat. Med. 2004, 10, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Hickey, R.D.; Paulk, N.K.; Culberson, A.J.; Olson, S.B.; Finegold, M.J.; Grompe, M. Ploidy reductions in murine fusion-derived hepatocytes. PLoS Genet. 2009, 5, e1000385. [Google Scholar] [CrossRef] [Green Version]
- Duncan, A.W.; Taylor, M.H.; Hickey, R.D.; Hanlon Newell, A.E.; Lenzi, M.L.; Olson, S.B.; Finegold, M.J.; Grompe, M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 2010, 467, 707–710. [Google Scholar] [CrossRef] [Green Version]
- Kirillova, A.; Han, L.; Liu, H.; Kühn, B. Polyploid cardiomyocytes: Implications for heart regeneration. Development 2021, 148, dev199401. [Google Scholar] [CrossRef]
- Derks, W.; Bergmann, O. Polyploidy in Cardiomyocytes: Roadblock to Heart Regeneration? Circ. Res. 2020, 126, 552–565. [Google Scholar] [CrossRef]
- Andreassen, P.R.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol. Biol. Cell 2001, 12, 1315–1328. [Google Scholar] [CrossRef] [Green Version]
- Margolis, R.L.; Lohez, O.D.; Andreassen, P.R. G1 tetraploidy checkpoint and the suppression of tumorigenesis. J. Cell Biochem. 2003, 88, 673–683. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Ebato, K.; Kato, H.; Arakawa, S.; Shimizu, S.; Miyajima, A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr. Biol. 2012, 22, 1166–1175. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, P.D.; Delgado, E.R.; Alencastro, F.; Leek, M.P.; Roy, N.; Weirich, M.P.; Stahl, E.C.; Otero, P.A.; Chen, M.I.; Brown, W.K.; et al. The Polyploid State Restricts Hepatocyte Proliferation and Liver Regeneration in Mice. Hepatology 2019, 69, 1242–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, A.W.; Hanlon Newell, A.E.; Bi, W.; Finegold, M.J.; Olson, S.B.; Beaudet, A.L.; Grompe, M. Aneuploidy as a mechanism for stress-induced liver adaptation. J. Clin. Investig. 2012, 122, 3307–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, K.; Al-Dhalimy, M.; Finegold, M.; Grompe, M. In vivo suppressor mutations correct a murine model of hereditary tyrosinemia type I. Proc. Natl. Acad. Sci. USA 1999, 96, 11928–11933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knouse, K.A.; Wu, J.; Whittaker, C.A.; Amon, A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 13409–13414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sladky, V.C.; Knapp, K.; Soratroi, C.; Heppke, J.; Eichin, F.; Rocamora-Reverte, L.; Szabo, T.G.; Bongiovanni, L.; Westendorp, B.; Moreno, E.; et al. E2F-Family Members Engage the PIDDosome to Limit Hepatocyte Ploidy in Liver Development and Regeneration. Dev. Cell 2020, 52, 335–349.e7. [Google Scholar] [CrossRef]
- Rancati, G.; Pavelka, N.; Fleharty, B.; Noll, A.; Trimble, R.; Walton, K.; Perera, A.; Staehling-Hampton, K.; Seidel, C.W.; Li, R. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 2008, 135, 879–893. [Google Scholar] [CrossRef] [Green Version]
- Sabat-Pośpiech, D.; Fabian-Kolpanowicz, K.; Prior, I.A.; Coulson, J.M.; Fielding, A.B. Targeting centrosome amplification, an Achilles’ heel of cancer. Biochem. Soc. Trans. 2019, 47, 1209–1222. [Google Scholar] [CrossRef]
- Godinho, S.A.; Pellman, D. Causes and consequences of centrosome abnormalities in cancer. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130467. [Google Scholar] [CrossRef] [Green Version]
- Boveri, T. Zur Frage der Entstehung Maligner Tumoren; Gustav Fischer Verlag: Frankfurt am Main, Germany, 1914. [Google Scholar]
- Ben-David, U.; Amon, A. Context is everything: Aneuploidy in cancer. Nat. Rev. Genet. 2020, 21, 44–62. [Google Scholar] [CrossRef]
- Levine, M.S.; Bakker, B.; Boeckx, B.; Moyett, J.; Lu, J.; Vitre, B.; Spierings, D.C.; Lansdorp, P.M.; Cleveland, D.W.; Lambrechts, D.; et al. Centrosome Amplification Is Sufficient to Promote Spontaneous Tumorigenesis in Mammals. Dev. Cell 2017, 40, 313–322.e5. [Google Scholar] [CrossRef] [Green Version]
- Lens, S.M.A.; Medema, R.H. Cytokinesis defects and cancer. Nat. Rev. Cancer 2019, 19, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, P.C.; Cowan, D.S.; Sanchez, C.A.; Barrett, M.T.; Emond, M.J.; Levine, D.S.; Rabinovitch, P.S.; Reid, B.J. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett’s esophagus. Proc. Natl. Acad. Sci. USA 1996, 93, 7081–7084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2,658 cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Nguyen, L.H.; Zhou, K.; Tu, H.C.; Sehgal, A.; Nassour, I.; Li, L.; Gopal, P.; Goodman, J.; Singal, A.G.; et al. Knockdown of Anillin Actin Binding Protein Blocks Cytokinesis in Hepatocytes and Reduces Liver Tumor Development in Mice Without Affecting Regeneration. Gastroenterology 2018, 154, 1421–1434. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Huang, Y.S.; Fustin, J.M.; Doi, M.; Chen, H.; Lai, H.H.; Lin, S.H.; Lee, Y.L.; King, P.C.; Hou, H.S.; et al. Hyperpolyploidization of hepatocyte initiates preneoplastic lesion formation in the liver. Nat. Commun. 2021, 12, 645. [Google Scholar] [CrossRef]
- Sansregret, L.; Vanhaesebroeck, B.; Swanton, C. Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Targa, A.; Rancati, G. Cancer: A CINful evolution. Curr. Opin. Cell Biol. 2018, 52, 136–144. [Google Scholar] [CrossRef]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Quinton, R.J.; DiDomizio, A.; Vittoria, M.A.; Kotýnková, K.; Ticas, C.J.; Patel, S.; Koga, Y.; Vakhshoorzadeh, J.; Hermance, N.; Kuroda, T.S.; et al. Whole-genome doubling confers unique genetic vulnerabilities on tumour cells. Nature 2021, 590, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsova, A.Y.; Seget, K.; Moeller, G.K.; de Pagter, M.S.; de Roos, J.A.; Dürrbaum, M.; Kuffer, C.; Müller, S.; Zaman, G.J.; Kloosterman, W.P.; et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 2015, 14, 2810–2820. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Sharir, Y.; McFarland, J.M.; Abdusamad, M.; Marquis, C.; Bernhard, S.V.; Kazachkova, M.; Tang, H.; Ippolito, M.R.; Laue, K.; Zerbib, J.; et al. Aneuploidy renders cancer cells vulnerable to mitotic checkpoint inhibition. Nature 2021, 590, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Schubert, M.; Tijhuis, A.E.; Requesens, M.; Roorda, M.; van den Brink, A.; Ruiz, L.A.; Bakker, P.L.; van der Sluis, T.; Pieters, W.; et al. cGAS-STING drives the IL-6-dependent survival of chromosomally instable cancers. Nature 2022, 607, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Balsells, J.; Caragol, I.; Allende, E.; Diaz, I.; Charco, R.; Lazaro, J.L.; Murio, E.; Margarit, C. DNA ploidy study of resected hepatocellular carcinoma in cirrhotic liver. J. Hepatol. 1996, 25, 854–858. [Google Scholar] [CrossRef]
- Ng, I.O.; Lai, E.C.; Ho, J.C.; Cheung, L.K.; Ng, M.M.; So, M.K. Flow cytometric analysis of DNA ploidy in hepatocellular carcinoma. Am. J. Clin. Pathol. 1994, 102, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, J.; Okamoto, E.; Yamanaka, N.; Toyosaka, A.; Mitsunobu, M. Flow cytometric DNA analysis of hepatocellular carcinoma. Cancer 1991, 67, 939–944. [Google Scholar] [CrossRef]
- Nagasue, N.; Kohno, H.; Hayashi, T.; Yamanoi, A.; Uchida, M.; Takemoto, Y.; Makino, Y.; Ono, T.; Hayashi, J.; Nakamura, T. Lack of intratumoral heterogeneity in DNA ploidy pattern of hepatocellular carcinoma. Gastroenterology 1993, 105, 1449–1454. [Google Scholar] [CrossRef]
- Bou-Nader, M.; Caruso, S.; Donne, R.; Celton-Morizur, S.; Calderaro, J.; Gentric, G.; Cadoux, M.; L’Hermitte, A.; Klein, C.; Guilbert, T.; et al. Polyploidy spectrum: A new marker in HCC classification. Gut 2020, 69, 355–364. [Google Scholar] [CrossRef]
- Frade, J.; Nakagawa, S.; Cortes, P.; di Vicino, U.; Romo, N.; Lluis, F.; Cosma, M.P. Controlled ploidy reduction of pluripotent 4n cells generates 2n cells during mouse embryo development. Sci. Adv. 2019, 5, eaax4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsumoto, T. Implications of Polyploidy and Ploidy Alterations in Hepatocytes in Liver Injuries and Cancers. Int. J. Mol. Sci. 2022, 23, 9409. https://doi.org/10.3390/ijms23169409
Matsumoto T. Implications of Polyploidy and Ploidy Alterations in Hepatocytes in Liver Injuries and Cancers. International Journal of Molecular Sciences. 2022; 23(16):9409. https://doi.org/10.3390/ijms23169409
Chicago/Turabian StyleMatsumoto, Tomonori. 2022. "Implications of Polyploidy and Ploidy Alterations in Hepatocytes in Liver Injuries and Cancers" International Journal of Molecular Sciences 23, no. 16: 9409. https://doi.org/10.3390/ijms23169409
APA StyleMatsumoto, T. (2022). Implications of Polyploidy and Ploidy Alterations in Hepatocytes in Liver Injuries and Cancers. International Journal of Molecular Sciences, 23(16), 9409. https://doi.org/10.3390/ijms23169409