
The Omnipresence of DYRK1A in Human Diseases


Abstract
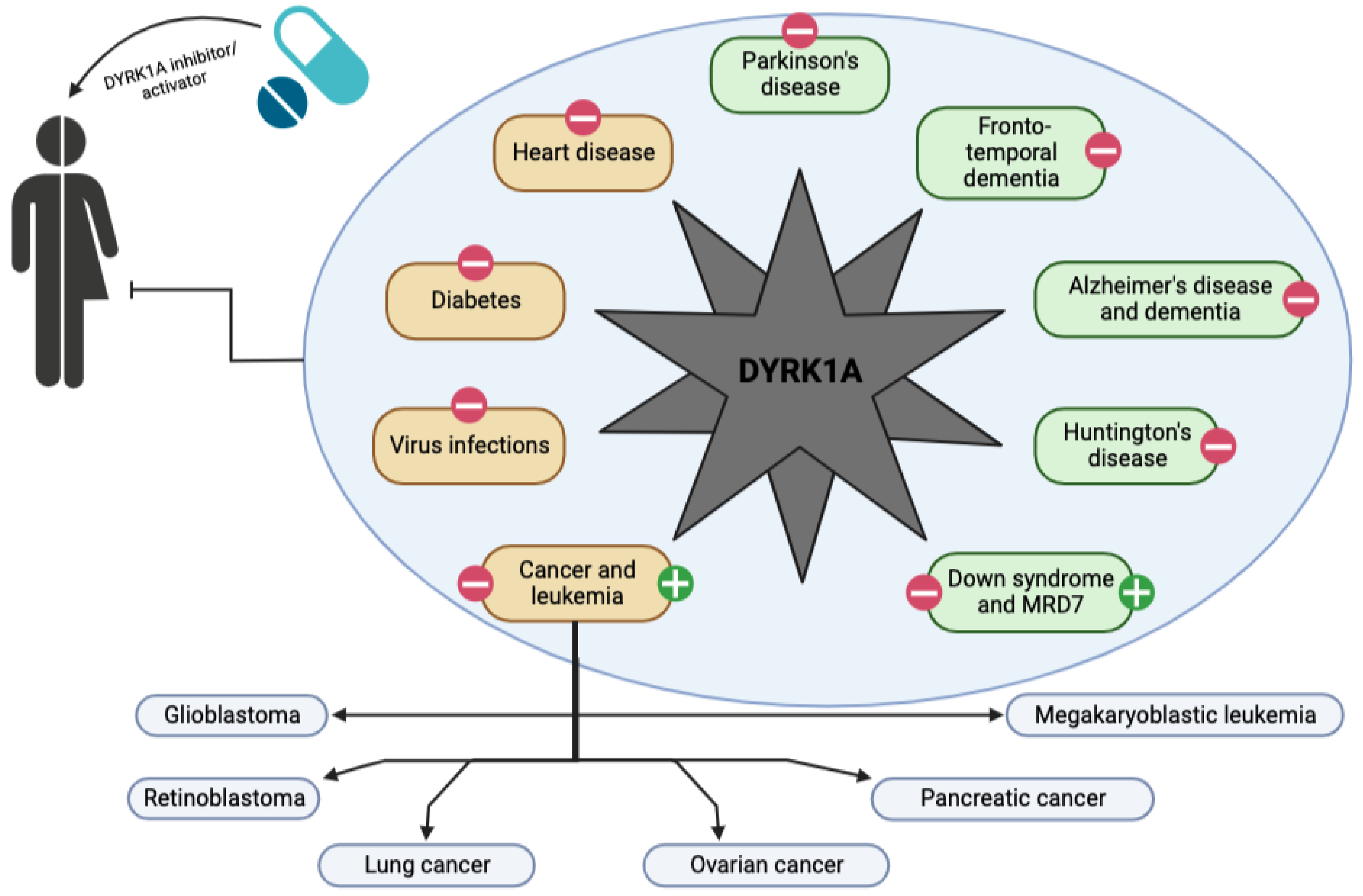
:1. Introduction
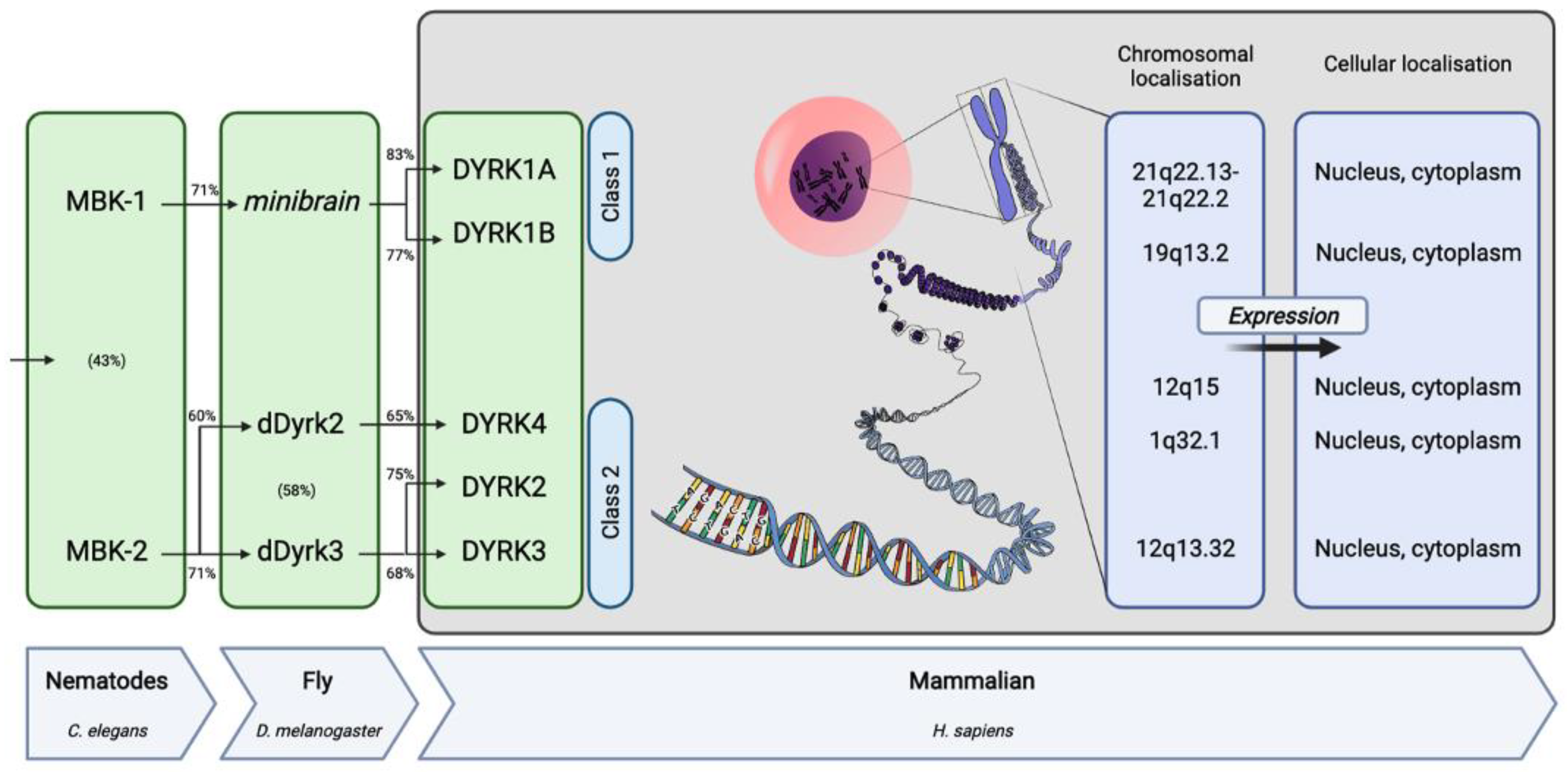
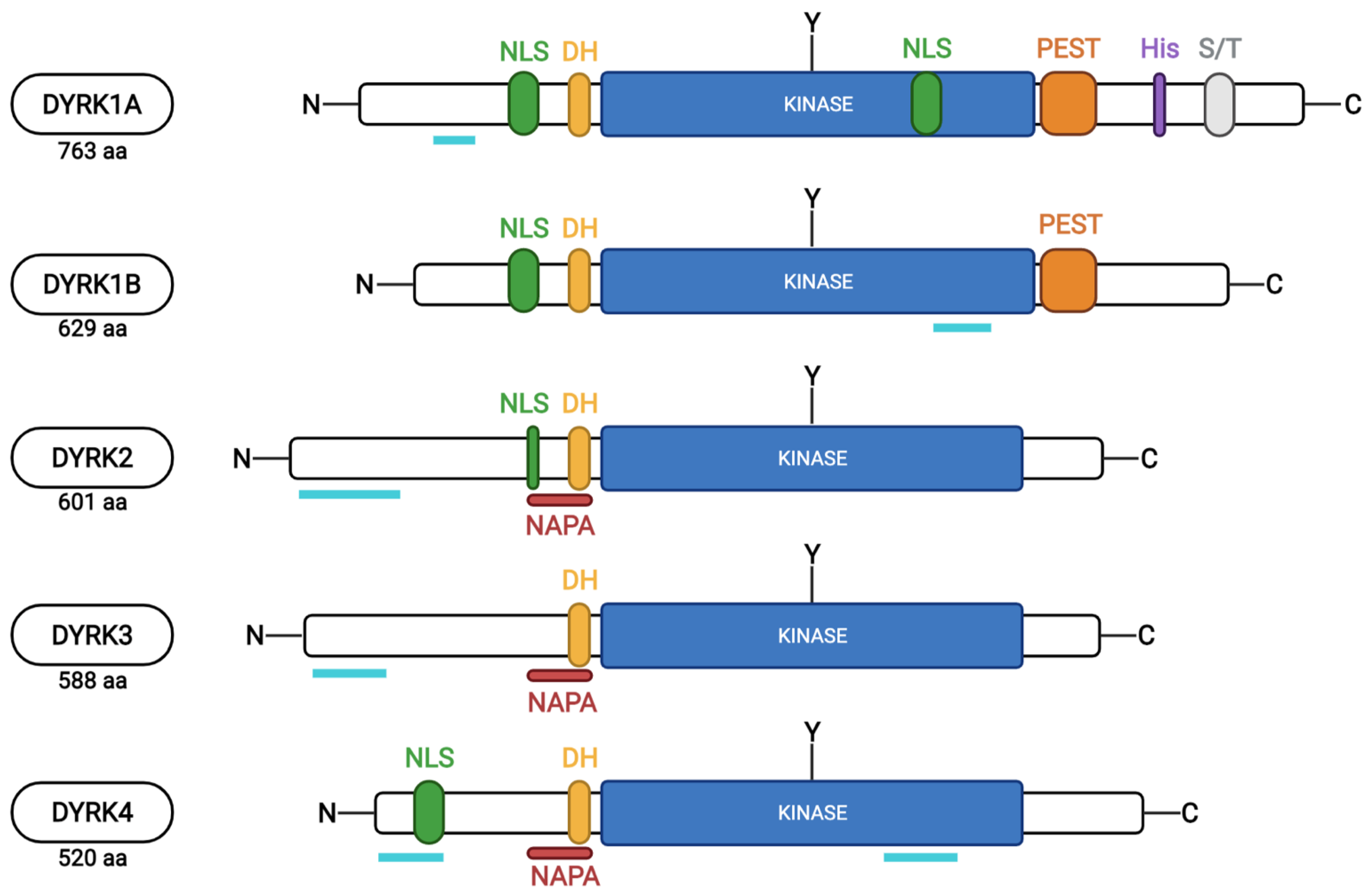
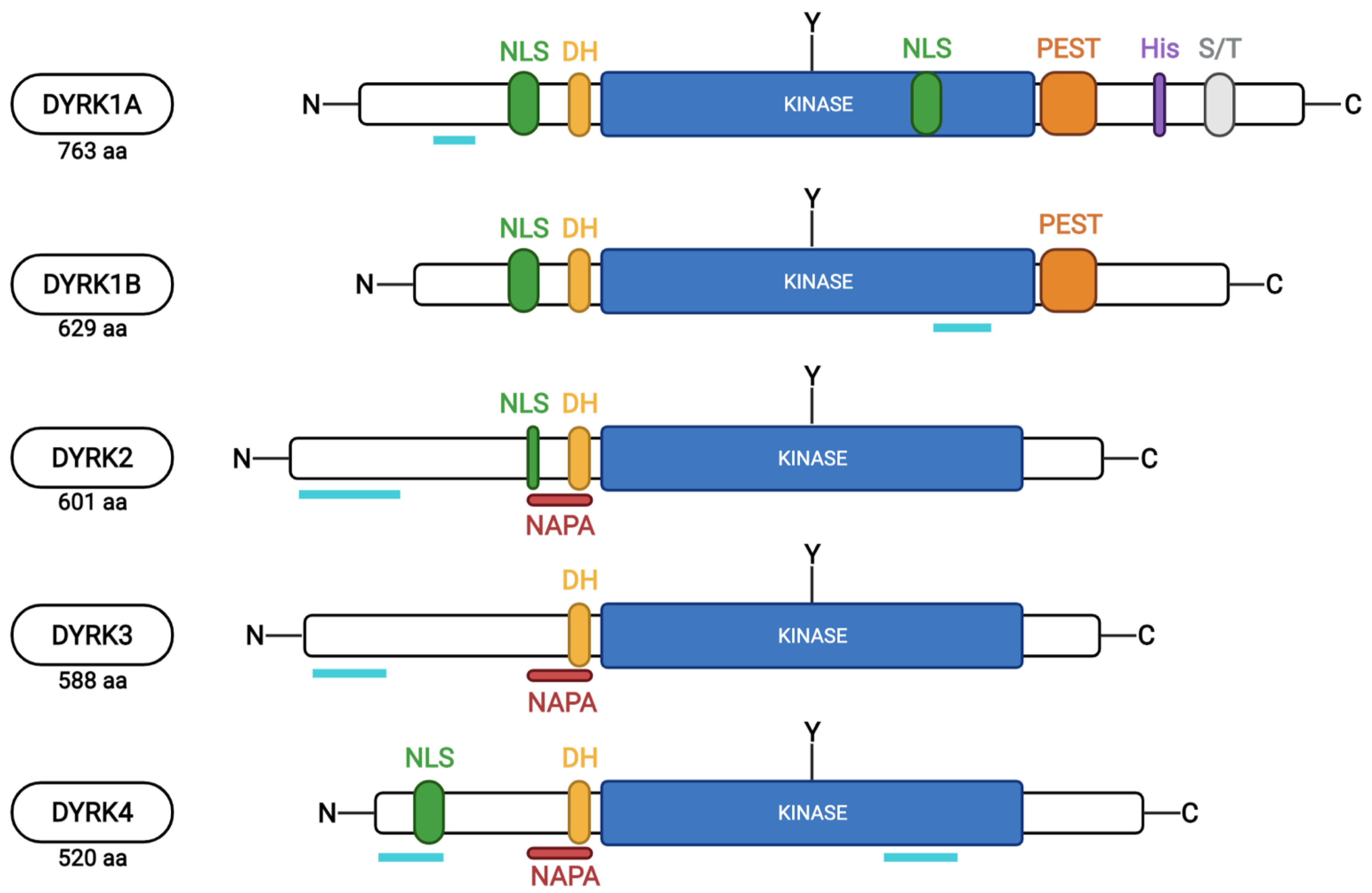
2. The DYRK Family
3. DYRK1A Expression and Enzymatic Activity
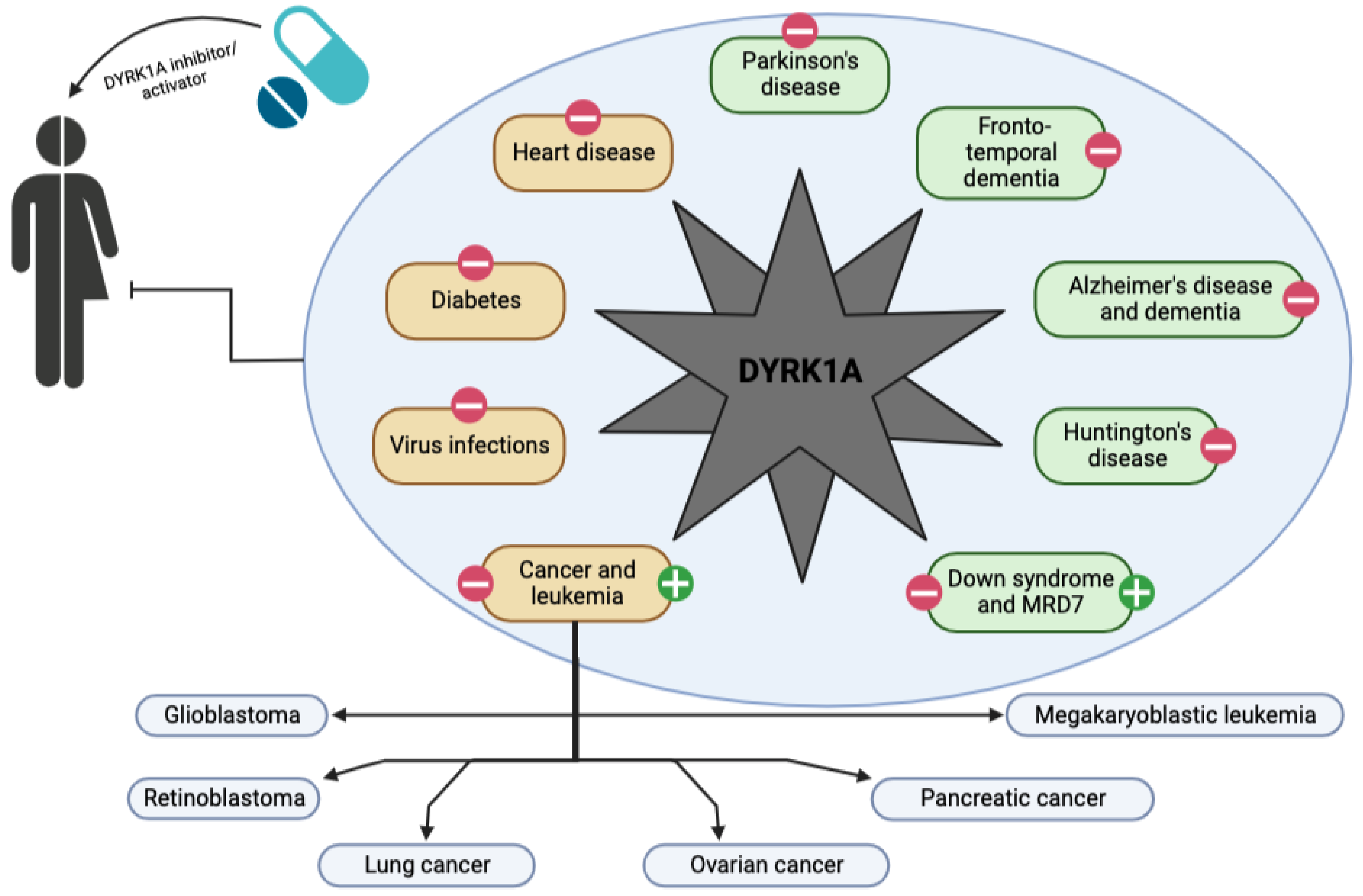
4. DYRK1A and Neurological Diseases
4.1. Down Syndrome (DS) and Intellectual Developmental Disorder Autosomal Dominant 7 (MRD7)
4.2. Dementia and Alzheimer’s Disease (AD)
4.3. Parkinson’s (PD) and Huntington’s (HD) Diseases and Fronto-Temporal Degeneration (FTD)
5. DYRK1A and Other Diseases
5.1. Diabetes
5.2. Solid Cancers and Leukemias
5.3. Viral Infections
5.4. Heart Diseases
6. DYRK1A, a Target for the Patient’s Future
7. Concluding Remarks and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Adam, D. How far will global population rise? Nature 2021, 597, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Martin Prince, A.; Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015 The Global Impact of Dementia an Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A brief history of human disease genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I.; Bologa, C.G.; Brunak, S.; Campbell, A.; Gan, G.N.; Gaulton, A.; Gomez, S.M.; Guha, R.; Hersey, A.; Holmes, J.; et al. Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discov. 2018, 17, 317–332. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein Kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Zhang, H.; Cao, X.; Tang, M.; Zhong, G.; Si, Y.; Li, H.; Zhu, F.; Liao, Q.; Li, L.; Zhao, J.; et al. A subcellular map of the human kinome. Elife 2021, 10, e64943. [Google Scholar] [CrossRef]
- Enjalbert, A.; le Pechon-Vallee, C. Protein Kinases. In Encyclopedia of Hormones; Academic Press: Cambridge, MA, USA, 2003; pp. 277–285. ISBN 4108641094. [Google Scholar]
- Wilson, L.J.; Linley, A.; Hammond, D.E.; Hood, F.E.; Coulson, J.M.; MacEwan, D.J.; Ross, S.J.; Slupsky, J.R.; Smith, P.D.; Eyers, P.A.; et al. New perspectives, opportunities, and challenges in exploring the human protein kinome. Cancer Res. 2018, 78, 15–29. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in Kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef] [PubMed]
- Aranda, S.; Laguna, A.; de la Luna, S. DYRK Family of protein Kinases: Evolutionary relationships, biochemical properties, and functional roles. FASEB J. 2011, 25, 449–462. [Google Scholar] [CrossRef]
- Lindberg, M.F.; Meijer, L. Dual-specificity, tyrosine phosphorylation-regulated Kinases (Dyrks) and Cdc2-like Kinases (Clks) in human disease, an overview. Int. J. Mol. Sci. 2021, 22, 6047. [Google Scholar] [CrossRef]
- Tejedor, F.J.; Hämmerle, B. MNB/DYRK1A as a multiple regulator of neuronal development. FEBS J. 2011, 278, 223–235. [Google Scholar] [CrossRef]
- Son, M.; Kim, D.Y.; Kim, C.-H. Disease modeling of rare neurological disorders in zebrafish. Int. J. Mol. Sci. 2022, 23, 3946. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Arato, K.; Lilienthal, E.; Zerweck, J.; Schutkowski, M.; Chatain, N.; Müller-Newen, G.; Becker, W.; de la Luna, S. Splice variants of the dual specificity tyrosine phosphorylation-regulated Kinase 4 (DYRK4) differ in their subcellular localization and catalytic activity. J. Biol. Chem. 2011, 286, 5494–5505. [Google Scholar] [CrossRef]
- Taira, N.; Nihira, K.; Yamaguchi, T.; Miki, Y.; Yoshida, K. DYRK2 is targeted to the nucleus and controls P53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol. Cell 2007, 25, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Sacher, F.; Möller, C.; Bone, W.; Gottwald, U.; Fritsch, M. The expression of the testis-specific Dyrk4 Kinase is highly restricted to step 8 spermatids but is not required for male fertility in mice. Mol. Cell. Endocrinol. 2007, 267, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; West, K.L.; Tan, X.Y.; Li, J.; Ishibashi, T.; Yu, C.; Sy, S.M.H.; Leung, J.W.C.; Huen, M.S.Y. Screen identifies DYRK1B network as mediator oftranscription repression on damaged chromatin. Proc. Natl. Acad. Sci. USA 2020, 117, 17019–17030. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K. Nuclear trafficking of pro-apoptotic Kinases in response to DNA damage. Trends Mol. Med. 2008, 14, 305–313. [Google Scholar] [CrossRef]
- Guo, X.; Williams, J.G.; Schug, T.T.; Li, X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J. Biol. Chem. 2010, 285, 13223–13232. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Ewton, D.Z.; Li, S.; Naqvi, A.; Mercer, S.E.; Landas, S.; Friedman, E. The Kinase Mirk/Dyrk1B mediates cell survival in pancreatic ductal adenocarcinoma. Cancer Res. 2006, 66, 4149–4158. [Google Scholar] [CrossRef]
- Himpel, S.; Panzer, P.; Eirmbter, K.; Czajkowska, H.; Sayed, M.; Packman, L.C.; Blundell, T.; Kentrup, H.; Grötzinger, J.; Joost, H.-G.; et al. Identification of the Autophosphorylation Sites and Characterization of Their Effects in the Protein Kinase DYRK1A. Biochem. J. 2001, 359, 497–505. [Google Scholar] [CrossRef]
- Lochhead, P.A.; Sibbet, G.; Morrice, N.; Cleghon, V. Activation-loop autophosphorylation is mediated by a novel transitional intermediate form of DYRKs. Cell 2005, 121, 925–936. [Google Scholar] [CrossRef]
- Kentrup, H.; Becker, W.; Heukelbach, J.; Wilmes, A.; Schü, A.; Huppertz, C.; Kainulainen, H.; Joost, H.-G. Dyrk, a Dual Specificity Protein Kinase with Unique Structural Features Whose Activity Is Dependent on Tyrosine Residues between Subdomains VII and VIII. J. Biol. Chem. 1996, 271, 3488–3495. [Google Scholar] [CrossRef] [PubMed]
- Laham, A.J.; Saber-Ayad, M.; El-Awady, R. DYRK1A: A Down Syndrome-related dual protein kinase with a versatile role in tumorigenesis. Cell. Mol. Life Sci. 2021, 78, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Guimera, J.; Casas, C.; Estivill, X.; Pritchard, M. Human minibrain homologue (MNBH/DYRK1): Characterization, alternative splicing, differential tissue expression, and overexpression in Down Syndrome. Genomics 1999, 57, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Belgardt, B.F.; Lammert, E. DYRK1A: A promising drug target for islet transplant-based diabetes therapies. Diabetes 2016, 65, 1496–1498. [Google Scholar] [CrossRef]
- Martí, E.; Altafaj, X.; Dierssen, M.; de La Luna, S.; Fotaki, V.; Alvarez, M.; Pérez-Riba, M.; Ferrer, I.; Estivill, X. Dyrk1A Expression pattern supports specific roles of this Kinase in the adult central nervous system. Brain Res. 2003, 964, 250–263. [Google Scholar] [CrossRef]
- Murakami, N.; Bolton, D.; Hwang, Y.W. Dyrk1A binds to multiple endocytic proteins required for formation of clathrin-coated vesicles. Biochemistry 2009, 48, 9297–9305. [Google Scholar] [CrossRef]
- Vona, C.D.; Bezdan, D.; Islam, A.B.M.M.K.; Salichs, E.; López-Bigas, N.; Ossowski, S.; Luna, S.D.L. Chromatin-wide profiling of DYRK1A reveals a role as a gene-specific RNA polymerase II CTD Kinase. Mol. Cell 2015, 57, 506–520. [Google Scholar] [CrossRef]
- Arron, J.R.; Winslow, M.M.; Polleri, A.; Chang, C.P.; Wu, H.; Gao, X.; Neilson, J.R.; Chen, L.; Heit, J.J.; Kim, S.K.; et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 2006, 441, 595–600. [Google Scholar] [CrossRef]
- Fernandez-Martinez, J.; Vela, E.M.; Tora-Ponsioen, M.; Ocaña, O.H.; Nieto, M.A.; Galceran, J. Attenuation of notch signalling by the Down-Syndrome-associated Kinase DYRK1A. J. Cell Sci. 2009, 122, 1574–1583. [Google Scholar] [CrossRef]
- Qian, W.; Liang, H.; Shi, J.; Jin, N.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X.; Liu, F. Regulation of the alternative splicing of Tau Exon 10 by SC35 and Dyrk1A. Nucleic Acids Res. 2011, 39, 6161–6171. [Google Scholar] [CrossRef]
- Bescond, M.; Rahmani, Z. Dual-specificity Tyrosine-Phosphorylated and regulated Kinase 1A (DYRK1A) interacts with the Phytanoyl-CoA α-Hydroxylase associated protein 1 (PAHX-AP1), a brain specific protein. Int. J. Biochem. Cell Biol. 2005, 37, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Adayev, T.; Chen-Hwang, M.C.; Murakami, N.; Lee, E.; Bolton, D.C.; Hwang, Y.W. Dual-specificity tyrosine phosphorylation-regulated Kinase 1A does not require tyrosine phosphorylation for activity in vitro. Biochemistry 2007, 46, 7614–7624. [Google Scholar] [CrossRef] [PubMed]
- Guedj, F.; Pereira, P.L.; Najas, S.; Barallobre, M.J.; Chabert, C.; Souchet, B.; Sebrie, C.; Verney, C.; Herault, Y.; Arbones, M.; et al. DYRK1A: A master regulatory protein controlling brain growth. Neurobiol. Dis. 2012, 46, 190–203. [Google Scholar] [CrossRef]
- Arbones, M.L.; Thomazeau, A.; Nakano-Kobayashi, A.; Hagiwara, M.; Delabar, J.M. DYRK1A and cognition: A lifelong relationship. Pharmacol. Ther. 2019, 194, 199–221. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.R.; Balupuri, A.; Choi, K.E.; Kang, N.S. Small molecule inhibitors of DYRK1A identified by computational and experimental approaches. Int. J. Mol. Sci. 2020, 21, 6826. [Google Scholar] [CrossRef]
- Becker, W.; Sippl, W. Activation, regulation, and inhibition of DYRK1A. FEBS J. 2011, 278, 246–256. [Google Scholar] [CrossRef]
- Ding, S.; Shi, J.; Qian, W.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X.; Liu, F. Regulation of alternative splicing of Tau exon 10 by 9G8 and Dyrk1A. Neurobiol. Aging 2012, 33, 1389–1399. [Google Scholar] [CrossRef]
- Murakami, N.; Xie, W.; Renne, C.L.; Chen-Hwang, M.C.; Wieraszko, A.; Yu, W.H. Phosphorylation of amphiphysin I by minibrain Kinase/dual-specificity tyrosine phosphorylation-regulated Kinase, a Kinase implicated in Down Syndrome. J. Biol. Chem. 2006, 281, 23712–23724. [Google Scholar] [CrossRef]
- Ryoo, S.R.; Cho, H.J.; Lee, H.W.; Jeong, H.K.; Radnaabazar, C.; Kim, Y.S.; Kim, M.J.; Son, M.Y.; Seo, H.; Chung, S.H.; et al. Dual-specificity Tyrosine(Y)-Phosphorylation regulated Kinase 1A-mediated phosphorylation of amyloid precursor protein: Evidence for a functional link between Down Syndrome and Alzheimer’s Disease. J. Neurochem. 2008, 104, 1333–1344. [Google Scholar] [CrossRef]
- Sitz, J.H.; Tigges, M.; Baumgärtel, K.; Khaspekov, L.G.; Lutz, B. Dyrk1A Potentiates steroid hormone-induced transcription via the chromatin remodeling factor Arip4. Mol. Cell. Biol. 2004, 24, 5821–5834. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, T.; Zhou, C.; Chohan, M.O.; Gu, X.; Wegiel, J.; Zhou, J.; Hwang, Y.W.; Iqbal, K.; Grundke-Iqbal, I.; et al. Increased dosage of Dyrk1A alters Alternative Splicing Factor (ASF)-regulated alternative splicing of Tau in Down Syndrome. J. Biol. Chem. 2008, 283, 28660–28669. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.A.; Rahmani, Z. DYRK1A Enhances the mitogen-activated protein Kinase cascade in PC12 cells by forming a complex with Ras, B-Raf, and MEK1. Mol. Biol. Cell 2005, 16, 3562–3573. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.; Boni, J.; Cuatrecasas, M.; Bofill-De Ros, X.; Núñez-Manchón, E.; Gironella, M.; Vaquero, E.C.; Arbones, M.L.; de la Luna, S.; Fillat, C. DYRK1A modulates C-MET in Pancreatic Ductal Adenocarcinoma to drive tumour growth. Gut 2019, 68, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.J.; Ahn, Y.S.; Chung, K.C. Protein Kinase Dyrk1 activates CAMP response element-binding protein during neuronal differentiation in hippocampal progenitor cells. J. Biol. Chem. 2001, 276, 39819–39824. [Google Scholar] [CrossRef]
- Laguna, A.; Barallobre, M.J.; Marchena, M.Á.; Mateus, C.; Ramírez, E.; Martínez-Cue, C.C.; Delabar, J.M.; Castelo-Branco, M.; de la Villa, P.; Arbonés, M.L. Triplication of Dyrk1a causes retinal structural and functional alterations in Down Syndrome. Hum. Mol. Genet. 2013, 22, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Laguna, A.; Aranda, S.; Barallobre, M.J.; Barhoum, R.; Fernández, E.; Fotaki, V.; Delabar, J.M.; de la Luna, S.; de la Villa, P.; Arbonés, M.L. The protein Kinase DYRK1A regulates Caspase-9-mediated apoptosis during retina development. Dev. Cell 2008, 15, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Humphrey, S.J.; Ellis, M.; Hoque, M.; Abbassi, R.H.; Chen, B.; Longworth, M.; Needham, E.J.; James, D.E.; Johns, T.G.; et al. Global phosphoproteomics reveals DYRK1A regulates CDK1 activity in glioblastoma cells. Cell Death Discov. 2021, 7, 81. [Google Scholar] [CrossRef]
- Soppa, U.; Schumacher, J.; Ortiz, V.F.; Pasqualon, T.; Tejedor, F.J.; Becker, W. The Down Syndrome-related protein Kinase DYRK1A phosphorylates P27 Kip1and Cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle 2014, 13, 2084–2100. [Google Scholar] [CrossRef]
- Xiang, J.; Yang, S.; Xin, N.; Gaertig, M.A.; Reeves, R.H.; Li, S.; Li, X.J. DYRK1A regulates Hap1-Dcaf7/WDR68 binding with implication for delayed growth in Down Syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, E1224–E1233. [Google Scholar] [CrossRef]
- Glenewinkel, F.; Cohen, M.J.; King, C.R.; Kaspar, S.; Bamberg-Lemper, S.; Mymryk, J.S.; Becker, W. The adaptor protein DCAF7 mediates the interaction of the Adenovirus E1A oncoprotein with the protein Kinases DYRK1A and HIPK2. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Guiley, K.Z.; Liban, T.J.; Felthousen, J.G.; Ramanan, P.; Litovchick, L.; Rubin, S.M. Structural mechanisms of DREAM complex assembly and regulation. Genes Dev. 2015, 29, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Karakose, E.; Argmann, C.; Wang, H.; Balev, M.; Brody, R.I.; Rivas, H.G.; Liu, X.; Wood, O.; Liu, H.; et al. Disrupting the DREAM complex enables proliferation of adult human pancreatic beta cells. J. Clin. Investig. 2022, 132, e157086. [Google Scholar] [CrossRef]
- Litovchick, L.; Florens, L.A.; Swanson, S.K.; Washburn, M.P.; Decaprio, J.A. DYRK1A protein Kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011, 25, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Pozo, N.; Zahonero, C.; Fernández, P.; Liñares, J.M.; Ayuso, A.; Hagiwara, M.; Pérez, A.; Ricoy, J.R.; Hernández-Laín, A.; Sepúlveda, J.M.; et al. Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J. Clin. Investig. 2013, 123, 2475–2487. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-l.; Ding, K.; Hu, X.; Wu, L.W.; Zhou, D.M.; Rao, M.J.; Lin, N.M.; Zhang, C. DYRK1A inhibition suppresses STAT3/EGFR/Met signalling and sensitizes EGFR wild-type NSCLC cells to AZD9291. J. Cell. Mol. Med. 2019, 23, 7427–7437. [Google Scholar] [CrossRef]
- Bhansali, R.S.; Rammohan, M.; Lee, P.; Laurent, A.P.; Wen, Q.; Suraneni, P.; Yip, B.H.; Tsai, Y.C.; Jenni, S.; Bornhauser, B.; et al. DYRK1A regulates B cell acute Lymphoblastic Leukemia through phosphorylation of FOXO1 and STAT3. J. Clin. Investig. 2021, 131, e135937. [Google Scholar] [CrossRef]
- Von Groote-Bidlingmaier, F.; Schmoll, D.; Orth, H.M.; Joost, H.G.; Becker, W.; Barthel, A. DYRK1 Is a co-activator of FKHR (FOXO1a)-Dependent Glucose-6-Phosphatase gene expression. Biochem. Biophys. Res. Commun. 2003, 300, 764–769. [Google Scholar] [CrossRef]
- Ehe, B.K.; Lamson, D.R.; Tarpley, M.; Onyenwoke, R.U.; Graves, L.M.; Williams, K.P. Identification of a DYRK1A-mediated phosphorylation site within the nuclear localization sequence of the hedgehog transcription factor GLI1. Biochem. Biophys. Res. Commun. 2017, 491, 767–772. [Google Scholar] [CrossRef]
- Grau, C.; Arató, K.; Fernández-Fernández, J.M.; Valderrama, A.; Sindreu, C.; Fillat, C.; Ferrer, I.; de la Luna, S.; Altafaj, X. DYRK1A-mediated phosphorylation of GluN2A at Ser1048 regulates the surface expression and channel activity of GluN1/GluN2A receptors. Front. Cell. Neurosci. 2014, 8, 331. [Google Scholar] [CrossRef]
- Song, W.J.; Song, E.A.C.; Jung, M.S.; Choi, S.H.; Baik, H.H.; Jin, B.K.; Kim, J.H.; Chung, S.H. Phosphorylation and inactivation of glycogen synthase Kinase 3β (GSK3β) by dual-specificity Tyrosine Phosphorylation-Regulated Kinase 1A (Dyrk1A). J. Biol. Chem. 2015, 290, 2321–2333. [Google Scholar] [CrossRef]
- Kang, J.E.; Choi, S.A.; Park, J.B.; Chung, K.C. Regulation of the proapoptotic activity of huntingtin interacting protein 1 by Dyrk1 and Caspase-3 in hippocampal neuroprogenitor cells. J. Neurosci. Res. 2005, 81, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Xavier, A.C.; Ge, Y.; Taub, J.W. Down Syndrome and Malignancies: A unique clinical relationship. J. Mol. Diagn. 2009, 11, 371–380. [Google Scholar] [CrossRef]
- Jang, S.M.; Azebi, S.; Soubigou, G.; Muchardt, C. DYRK1A Phoshorylates Histone H3 to differentially regulate the binding of HP1 isoforms and antagonize HP1-mediated transcriptional repression. EMBO Rep. 2014, 15, 686–694. [Google Scholar] [CrossRef]
- Lee, S.B.; Frattini, V.; Bansal, M.; Castano, A.M.; Sherman, D.; Hutchinson, K.; Bruce, J.N.; Califano, A.; Liu, G.; Cardozo, T.; et al. An ID2-dependent mechanism for VHL inactivation in cancer. Nature 2016, 529, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Dirice, E.; Walpita, D.; Vetere, A.; Meier, B.C.; Kahraman, S.; Hu, J.; Dančík, V.; Burns, S.M.; Gilbert, T.J.; Olson, D.E.; et al. Inhibition of DYRK1A stimulates human β-cell proliferation. Diabetes 2016, 65, 1660–1671. [Google Scholar] [CrossRef] [PubMed]
- Booiman, T.; Loukachov, V.V.; van Dort, K.A.; van’t Wout, A.B.; Kootstra, N.A. DYRK1A controls HIV-1 replication at a transcriptional level in an NFAT dependent manner. PLoS ONE 2015, 10, e0144229. [Google Scholar] [CrossRef]
- Raaf, L.; Noll, C.; Cherifi, M.; Benazzoug, Y.; Delabar, J.-M.; Janel, N. Hyperhomocysteinemia-Induced Dyrk1a Downregulation Results in Cardiomyocytehypertrophy in rats. Int. J. Cardiol. 2010, 145, 305–306. [Google Scholar] [CrossRef]
- Kawakubo, T.; Mori, R.; Shirotani, K.; Iwata, N.; Asai, M. Neprilysin is suppressed by dual-specificity Tyrosine-Phosphorylation regulated Kinase 1A (DYRK1A) in Down-Syndrome-derived fibroblasts. Biol. Pharm. Bull. 2017, 40, 327–333. [Google Scholar] [CrossRef]
- Lepagnol-Bestel, A.M.; Zvara, A.; Maussion, G.; Quignon, F.; Ngimbous, B.; Ramoz, N.; Imbeaud, S.; Loe-Mie, Y.; Benihoud, K.; Agier, N.; et al. DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down Syndrome. Hum. Mol. Genet. 2009, 18, 1405–1414. [Google Scholar] [CrossRef]
- Park, J.; Oh, Y.; Yoo, L.; Jung, M.S.; Song, W.J.; Lee, S.H.; Seo, H.; Chung, K.C. Dyrk1A Phosphorylates P53 and inhibits proliferation of embryonic neuronal cells. J. Biol. Chem. 2010, 285, 31895–31906. [Google Scholar] [CrossRef]
- Im, E.; Chung, K.C. Dyrk1A Phosphorylates Parkin at Ser-131 and negatively regulates its Ubiquitin E3 Ligase activity. J. Neurochem. 2015, 134, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Ryu, Y.S.; Park, S.Y.; Jung, M.S.; Yoon, S.H.; Kwen, M.Y.; Lee, S.Y.; Choi, S.H.; Radnaabazar, C.; Kim, M.K.; Kim, H.; et al. Dyrk1A-mediated phosphorylation of presenilin 1: A functional link between Down Syndrome and Alzheimer’s Disease. J. Neurochem. 2010, 115, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Qiang, F.; Wu, Y. RCAN1 in the inverse association between Alzheimer’s Disease and cancer. Oncotarget 2018, 9, 54–66. [Google Scholar]
- Jung, M.S.; Park, J.H.; Ryu, Y.S.; Choi, S.H.; Yoon, S.H.; Kwen, M.Y.; Oh, J.Y.; Song, W.J.; Chung, S.H. Regulation of RCAN1 protein activity by Dyrk1A protein-mediated phosphorylation. J. Biol. Chem. 2011, 286, 40401–40412. [Google Scholar] [CrossRef]
- Sitz, J.H.; Baumgärtel, K.; Hämmerle, B.; Papadopoulos, C.; Hekerman, P.; Tejedor, F.J.; Becker, W.; Lutz, B. The Down Syndrome candidate dual-specificity Tyrosine Phosphorylation-regulated Kinase 1A Phosphorylates the neurodegeneration-related Septin 4. Neuroscience 2008, 157, 596–605. [Google Scholar] [CrossRef]
- Kimura, R.; Kamino, K.; Yamamoto, M.; Nuripa, A.; Kida, T.; Kazui, H.; Hashimoto, R.; Tanaka, T.; Kudo, T.; Yamagata, H.; et al. The DYRK1A gene, encoded in chromosome 21 Down Syndrome critical region, bridges between β-Amyloid production and Tau phosphorylation in Alzheimer Disease. Hum. Mol. Genet. 2007, 16, 15–23. [Google Scholar] [CrossRef]
- Walter, C.; Marada, A.; Suhm, T.; Ernsberger, R.; Muders, V.; Kücükköse, C.; Sánchez-Martín, P.; Hu, Z.; Aich, A.; Loroch, S.; et al. Global kinome profiling reveals DYRK1A as critical activator of the human mitochondrial import machinery. Nat. Commun. 2021, 12, 4284. [Google Scholar] [CrossRef]
- Li, Y.; Xie, X.; Jie, Z.; Zhu, L.; Yang, J.-Y.; Ko, C.-J.; Gao, T.; Jain, A.; Jung, S.Y.; Baran, N.; et al. DYRK1a Mediates BAFF-induced noncanonical NF-ΚB activation to promote autoimmunity and B-cell leukemogenesis. Blood 2021, 138, 2360–2371. [Google Scholar] [CrossRef]
- Eun, J.K.; Jee, Y.S.; Hyun, J.L.; Rhim, H.; Hasegawa, M.; Iwatsubo, T.; Do, S.M.; Kim, J.; Paik, S.R.; Kwang, C.C. Dyrk1A phosphorylates α-Synuclein and enhances intracellular inclusion formation. J. Biol. Chem. 2006, 281, 33250–33257. [Google Scholar] [CrossRef]
- Wegiel, J.; Gong, C.X.; Hwang, Y.W. The role of DYRK1A in neurodegenerative diseases. FEBS J. 2011, 278, 236–245. [Google Scholar] [CrossRef]
- Duchon, A.; Herault, Y. DYRK1A, a dosage-sensitive gene involved in neurodevelopmental disorders, is a target for drug development in Down Syndrome. Front. Behav. Neurosci. 2016, 10, 104. [Google Scholar] [CrossRef]
- Courraud, J.; Chater-Diehl, E.; Durand, B.; Vincent, M.; del Mar, M.; Moreno, M.; Boujelbene, I.; Drouot, N.; Genschik, L.; Schaefer, E.; et al. Integrative approach to interpret DYRK1A variants, leading to a frequent neurodevelopmental disorder. Genet. Med. 2021, 23, 2150–2159. [Google Scholar] [CrossRef]
- Van Bon, B.W.M.; Coe, B.P.; Bernier, R.; Green, C.; Gerdts, J.; Witherspoon, K.; Kleefstra, T.; Willemsen, M.H.; Kumar, R.; Bosco, P.; et al. Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol. Psychiatry 2016, 21, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Arranz, J.; Balducci, E.; Arató, K.; Sánchez-Elexpuru, G.; Najas, S.; Parras, A.; Rebollo, E.; Pijuan, I.; Erb, I.; Verde, G.; et al. Impaired Development of Neocortical Circuits Contributes to the Neurological Alterations in DYRK1A haploinsufficiency syndrome. Neurobiol. Dis. 2019, 127, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Brault, V.; Nguyen, T.L.; Flores-Gutiérrez, J.; Iacono, G.; Birling, M.C.; Lalanne, V.; Meziane, H.; Manousopoulou, A.; Pavlovic, G.; Lindner, L.; et al. Dyrk1a gene dosage in glutamatergic neurons has key effects in cognitive deficits observed in mouse models of MRD7 and Down Syndrome. PLoS Genet. 2021, 17, e1009777. [Google Scholar] [CrossRef] [PubMed]
- Najas, S.; Arranz, J.; Lochhead, P.A.; Ashford, A.L.; Oxley, D.; Delabar, J.M.; Cook, S.J.; Barallobre, M.J.; Arbonés, M.L. DYRK1A-mediated cyclin D1 degradation in neural stem cells contributes to the neurogenic cortical defects in Down Syndrome. EBioMedicine 2015, 2, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Bellmaine, S.F.; Ovchinnikov, D.A.; Manallack, D.T.; Cuddy, C.E.; Elefanty, A.G.; Stanley, E.G.; Wolvetang, E.J.; Williams, S.J.; Pera, M.; Hall, E. Inhibition of DYRK1A disrupts neural lineage specificationin human pluripotent stem cells. Elife 2017, 6, e24502. [Google Scholar] [CrossRef]
- Raveau, M.; Shimohata, A.; Amano, K.; Miyamoto, H.; Yamakawa, K. DYRK1A-haploinsufficiency in mice causes autistic-like features and febrile seizures. Neurobiol. Dis. 2018, 110, 180–191. [Google Scholar] [CrossRef]
- Fotaki, V.; Dierssen, M.; Alcántara, S.; Martínez, S.; Martí, E.; Casas, C.; Visa, J.; Soriano, E.; Estivill, X.; Arbonés, M.L. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell. Biol. 2002, 22, 6636–6647. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer Disease. Nat. Rev. 2021, 7, 33. [Google Scholar] [CrossRef]
- Fernandez Bessone, I.; Navarro, J.; Martinez, E.; Karmirian, K.; Holubiec, M.; Alloatti, M.; Goto-Silva, L.; Arnaiz Yepez, C.; Martins-de-Souza, D.; Nascimento, J.M.; et al. DYRK1A regulates the bidirectional axonal transport of APP in human-derived neurons. J. Neurosci. 2022, 42, 6344–6358. [Google Scholar] [CrossRef] [PubMed]
- Branca, C.; Shaw, D.M.; Belfiore, R.; Gokhale, V.; Shaw, A.Y.; Foley, C.; Smith, B.; Hulme, C.; Dunckley, T.; Meechoovet, B.; et al. Dyrk1 inhibition improves Alzheimer’s disease-like pathology. Aging Cell 2017, 16, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, R.; Meechoovet, B.; Ow, A.; Foley, C.; Shaw, A.; Smith, B.; Oddo, S.; Hulme, C.; Dunckley, T. Chronic Dyrk1 inhibition delays the onset of AD-Like pathology in 3xTg-AD mice. Mol. Neurobiol. 2019, 56, 8364–8375. [Google Scholar] [CrossRef] [PubMed]
- García-Cerro, S.; Rueda, N.; Vidal, V.; Lantigua, S.; Martínez-Cué, C. Normalizing the Gene Dosage of Dyrk1A in a mouse model of Down Syndrome rescues several Alzheimer’s Disease phenotypes. Neurobiol. Dis. 2017, 106, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Mumford, P.; Tosh, J.; Anderle, S.; Gkanatsiou Wikberg, E.; Noy, S.; Cleverley, K.; Saito, T.; Saido, T.C.; Yu, E.; Brinkmalm, G.; et al. Genetic mapping of APP and Amyloid-β biology modulation by trisomy 21. J. Neurosci. 2022, 42, 6453–6468. [Google Scholar] [CrossRef] [PubMed]
- Stotani, S.; Giordanetto, F.; Medda, F. DYRK1A inhibition as potential treatment for Alzheimer’s Disease. Future Med. Chem. 2016, 8, 681–696. [Google Scholar] [CrossRef]
- Melchior, B.; Mittapalli, G.K.; Lai, C.; Duong-Polk, K.; Stewart, J.; Güner, B.; Hofilena, B.; Tjitro, A.; Anderson, S.D.; Herman, D.S.; et al. Tau Pathology Reduction with SM07883, a Novel, Potent, and Selective Oral DYRK1A Inhibitor: A potential therapeutic for Alzheimer’s Disease. Aging Cell 2019, 18, e13000. [Google Scholar] [CrossRef]
- Lee, Y.H.; Im, E.; Hyun, M.; Park, J.; Chung, K.C. Protein phosphatase PPM1B inhibits DYRK1A Kinase through dephosphorylation of PS258 and reduces toxic Tau aggregation. J. Biol. Chem. 2021, 296, 100245. [Google Scholar] [CrossRef]
- Yin, X.; Jin, N.; Gu, J.; Shi, J.; Zhou, J.; Gong, C.X.; Iqbal, K.; Grundke-Iqbal, I.; Liu, F. Dual-specificity tyrosine phosphorylation-regulated Kinase 1A (Dyrk1A) modulates serine/arginine-rich protein 55 (SRp55)-promoted Tau Exon 10 inclusion. J. Biol. Chem. 2012, 287, 30497–30506. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and treatment of Parkinson Disease: A Review. JAMA J. Am. Med. Assoc. 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Young, J.J.; Lavakumar, M.; Tampi, D.; Balachandran, S.; Tampi, R.R. Frontotemporal Dementia: Latest evidence and clinical implications. Ther. Adv. Psychopharmacol. 2018, 8, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, A. Frontotemporal dementia, Pick disease, and corticobasal degeneration: One entity or 3? Arch. Neurol. 1997, 54, 1427–1429. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef] [PubMed]
- Kargbo, R.B. Selective DYRK1A inhibitor for the treatment of neurodegenerative diseases: Alzheimer, Parkinson, Huntington, and Down Syndrome. ACS Med. Chem. Lett. 2020, 11, 1795–1796. [Google Scholar] [CrossRef]
- Ferrer, I.; Barrachina, M.; Puig, B.; Martínez De Lagrán, M.; Martí, E.; Avila, J.; Dierssen, M. Constitutive Dyrk1A is abnormally expressed in Alzheimer Disease, Down Syndrome, Pick Disease, and related transgenic models. Neurobiol. Dis. 2005, 20, 392–400. [Google Scholar] [CrossRef]
- Barallobre, M.J.; Perier, C.; Bové, J.; Laguna, A.; Delabar, J.M.; Vila, M.; Arbonés, M.L. DYRK1A promotes dopaminergic neuron survival in the developing brain and in a mouse model of Parkinson’s Disease. Cell Death Dis. 2014, 5, e1289. [Google Scholar] [CrossRef]
- Chiu, C.C.; Yeh, T.H.; Chen, R.S.; Chen, H.C.; Huang, Y.Z.; Weng, Y.H.; Cheng, Y.C.; Liu, Y.C.; Cheng, A.J.; Lu, Y.C.; et al. Upregulated expression of MicroRNA-204-5p leads to the death of dopaminergic cells by targeting DYRK1A-mediated apoptotic signaling cascade. Front. Cell. Neurosci. 2019, 13, 399. [Google Scholar] [CrossRef]
- Feki, A.; Hibaoui, Y. DYRK1A protein, a promising therapeutic target to improve cognitive deficits in Down Syndrome. Brain Sci. 2018, 8, 187. [Google Scholar] [CrossRef]
- Demuro, S.; di Martino, R.M.C.; Ortega, J.A.; Cavalli, A. Gsk-3β, Fyn, and Dyrk1a: Master regulators in neurodegenerative pathways. Int. J. Mol. Sci. 2021, 22, 9098. [Google Scholar] [CrossRef]
- Deshpande, A.D.; Harris-Hayes, M.; Schootman, M. Epidemiology of Diabetes and Diabetes-related complications. Phys. Ther. 2008, 88, 1254–1264. [Google Scholar] [CrossRef]
- Arneth, B.; Arneth, R.; Shams, M. Metabolomics of Type 1 and Type 2 Diabetes. Int. J. Mol. Sci. 2019, 20, 2467. [Google Scholar] [CrossRef] [PubMed]
- Ackeifi, C.; Wang, P.; Karakose, E.; Manning Fox, J.E.; González, B.J.; Liu, H.; Wilson, J.; Swartz, E.; Berrouet, C.; Li, Y.; et al. GLP-1 receptor agonists synergize with DYRK1A inhibitors to potentiate functional human β cell regeneration. Sci. Transl. Med. 2020, 12, 9996. [Google Scholar] [CrossRef]
- Scavuzzo, M.A.; Borowiak, M. Two drugs converged in a pancreatic β cell. Sci. Transl. Med. 2020, 12, eaba7359. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.K. Small-molecule discovery in the pancreatic beta cell. Curr. Opin. Chem. Biol. 2022, 68, 102150. [Google Scholar] [CrossRef] [PubMed]
- Pucelik, B.; Barzowska, A.; Dąbrowski, J.M.; Czarna, A. Diabetic kinome inhibitors—A new opportunity for β-cells restoration. Int. J. Mol. Sci. 2021, 22, 9083. [Google Scholar] [CrossRef]
- Barzowska, A.; Pucelik, B.; Pustelny, K.; Matsuda, A.; Martyniak, A.; Stępniewski, J.; Maksymiuk, A.; Dawidowski, M.; Rothweiler, U.; Dulak, J.; et al. DYRK1A Kinase inhibitors promote β-cell survival and insulin homeostasis. Cells 2021, 10, 2263. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Gilliland, D.G.; Jordan, C.T.; Felix, C.A. The molecular basis of Leukemia. Hematology 2004, 2004, 80–97. [Google Scholar] [CrossRef]
- Rammohan, M.; Harris, E.; Bhansali, R.S.; Zhao, E.; Li, L.S.; Crispino, J.D. The chromosome 21 Kinase DYRK1A: Emerging roles in cancer biology and potential as a therapeutic target. Oncogene 2022, 41, 2003–2011. [Google Scholar] [CrossRef]
- Baek, K.H.; Zaslavsky, A.; Lynch, R.C.; Britt, C.; Okada, Y.; Siarey, R.J.; Lensch, M.W.; Park, I.H.; Yoon, S.S.; Minami, T.; et al. Down’s Syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature 2009, 459, 1126–1130. [Google Scholar] [CrossRef]
- Malinge, S.; Bliss-Moreau, M.; Kirsammer, G.; Diebold, L.; Chlon, T.; Gurbuxani, S.; Crispino, J.D. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes Megakaryoblastic Leukemia in a murine model of Down Syndrome. J. Clin. Investig. 2012, 122, 948–962. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F.; Shkreta, L.; Chabot, B.; Durantel, D.; Salvetti, A. Interplay Between CMGC Kinases Targeting SR proteins and viral replication: Splicing and beyond. Front. Microbiol. 2021, 12, 658721. [Google Scholar] [CrossRef] [PubMed]
- Kisaka, J.K.; Ratner, L.; Kyei, G.B. The dual-specificity Kinase DYRK1A modulates the levels of cyclin L2 to control HIV replication in macrophages. J. Virol. 2020, 94, e01583-19. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.J.; Chang, H.S.; Wang, C.Y.; Yu, W.C.Y. DYRK1A stabilizes HPV16E7 oncoprotein through phosphorylation of the Threonine 5 and Threonine 7 residues. Int. J. Biochem. Cell Biol. 2008, 40, 2431–2441. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Alfajaro, M.M.; DeWeirdt, P.C.; Hanna, R.E.; Lu-Culligan, W.J.; Cai, W.L.; Strine, M.S.; Zhang, S.M.; Graziano, V.R.; Schmitz, C.O.; et al. Genome-wide CRISPR screens reveal host factors critical for SARS-CoV-2 infection. Cell 2021, 184, 76–91.e13. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart disease and stroke statistics-2019 update: A report from the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Hille, S.; Dierck, F.; Kühl, C.; Sosna, J.; Adam-Klages, S.; Adam, D.; Lüllmann-Rauch, R.; Frey, N.; Kuhn, C. Dyrk1a regulates the cardiomyocyte cell cycle via D-Cyclin-Dependent Rb/E2f-signalling. Cardiovasc. Res. 2016, 110, 381–394. [Google Scholar] [CrossRef]
- Liu, T.; Wang, Y.; Wang, J.; Ren, C.; Chen, H.; Zhang, J. DYRK1A Inhibitors for Disease Therapy: Current status and perspectives. Eur. J. Med. Chem. 2022, 229, 114062. [Google Scholar] [CrossRef]
- Weber, C.; Sipos, M.; Paczal, A.; Balint, B.; Kun, V.; Foloppe, N.; Dokurno, P.; Massey, A.J.; Walmsley, D.L.; Hubbard, R.E.; et al. Structure-guided discovery of potent and selective DYRK1A inhibitors. J. Med. Chem. 2021, 64, 6745–6764. [Google Scholar] [CrossRef]
- Smith, B.; Medda, F.; Gokhale, V.; Dunckley, T.; Hulme, C. Recent advances in the design, synthesis, and biological evaluation of selective DYRK1A inhibitors: A new avenue for a disease modifying treatment of Alzheimers? ACS Chem. Neurosci. 2012, 3, 857–872. [Google Scholar] [CrossRef]
- AlNajjar, Y.T.; Gabr, M.; ElHady, A.K.; Salah, M.; Wilms, G.; Abadi, A.H.; Becker, W.; Abdel-Halim, M.; Engel, M. Discovery of novel 6-Hydroxybenzothiazole Urea Derivatives as Dual Dyrk1A/α-Synuclein aggregation inhibitors with neuroprotective effects. Eur. J. Med. Chem. 2022, 227, 113911. [Google Scholar] [CrossRef] [PubMed]
- Shahroz, M.M.; Sharma, H.K.; Altamimi, A.S.A.; Alamri, M.A.; Ali, A.; Ali, A.; Alqahtani, S.; Altharawi, A.; Alabbas, A.B.; Alossaimi, M.A.; et al. Novel and potential small molecule scaffolds as DYRK1A inhibitors by integrated molecular docking-based virtual screening and dynamics simulation study. Molecules 2022, 27, 1159. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.; Fruit, C.; Hérault, Y.; Meijer, L.; Besson, T. Dual-specificity Tyrosine Phosphorylation-Regulated Kinase 1A (DYRK1A) inhibitors: A survey of recent patent literature. Expert Opin. Ther. Pat. 2017, 27, 1183–1199. [Google Scholar] [CrossRef] [PubMed]
- Neumann, F.; Gourdain, S.; Albac, C.; Dekker, A.D.; Bui, L.C.; Dairou, J.; Schmitz-Afonso, I.; Hue, N.; Rodrigues-Lima, F.; Delabar, J.M.; et al. DYRK1A inhibition and cognitive rescue in a Down Syndrome mouse model are induced by new Fluoro-DANDY derivatives. Sci. Rep. 2018, 8, 2859. [Google Scholar] [CrossRef]
- Guard, S.E.; Poss, Z.C.; Ebmeier, C.C.; Pagratis, M.; Simpson, H.; Taatjes, D.J.; Old, W.M. The nuclear interactome of DYRK1A reveals a functional role in DNA damage repair. Sci. Rep. 2019, 9, 6539. [Google Scholar] [CrossRef]
- Lan, B.; Zeng, S.; Zhang, S.; Ren, X.; Xing, Y.; Kutschick, I.; Pfeffer, S.; Frey, B.; Britzen-Laurent, N.; Grützmann, R.; et al. CRISPR-Cas9 screen identifies DYRK1A as a target for radiotherapy sensitization in pancreatic cancer. Cancers 2022, 14, 326. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–221. [Google Scholar] [CrossRef]
- Da Costa Martins, P.A.; Salic, K.; Gladka, M.M.; Armand, A.S.; Leptidis, S.; el Azzouzi, H.; Hansen, A.; Coenen-De Roo, C.J.; Bierhuizen, M.F.; van der Nagel, R.; et al. MicroRNA-199b targets the nuclear Kinase Dyrk1a in an auto-amplification loop promoting Calcineurin/NFAT signalling. Nat. Cell Biol. 2010, 12, 1220–1227. [Google Scholar] [CrossRef]
- Zhang, S.; Cheng, Z.; Wang, Y.; Han, T. The risks of MiRNA therapeutics: In a drug target perspective. Drug Des. Dev. Ther. 2021, 15, 721–733. [Google Scholar] [CrossRef]
- He, Y.; Khan, S.; Huo, Z.; Lv, D.; Zhang, X.; Liu, X.; Yuan, Y.; Hromas, R.; Xu, M.; Zheng, G.; et al. Proteolysis targeting chimeras (PROTACs) are emerging therapeutics for hematologic malignancies. J. Hematol. Oncol. 2020, 13, 1–24. [Google Scholar] [CrossRef]
- Menon, V.; Ananthapadmanabhan, V.; Swanson, S.; Saini, S.; Sesay, F.; Yakovlev, V.; Florens, L.; DeCaprio, J.A.; Washburn, M.; Dozmorov, M.; et al. DYRK1A Regulates the Recruitment of 53BP1 to the Sites of DNA damage in part through interaction with RNF169. Cell Cycle 2019, 18, 531–551. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Symbols | Full Names | Functions | Effects | Human Diseases | References |
|---|---|---|---|---|---|
| SRDF7/9G8 | / | I, S | Inhibition of Tau exon 10 inclusion promotion, imbalance of 3R-tau and 4R-tau expression and neurofibrillary degeneration | Down syndrome, Alzheimer’s and Parkinson’s disease, Fronto-temporal dementia with Parkinsonism linked to chromosome 17 | [39] |
| AMPH | Amphisin | I, S | Depolarization and polarization of isolated synaptosomes and regulation of general neuronal plasticity | Down syndrome (potential) | [40] |
| APP | Amyloid precursor protein | I, S | Accumulation of β- amyloid peptides (Aβ) in plaques, interference with nMDA receptor function, abnormal calcium influx and neuronal oxidative stress and activation of GSK-3β | Down syndrome, Alzheimer’s disease | [41] |
| RAD54L2/Arip4 | Androgen Receptor Interacting Protein 4 | I | Changes in the homeostasis of steroid hormone-controlled cellular events | Down syndrome (potential) | [42] |
| SRSF1/ASF | Alternative splicing factor | I, S | Increase in 3R-tau level, tau hyperphosphorylation and aggregation in neurofibrillary | Down syndrome, Alzheimer’s, and Parkinson’s disease | [43] |
| B-Raf | Braf transforming gene | I | Inhibition of neuroprogenitor cells proliferation and premature differentiation | Down syndrome (potential) | [44] |
| c-MET | / | I | Pancreatic malignant cell proliferation | Pancreatic ductal adenocarcinoma, lung cancer | [45,46] |
| CASP-9 | Cystein aspartyl protease Caspase 9 | I, S | Increased retinal size and abnormal retinal function, apoptosis | Down syndrome | [47,48] |
| CDC23 | / | I, S | Degradation of cyclin B, deactivation of CDK1 and retinoblastoma cell proliferation | Glioblastoma | [49] |
| CREB1 | cAMP response element-binding protein | I, S | Inhibition of hippocampal progenitor cells differentiation | Down syndrome (potential) | [46] |
| Ccnd 1,2 & 3 Ccln L2 | Cyclin D1, D2 and D3 Cyclin L2 | I, S | Cardiomyocyte proliferation and premature differentiation, inhibition of transcription factors and arresting cell cycle Phosphorylation of Cyclin L2 and cellular degradation | Cardiomyopathy and heart failure associated with Down syndrome, cancer (tumorsupressor) Viral infection (HIV-1) | [24,50] |
| Dcaf7 | DDB1 and CUL4 associated factor 7 | I | Huntingtin-associated protein 1 association reduction and growth retardation Mediation of the interaction of the adenovirus E1A oncoprotein | Down syndrome (potential), viral infection | [51,52] |
| DREAM complex | / | I | Transcription inhibition of cell cycle genes in the G0/G1 phase Proliferation of human pancreatic beta cells | Down syndrome (potential), ovarian cancer, diabetes | [53,54,55] |
| EGFR | Epidermal growth factor receptor | I | Malignant cell proliferation | Lung cancer, glioblastoma | [56,57] |
| FOXO1 | Forkhead transcription factor FKH R | I, S | Disruption of DNA damage, ROS regulation and cell death in leukemic B cells | Leukemia | [58,59] |
| GLI1 | Glioma-associated oncogene 1 | I, S | Cell growth promotion, differentiation, and tissue patterning | Cancer (oncogene) | [60] |
| GluN2A | Glutamate receptor, ionotropic, NMDA2A | I, S | Synaptic alteration | Down syndrome | [61] |
| GSK-3β | Glycogen synthase kinase 3beta | I, S | Downregulation of Nrf2, disequilibrium between cellular oxidants and the antioxidative processes, phosphorylation of α-synuclein and adipogenic proteins expression reduction | Alzheimer’s and Parkinson’s disease, obesity (potential) | [62] |
| HAP1 | Huntingtin interacting protein 1 | I | Dcaf7 association reduction and hypothalamus growth retardation, neuronal differentiation inhibition and cell death | Down syndrome (potential) | [51,63] |
| HP1 | Heterochromatin protein 1 | I, S | Repression of HP-mediated transcription and abnormal activation of cytokine genes | Down syndrome-associated megakaryoblastic leukemia | [64,65] |
| ID2 | / | I, S | Destabilization of transcription factors, loss of gliomna stemness and inhibition of tumour growth | Cancer and glioblastoma | [66] |
| MEK | Dual specificity mitogen-activated protein kinase | I | Inhibition of neuroprogenitor cells proliferation and premature differentiation | Down syndrome (potential) | [44] |
| NAFTc | Nuclear factor of activated T cells | I | Angiogenesis promotion, neuroprogenitor cells proliferation inhibition and β-cell proliferation attenuation | Diabetes, heart diseases, cancer (oncogene), viral infection and Down Syndrome | [30,67,68,69] |
| MME/NEP | Neprilysin | I | Accumulation of β- amyloid peptides (Aβ) in plaques | Alzheimer’s disease | [70] |
| Notch | Notch Signaling Pathway | I, S | Neural cells signaling attenuation | Down syndrome (potential) | [31] |
| NRSF/REST | RE1-silencing transcription factor | I | Inhibition of cells proliferation and differentiation | Cancer (tumorsupressor), MRD7 (potential) | [71] |
| CDKN1B/P27 | / | I, S | Inhibition of neuroprogenitor cells proliferation and premature differentiation | Down syndrome (potential), cancer (tumorsupressor) | [50] |
| P53 | Transformation related protein53 | I, S | Neuronal proliferation, increase of p21 expression, cell cycle arrest or apoptosis | Cancer (tumorsupressor) and Down syndrome (potential) | [15,72] |
| PAHX-AP1 | Phytanoyl-CoA α-hydroxylase-associated protein 1 | I, S | Facilitate DYRK1A-CREB interaction and development of neurological abnormalities | Down syndrome (potential) | [33] |
| Prkn | Parkin | I, S | E3 ubiquitin ligase activity and neuronal protection inhibition and loss of dopaminergic neurons | Parkinson’s disease | [73] |
| PS1 | Presenilin1 | I, S | Increase γ-secretase activity and accumulation of β- amyloid peptides (Aβ) in plaques | Down syndrome and Alzheimer’s disease | [74] |
| RCAN1 | Regulator of calcineurin 1 | I | Dysregulation of calcineurin, inhibition of signaling pathways that are controlled by NFAT and thus tau dysregulation, neuronal apoptosis, cell proliferation and development, etc. | Down syndrome, Alzheimer’s disease, and cancer | [75,76] |
| SRSF2/SC35 | Splicing factor 35 | I, S | Dysregulation of tau exon 10 splicing, imbalance of 3R-tau and 4R-tau expression and neurofibrillary degeneration | Down syndrome, Alzheimer’s, and Parkinson’s disease | [32] |
| SEPT4 | Septin 4 | I, S | Accumulation of β- amyloid peptides (Aβ) in plaques, tau self-aggregation and fibrillation, aggregation/inclusion formation of α-synuclein (Lewy bodies), loss of dopaminergic neurons. | Down syndrome (potential), Alzheimer’s, and Parkinson’s diseases (potential) | [77] |
| SIRT1 | Sirtuin 1 | I, S | Cell apoptosis inhibition | Down syndrome (potential), cancer (oncogene) | [19] |
| STAT3 | / | I, S | Disruption of DNA damage, ROS regulation and cell death in leukemic B cells | Leukemia | [57,58] |
| τ/Tau | Tau protein | I, S | Reduction of tau biological activity and tau self-aggregation and fibrillation | Alzheimer’s disease, Parkinson’s disease, Fronto-temporal dementia | [78] |
| TOM70 | Translocase of the outer mambrane | I | Decrease in import capacity of metabolite carriers and critical problem in mitochondrial machinery | Metabolic diseases, MRD7 and Down syndrome (potential) | [79] |
| TRAF3 | I, S | Degradation of the noncanonical nuclear factor (NF)-κB–inducing kinase (NIK) | Autoimmune disease and leukemia (potential) | [80] | |
| α-syn | α-synuclein | I, S | Aggregation/inclusion formation of α-synuclein (Lewy bodies) and loss of dopaminergic neurons | Parkinson’s disease | [81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deboever, E.; Fistrovich, A.; Hulme, C.; Dunckley, T. The Omnipresence of DYRK1A in Human Diseases. Int. J. Mol. Sci. 2022, 23, 9355. https://doi.org/10.3390/ijms23169355
Deboever E, Fistrovich A, Hulme C, Dunckley T. The Omnipresence of DYRK1A in Human Diseases. International Journal of Molecular Sciences. 2022; 23(16):9355. https://doi.org/10.3390/ijms23169355
Chicago/Turabian StyleDeboever, Estelle, Alessandra Fistrovich, Christopher Hulme, and Travis Dunckley. 2022. "The Omnipresence of DYRK1A in Human Diseases" International Journal of Molecular Sciences 23, no. 16: 9355. https://doi.org/10.3390/ijms23169355
APA StyleDeboever, E., Fistrovich, A., Hulme, C., & Dunckley, T. (2022). The Omnipresence of DYRK1A in Human Diseases. International Journal of Molecular Sciences, 23(16), 9355. https://doi.org/10.3390/ijms23169355