
Molecular Epidemiology and Virulence of Non-Typhoidal Salmonella in Armenia

 , , and
, , and 
Abstract
1. Introduction
2. Results
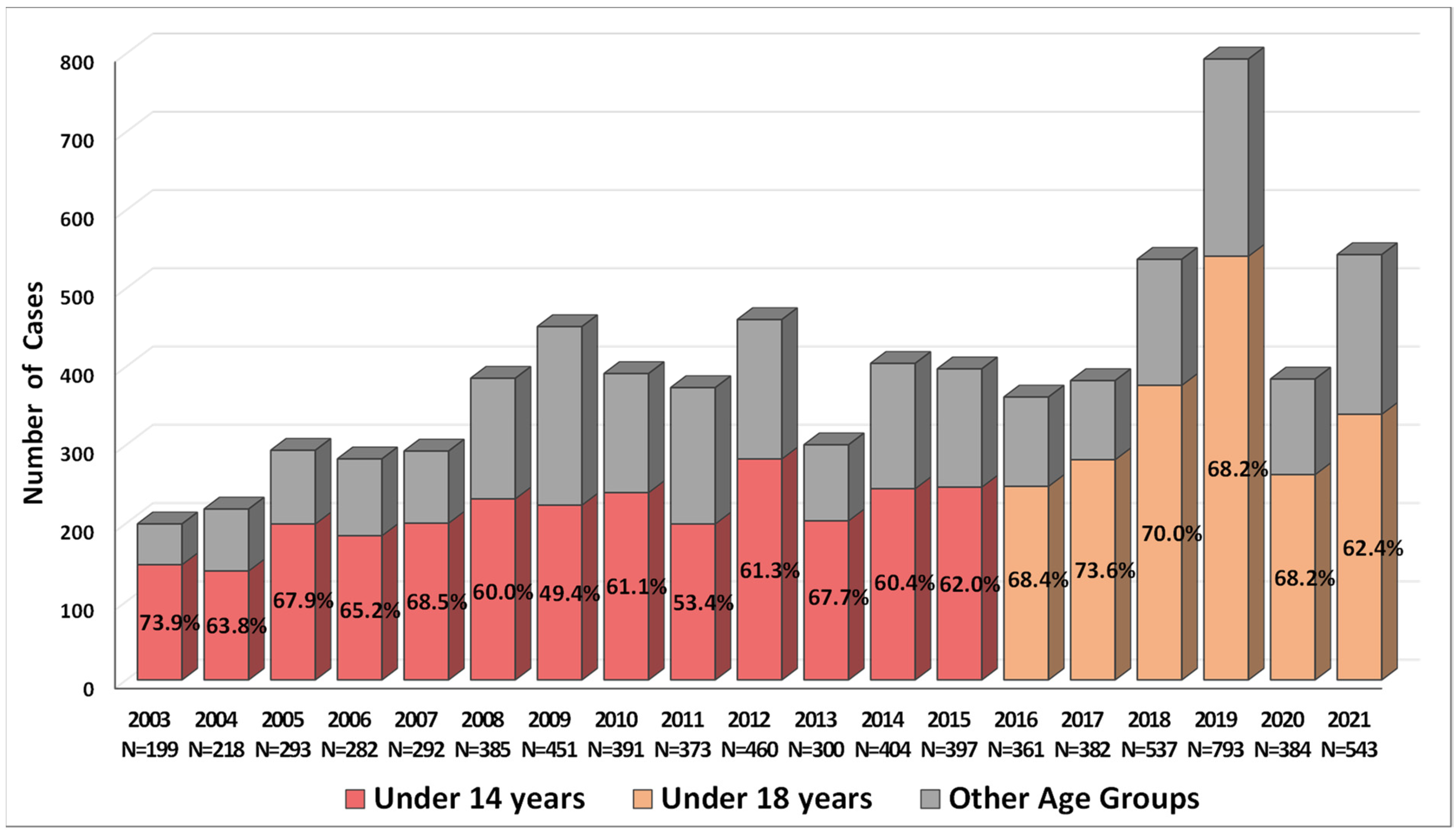
2.1. Incidence of Salmonellosis in Armenia in 2003–2021
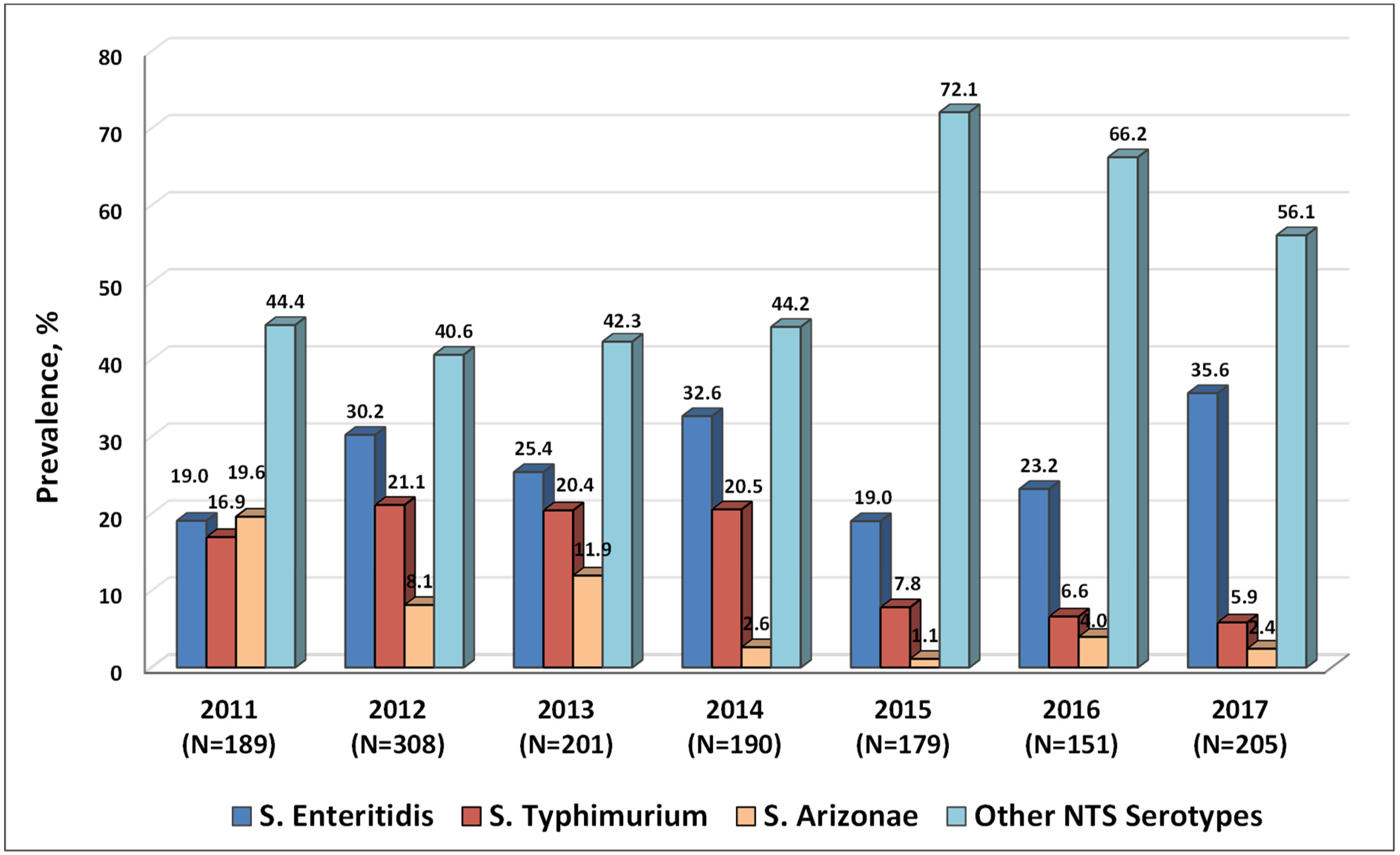
2.2. Serotypes of NTS Circulating in Armenia in 2011–2017
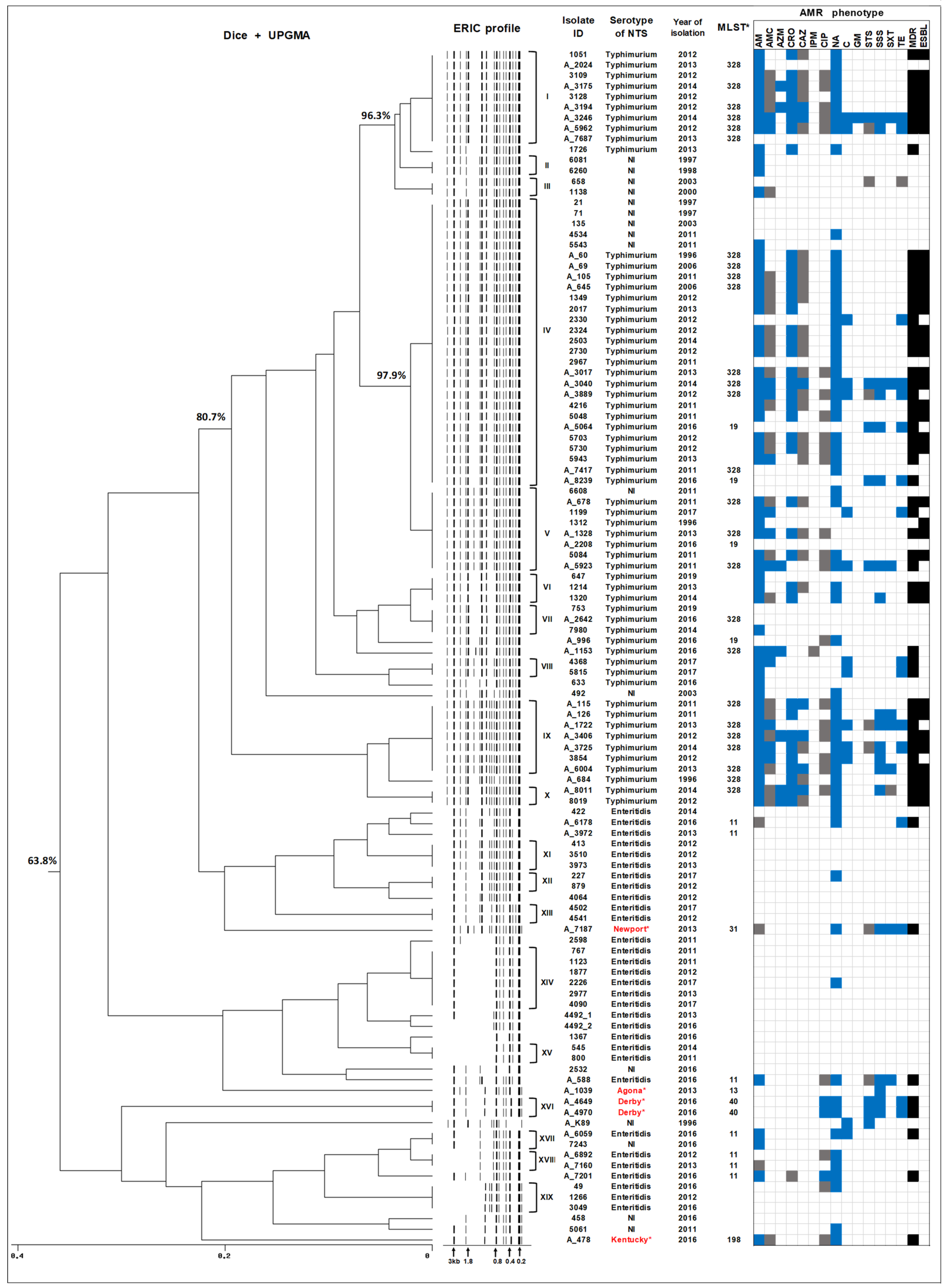
2.3. ERIC-PCR Subtyping of Human NTS Isolates Circulating in Armenia
2.4. Whole Genome Sequencing of Human NTS Isolates
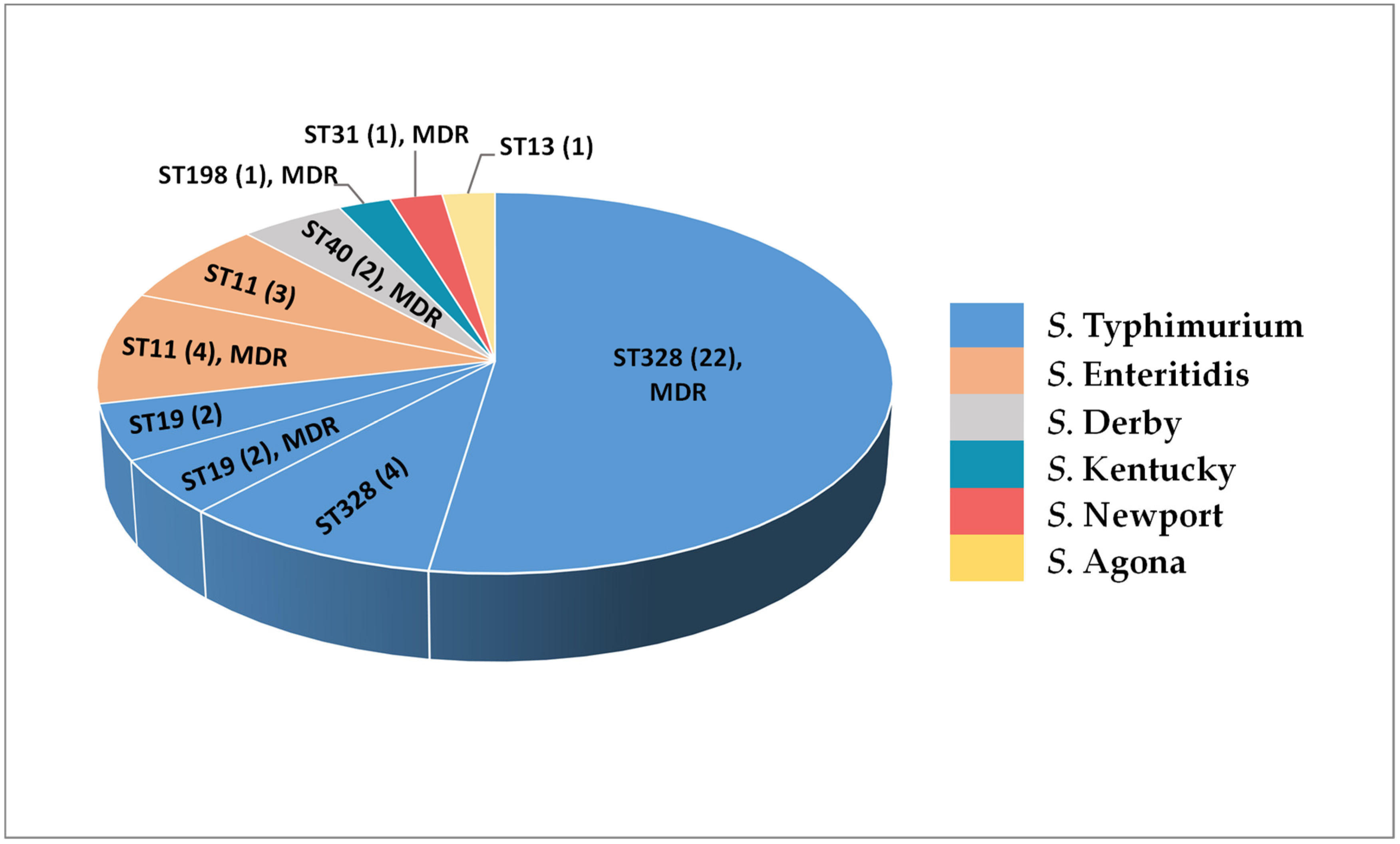
2.5. In Silico Serotyping and MLST Analysis of NTS Isolates
2.6. Virulence-Related Traits and Their Genetic Constituents in NTS Isolates
2.6.1. Virulence Genes
2.6.2. Salmonella Pathogenicity Islands
2.6.3. Salmonella Virulence Plasmids
2.6.4. Prophages Carrying Virulence-Related Genes
3. Discussion
4. Materials and Methods
4.1. Human Isolates of NTS
4.2. Antimicrobial Susceptibility Testing
4.3. Bacterial DNA Extraction
4.4. ERIC-PCR Typing/Analysis
4.5. Whole Genome Sequencing of NTS Isolates
4.6. Bioinformatics Analyses
4.7. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brenner, F.; Villar, R.; Angulo, F.; Tauxe, R.; Swaminathan, B. Salmonella nomenclature. J. Clin. Microbiol. 2000, 38, 2465–2467. [Google Scholar] [CrossRef] [PubMed]
- Pearce, M.E.; Langridge, G.C.; Lauer, A.C.; Grant, K.; Maiden, M.; Chattaway, M.A. An evaluation of the species and subspecies of the genus Salmonella with whole genome sequence data: Proposal of type strains and epithets for novel S. enterica subspecies VII, VIII, IX, X and XI. Genomics 2021, 113, 3152–3162. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.A.; Eade, C.R.; Wiedmann, M. Embracing Diversity: Differences in Virulence Mechanisms, Disease Severity, and Host Adaptations Contribute to the Success of Nontyphoidal Salmonella as a Foodborne Pathogen. Front. Microbiol. 2019, 10, 1368. [Google Scholar] [CrossRef] [PubMed]
- Havelaar, A.H.; Kirk, M.D.; Torgerson, P.R.; Gibb, H.J.; Hald, T.; Lake, R.J.; Praet, N.; Bellinger, D.C.; de Silva, N.R.; Gargouri, N.; et al. World Health Organization global estimates and regional comparisons of the burden of foodborne disease in 2010. PLoS Med. 2015, 12, e1001923. [Google Scholar] [CrossRef]
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.-A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States—Major pathogens. Emerg. Infect. Dis. 2011, 17, 7–15. [Google Scholar] [CrossRef]
- OzFoodNet Working Group. Monitoring the incidence and causes of diseases potentially transmitted by food in Australia: Annual report of the OzFoodNet network, 2011. Commun. Dis. Intell. Q. Rep. 2015, 39, E236–E264. [Google Scholar] [PubMed]
- Centers for Disease Control and Prevention [CDC]. National Enteric Disease Surveillance: Salmonella Annual Report; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2016.
- European Food Safety Authority [EFSA]; European Centre for Disease Prevention, and Control [ECDC]. The European Union summary report on trends, and sources of zoonoses, zoonotic agents, and food-borne outbreaks in 2016. EFSA J. 2017, 15, e05077. [Google Scholar] [CrossRef]
- Stevens, M.P.; Humphrey, T.J.; Maskell, D.J. Molecular insights into farm animal and zoonotic Salmonella infections. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 2709–2723. [Google Scholar] [CrossRef]
- Stevens, M.P.; Kingsley, R.A. Salmonella pathogenesis and host-adaptation in farmed animals. Curr. Opin. Microbiol. 2021, 63, 52–58. [Google Scholar] [CrossRef]
- Chu, C.; Chiu, C.H. Evolution of the virulence plasmids of non-typhoid Salmonella and its association with antimicrobial resistance. Microbes Infect. 2006, 8, 1931–1936. [Google Scholar] [CrossRef]
- Rychlik, I.; Gregorova, D.; Hradecka, H. Distribution and function of plasmids in Salmonella enterica. Vet. Microbiol. 2006, 112, 1–10. [Google Scholar] [CrossRef]
- Guerra, B.; Soto, S.; Helmuth, R.; Mendoza, M.C. Characterization of a self-transferable plasmid from Salmonella enterica serotype Typhimurium clinical isolates carrying two integron-borne gene cassettes together with virulence and drug resistance genes. Antimicrob. Agents Chemother. 2009, 46, 2977–2981. [Google Scholar] [CrossRef]
- Dos Santos, A.M.P.; Ferrari, R.G.; Conte-Junior, C.A. Virulence Factors in Salmonella Typhimurium: The Sagacity of a Bacterium. Curr. Microbiol. 2019, 76, 762–773. [Google Scholar] [CrossRef]
- Rivera-Chavez, F.; Baumler, A.J. The pyromaniac inside you: Salmonella metabolism in the host gut. Annu. Rev. Microbiol. 2015, 69, 31–48. [Google Scholar] [CrossRef]
- Cheng, R.A.; Orsi, R.H.; Wiedmann, M. The Number and Type of Chaperone-Usher Fimbriae Reflect Phylogenetic Clade Rather than Host Range in Salmonella. mSystems 2022, 7, e0011522. [Google Scholar] [CrossRef]
- Varteresian, I.; Gevorkian, Z.; Sarkisian, N.; Sedrakian, A.; Arakelova, K.; Ktsoian, Z. Conjugative plasmids of Salmonella strains resistant to antibiotics. J. Microbiol. Epidemiol. Immunol. 2002, 1, 78–79. (In Russian) [Google Scholar]
- Gevorgyan, Z.U.; Ktsoyan, L.A.; Sarkisyan, N.N.; Sedrakian, A.M.; Arakelova, K.A.; Ktsoyan, Z.A.; Asoyan, A.V.; Kazaryan, K.A.; Karageuzyan, K.G.; Aminov, R.I. Clinical presentation and immune status of children infected by multi-drug-resistant Salmonella strains. Electron. J. Nat. Sci. 2004, 1, 46–50. [Google Scholar]
- Sedrakyan, A.M.; Mnacakanyan, A.A.; Gevorgyan, Z.U.; Arakelova, K.A. Monitoring of Antibiotic Resistance of Salmonella Strains Circulating in Armenia. Rep. NAS RA 2007, 107, 87–93. Available online: http://elib.sci.am/2007_1/12_1_2007.pdf (accessed on 15 May 2022). (In Russian).
- Sedrakyan, A.; Arakelova, K.; Gevorgyan, Z.; Mnatsakanyan, A.; Zakharyan, M.; Hovhannisyan, A.; Asoyan, A.; Ktsoyan, Z. Multidrug resistance in clinical isolates of epidemiologically important serotypes of nontyphoidal Salmonella. Biol. J. Armen. 2013, 65, 13–19. Available online: http://arar.sci.am/Content/238111/file_0.pdf (accessed on 15 May 2022). (In Russian).
- Sedrakyan, A.M.; Arakelova, K.A.; Zakaryan, M.K.; Hovhanisyan, A.I.; Asoyan, A.V.; Gevorkyan, Z.U.; Mnatsakanyan, A.A.; Ktsyan, Z.A.; Boyajyan, A.A.; Aminov, R. Multidrug-resistance and presence of class 1 integrons in clinical isolates of Salmonella enterica serotype Enteritidis, circulating in Armenia. Russ. J. Infect. Immun. 2013, 3, 355–358. [Google Scholar] [CrossRef]
- Sedrakyan, A.M.; Ktsoyan, Z.A.; Arakelova, K.A.; Zakharyan, M.K.; Hovhannisyan, A.I.; Gevorgyan, Z.U.; Mnatsakanyan, A.A.; Kakabadze, E.G.; Makalatia, K.B.; Chanishvili, N.A.; et al. Extended-Spectrum β-Lactamases in Human Isolates of Multidrug-Resistant Non-typhoidal Salmonella enterica. Front. Microbiol. 2020, 11, 592223. [Google Scholar] [CrossRef] [PubMed]
- Statistical Committee of the Republic of Armenia/Socio-Economic Situation of RA, January–December/5.3. Public Health. Available online: https://www.armstat.am/en/?nid=82 (accessed on 23 June 2022).
- McClelland, M.; Sanderson, K.E.; Spieth, J.; Clifton, S.W.; Latreille, P.; Courtney, L.; Porwollik, S.; Ali, J.; Dante, M.; Du, F.; et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 2001, 413, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yin, Y.; Jones, M.B.; Zhang, Z.; Deatherage Kaiser, B.L.; Dinsmore, B.A.; Fitzgerald, C.; Fields, P.I.; Deng, X. Salmonella serotype determination utilizing high-throughput genome sequencing data. J. Clin. Microbiol. 2015, 53, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; den Bakker, H.C.; Li, S.; Chen, J.; Dinsmore, B.A.; Lane, C.; Lauer, A.C.; Fields, P.I.; Deng, X. SeqSero2: Rapid and improved Salmonella serotype determination using whole-genome sequencing data. Appl. Environ. Microbiol. 2019, 85, e01746-19. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications [version 1; referees: 2 approved]. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.D.; Jin, Q.; Chen, L.H.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Roer, L.; Hendriksen, R.S.; Leekitcharoenphon, P.; Lukjancenko, O.; Kaas, R.S.; Hasman, H.; Aarestrup, F.M. Is the Evolution of Salmonella enterica subsp. enterica Linked to Restriction-Modification Systems? mSystems 2016, 1, e00009-16. [Google Scholar] [CrossRef]
- Petermann, S.R.; Sherwood, J.S.; Logue, C.M. The Yersinia high pathogenicity island is present in Salmonella enterica Subspecies I isolated from turkeys. Microb. Pathog. 2008, 45, 110–114. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Lim, J.M.; Kwon, S.J.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Jackson, B.R.; Griffin, P.M.; Cole, D.; Walsh, K.A.; Chai, S.J. Outbreak-associated Salmonella enterica Serotypes and Food Commodities, United States, 1998–2008. Emerg. Infect. Dis. 2013, 19, 1239–1244. [Google Scholar] [CrossRef]
- Baumler, A.J.; Hargis, B.M.; Tsolis, R.M. Tracing the origins of Salmonella outbreaks. Science 2000, 287, 50–52. [Google Scholar] [CrossRef]
- Rodrigue, D.C.; Tauxe, R.V.; Rowe, B. International increase in Salmonella enteritidis: A new pandemic? Epidemiol. Infect. 1990, 105, 21–27. [Google Scholar] [CrossRef]
- Kobayashi, A.; Takahashi, S.; Ono, M.; Tanaka, K.; Kishima, M.; Akiba, M.; Uchida, I. Molecular typing of Salmonella enterica serovar Enteritidis isolates from food-producing animals in Japan by multilocus variable-number tandem repeat analysis: Evidence of clonal dissemination and replacement. Acta Vet. Scand. 2014, 56, 31. [Google Scholar] [CrossRef][Green Version]
- Matope, G.; Schlundt, J.; Makaya, P.V.; Aabo, L.; Baggesen, D.L. Salmonella enteritidis infection in poultry: An emerging zoonosis in Zimbabwe. Zimbabwe Vet. J. 1998, 29, 132–138. [Google Scholar] [CrossRef]
- Li, S.; He, Y.; Mann, D.A.; Deng, X. Global spread of Salmonella Enteritidis via centralized sourcing and international trade of poultry breeding stocks. Nat. Commun. 2021, 12, 5109. [Google Scholar] [CrossRef]
- Krauland, M.G.; Marsh, J.W.; Paterson, D.L.; Harrison, L.H. Integron-mediated Multidrug Resistance in a Global Collection of Nontyphoidal Salmonella enterica Isolates. Emerg. Infect. Dis. 2009, 15, 388–396. [Google Scholar] [CrossRef]
- Sun, H.; Wan, Y.; Du, P.; Bai, L. The Epidemiology of Monophasic Salmonella Typhimurium. Foodborne Pathog. Dis. 2020, 17, 87–97. [Google Scholar] [CrossRef]
- Edwards, R.A.; Schifferli, D.M.; Maloy, S.R. A role for Salmonella fimbriae in intraperitoneal infections. Proc. Natl. Acad. Sci. USA 2000, 97, 1258–1262. [Google Scholar] [CrossRef]
- Morales, C.A.; Guard, J.; Sanchez-Ingunza, R.; Shah, D.H.; Harrison, M. Virulence and metabolic characteristics of Salmonella enterica serovar Enteritidis strains with different sefD variants in hens. Appl. Environ. Microbiol. 2012, 78, 6405–6412. [Google Scholar] [CrossRef]
- Mikoleit, M.L. A WHO network building capacity to detect, control and prevent foodborne and other enteric infections from farm to table. In Laboratory Protocol: Biochemical Identification of Salmonella and Shigella Using an Abbreviated Panel of Tests; Enteric Diseases Laboratory Branch, Centers for Disease Control and Prevention: Atlanta, GA, USA, 2015; Available online: http://antimicrobialresistance.dk/CustomerData/Files/Folders/2-newsletter-pdf/20_07-gfn-biochem-v002-final-16oct2015.pdf (accessed on 26 June 2022).
- Grimont, P.A.D.; Weill, F.X. Antigenic Formulae of the Salmonella Serovars, 9th ed.; WHO Collaborating Centre for Reference and Research on Salmonella; Institut Pasteur: Paris, France, 2007; Available online: https://www.pasteur.fr/sites/default/files/veng_0.pdf (accessed on 26 June 2022).
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, 28th ed.; CLSI supplement M100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018; Available online: https://file.qums.ac.ir/repository/mmrc/CLSI-2018-M100-S28.pdf (accessed on 26 June 2022).
- CLSI. CLSI Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Tenth Edition; CLSI document M07-A10; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2015. [Google Scholar]
- Magiorakos, A.-P.; Srinivasan, A.; Carey, R.; Carmeli, Y.; Falagas, M.; Giske, C.; Harbarth, S.; Hindler, J.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef]
- Pui, C.F.; Wong, W.C.; Chai, L.C.; Nillian, E.; Ghazali, F.M.; Cheah, Y.K.; Nakaguchi, Y.; Nishibuchi, M.; Radu, S. Simultaneous detection of Salmonella spp., Salmonella Typhi and Salmonella Typhimurium in sliced fruits using multiplex PCR. Food Control 2011, 22, 337–342. [Google Scholar] [CrossRef]
- Versalovic, J.; Koeuth, T.; Lupski, R. Distribution of repetitive DNA sequences in eubacteria and application to fingerprinting of bacterial genomes. Nucleic Acids Res. 1991, 19, 6823–6831. [Google Scholar] [CrossRef]
- Colello, R.; Ruiz, M.J.; Padín, V.M.; Rogé, A.D.; Leotta, G.; Padola, N.L.; Etcheverría, A.I. Detection and Characterization of Salmonella Serotypes in the Production Chain of Two Pig Farms in Buenos Aires Province, Argentina. Front. Microbiol. 2018, 9, 1370. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Serotype of NTS (Number of Isolates) | Virulence Factor Class | |||||
|---|---|---|---|---|---|---|---|
| Typhimurium (n = 30) | Enteritidis (n = 7) | Derby (n = 2) | Agona (n = 1) | Kentucky (n = 1) | Newport (n = 1) | ||
| lpfABCDE | - | 7 | - | 1 | 1 | 1 | Fimbrial adherence determinants |
| peg | - | 7 | 2 | - | - | 1 | |
| pefABCD | 4 | 6 | - | - | - | - | |
| safA | 30 | 7 | 2 | - | 1 | 1 | |
| sefABCD | - | 7 | - | - | - | - | |
| staABCDEFG | - | - | 2 | 1 | - | - | |
| stcA | 30 | - | - | 1 | - | - | |
| stcB | 30 | - | - | 1 | 1 | - | |
| stcC | 30 | - | - | 1 | 1 | - | |
| stcD | 30 | - | - | 1 | - | - | |
| steABCDEF | - | 7 | 2 | 1 | 1 | 1 | |
| stjBC | 30 | - | - | 1 | 1 | 1 | |
| stkABCDEFG | - | - | - | - | 1 | - | |
| tcfABCD | - | - | - | - | 1 | - | |
| ratB | 30 | 7 | - | 1 | - | 1 | Nonfimbrial adherence determinants |
| sopE | - | 7 | - | - | - | 1 | T3SS1 translocated effector |
| gogB | 30 | - | - | - | - | - | T3SS2 translocated effector |
| sopD2 | - | 7 | 2 | 1 | 1 | 1 | |
| spvCD | 4 | 6 | - | - | - | - | |
| sseI/srfH | 30 | 7 | - | - | - | - | |
| sseK1 | 30 | 7 | 2 | 1 | 1 | - | |
| sseK2 | 30 | 7 | 2 | - | 1 | 1 | Macrophage inducible gene |
| mig-5 | 4 | 6 | - | - | - | - | |
| sodC1 | 30 | 7 | - | - | - | - | Stress adaptation |
| rck | 4 | 6 | - | - | - | - | Serum resistance |
| spvB | 4 | 6 | - | - | - | - | Toxin |
| upaG/ehaG | - | - | 2 | - | - | - | Autotransporter |
| ehaB | - | - | - | - | 1 | - | |
| faeCDEHIJ | - | - | - | - | 1 | - | Adherence/K88 fimbriae (Escherichia) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sedrakyan, A.; Ktsoyan, Z.; Arakelova, K.; Gevorgyan, Z.; Zakharyan, M.; Hakobyan, S.; Hovhannisyan, A.; Arakelyan, A.; Aminov, R. Molecular Epidemiology and Virulence of Non-Typhoidal Salmonella in Armenia. Int. J. Mol. Sci. 2022, 23, 9330. https://doi.org/10.3390/ijms23169330
Sedrakyan A, Ktsoyan Z, Arakelova K, Gevorgyan Z, Zakharyan M, Hakobyan S, Hovhannisyan A, Arakelyan A, Aminov R. Molecular Epidemiology and Virulence of Non-Typhoidal Salmonella in Armenia. International Journal of Molecular Sciences. 2022; 23(16):9330. https://doi.org/10.3390/ijms23169330
Chicago/Turabian StyleSedrakyan, Anahit, Zhanna Ktsoyan, Karine Arakelova, Zaruhi Gevorgyan, Magdalina Zakharyan, Shoghik Hakobyan, Alvard Hovhannisyan, Arsen Arakelyan, and Rustam Aminov. 2022. "Molecular Epidemiology and Virulence of Non-Typhoidal Salmonella in Armenia" International Journal of Molecular Sciences 23, no. 16: 9330. https://doi.org/10.3390/ijms23169330
APA StyleSedrakyan, A., Ktsoyan, Z., Arakelova, K., Gevorgyan, Z., Zakharyan, M., Hakobyan, S., Hovhannisyan, A., Arakelyan, A., & Aminov, R. (2022). Molecular Epidemiology and Virulence of Non-Typhoidal Salmonella in Armenia. International Journal of Molecular Sciences, 23(16), 9330. https://doi.org/10.3390/ijms23169330