
Sex as Biological Variable in Cardiac Mitochondrial Bioenergetic Responses to Acute Stress


Abstract
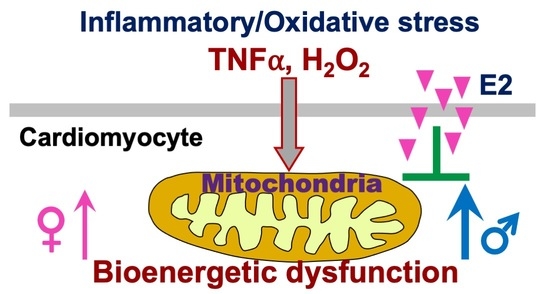
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
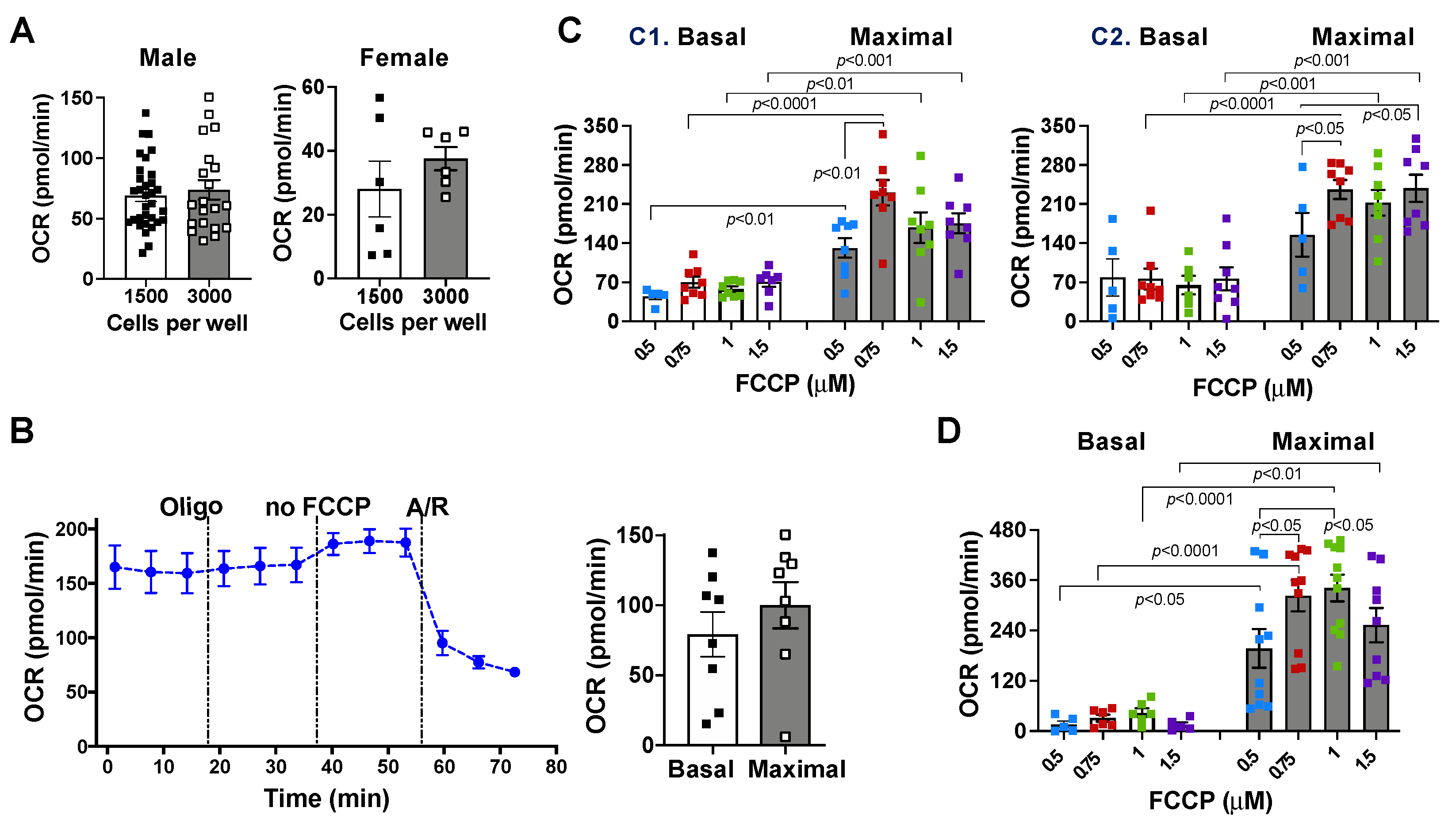
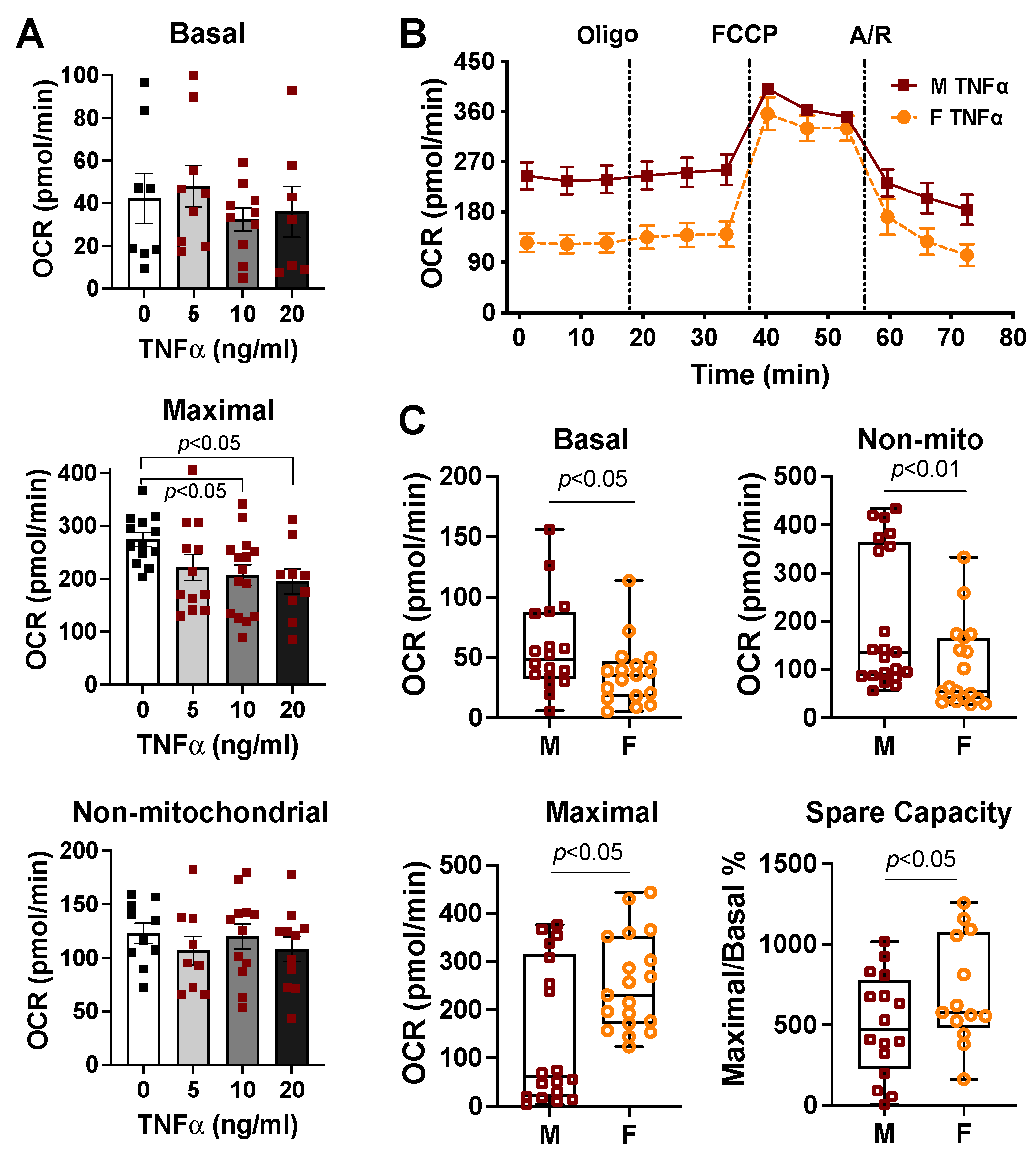
2.1. Effects of Cell Density and Carbonyl Cyanide-4 (Trifluoromethoxy) Phenylhydrazone (FCCP) Concentration on Mitochondrial Bioenergetic Response
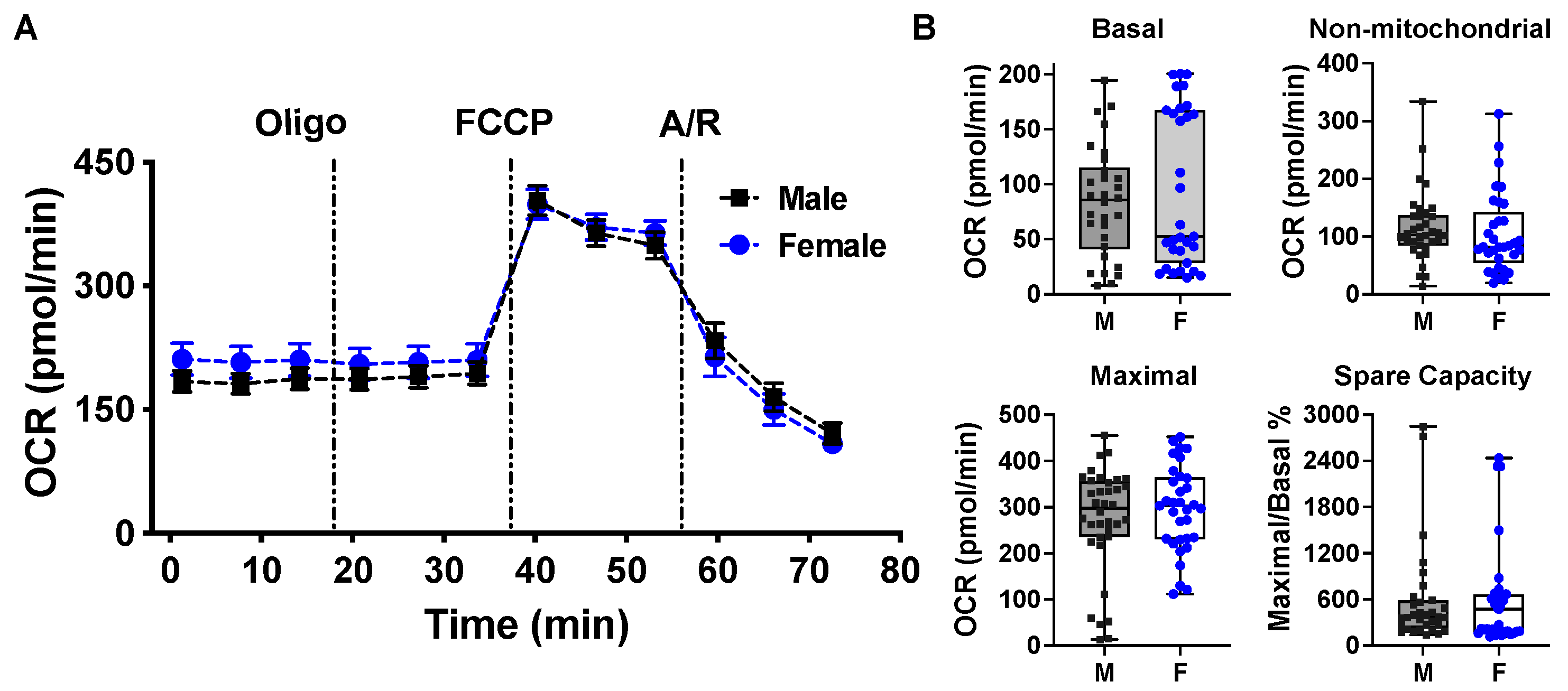
2.2. Mitochondrial Bioenergetic Function between Cardiomyocytes Isolated from Adult Male and Female Mice
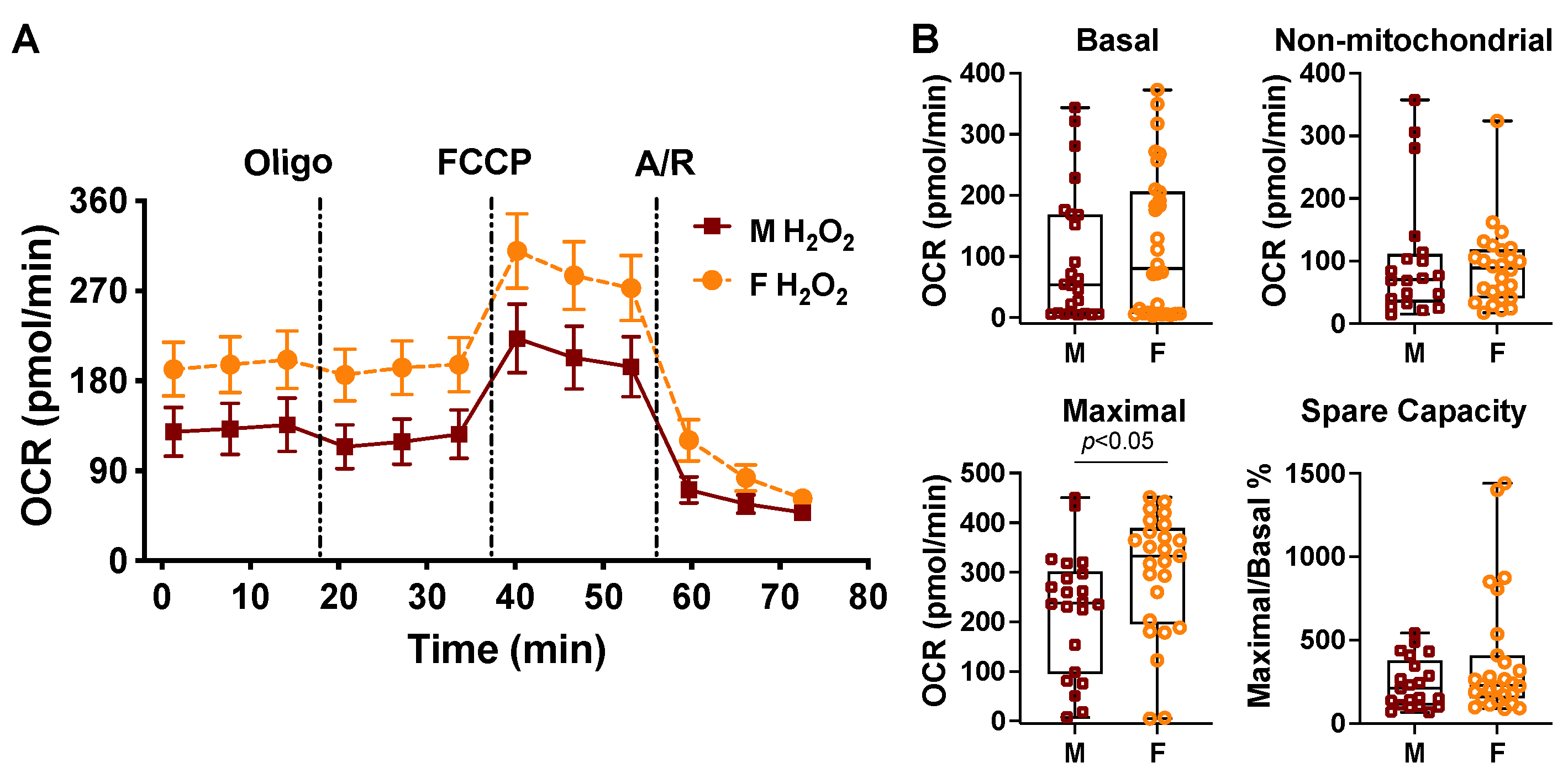
2.3. Sex Differences in Cardiac Mitochondrial Bioenergetic Response to TNFα or H2O2
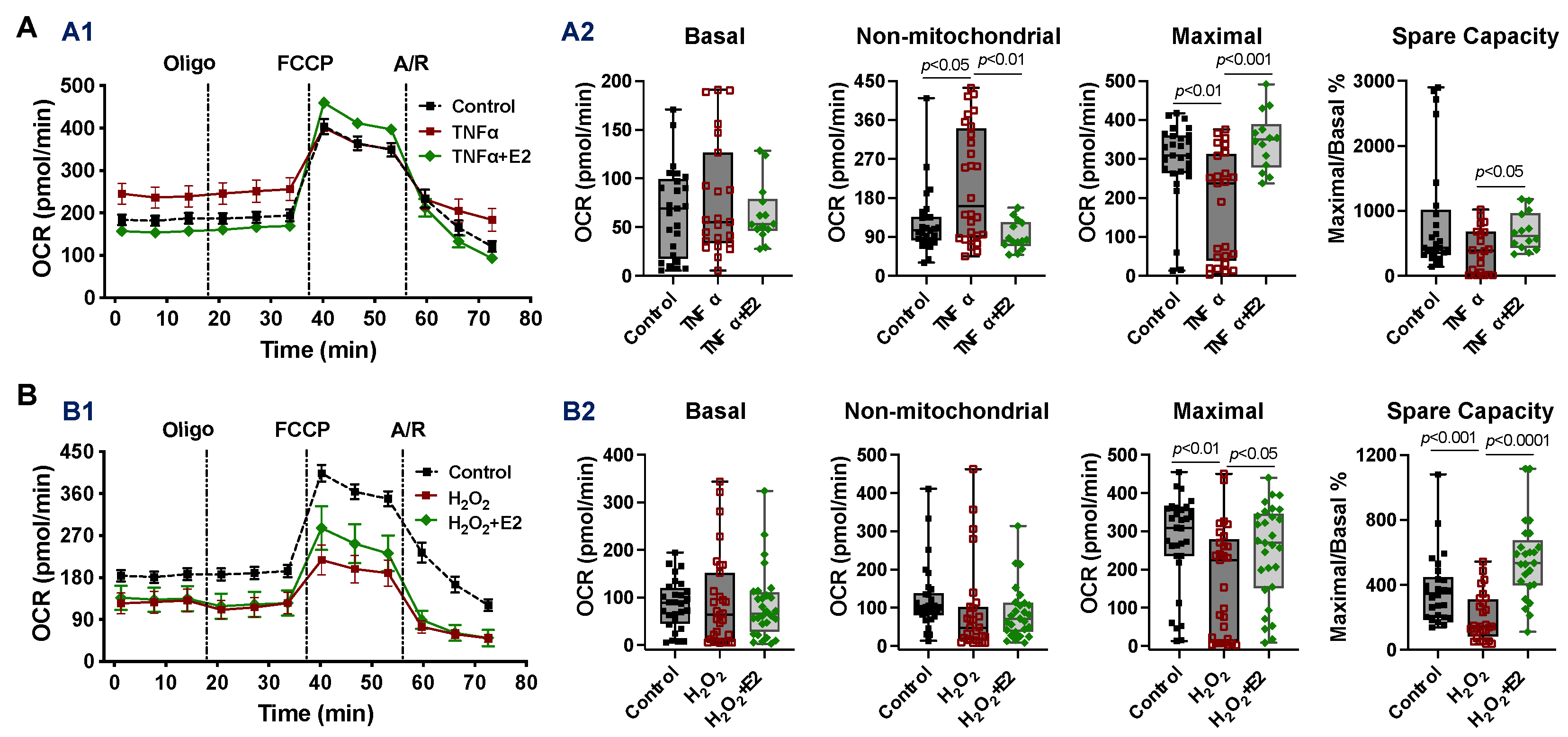
2.4. Effect of 17β-Estradiol (E2) on Mitochondrial Bioenergetic Function in Adult Cardiomyocytes Isolated from Male Mice
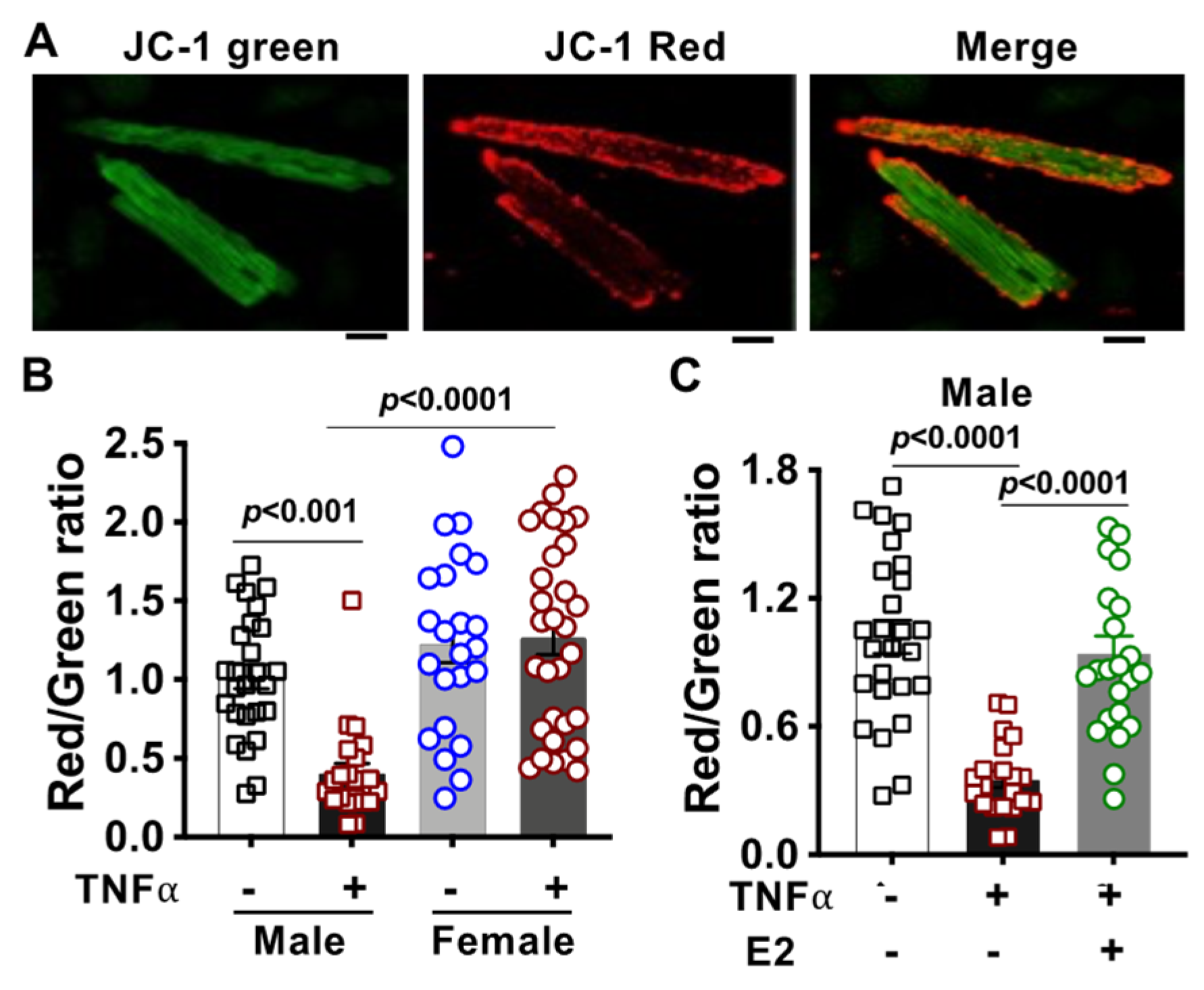
2.5. Alteration of Mitochondrial Membrane Potential in TNFα-Treated Cardiomyocytes
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Adult Mouse Cardiomyocyte Isolation and Preparation
4.3. TNFα Dose-Responsive Experiment of Mitochondrial Respiratory Function
4.4. Mitochondrial Bioenergetic Response by Seahorse XF96 Cell Mito Stress Test
4.5. Measurement of Mitochondrial Membrane Potential
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| A | antimycin A |
| BDM | 2,3-butanedione monoximoe |
| E2 | 17β-estradiol |
| ETC | electron transport chain |
| FCCP | Carbonyl cyanide-4 (trifluoromethoxy) Phenylhydrazone |
| F | female |
| H2O2 | hydrogen peroxide |
| ICU | intensive care unit |
| I/R | ischemia/reperfusion |
| M | male |
| OCR | oxygen consumption rate |
| Oligo | oligomycin |
| ROS | reactive oxygen species |
| R | rotenone |
| SC | supercomplex |
| TNFα | tumor necrosis factor α |
| ΔΨm | mitochondrial membrane potential |
References
- Klatte, K.; Chaitman, B.R.; Theroux, P.; Gavard, J.A.; Stocke, K.; Boyce, S.; Bartels, C.; Keller, B.; Jessel, A.; GUARDIAN Investigators. Increased mortality after coronary artery bypass graft surgery is associated with increased levels of postoperative creatine kinase-myocardial band isoenzyme release: Results from the GUARDIAN trial. J. Am. Coll. Cardiol. 2001, 38, 1070–1077. [Google Scholar] [CrossRef]
- van Dijk, D.; Nierich, A.P.; Jansen, E.W.; Nathoe, H.M.; Suyker, W.J.; Diephuis, J.C.; van Boven, W.J.; Borst, C.; Buskens, E.; Grobbee, D.E.; et al. Early outcome after off-pump versus on-pump coronary bypass surgery: Results from a randomized study. Circulation 2001, 104, 1761–1766. [Google Scholar] [CrossRef]
- Puskas, J.D.; Williams, W.H.; Duke, P.G.; Staples, J.R.; Glas, K.E.; Marshall, J.J.; Leimbach, M.; Huber, P.; Garas, S.; Sammons, B.H.; et al. Off-pump coronary artery bypass grafting provides complete revascularization with reduced myocardial injury, transfusion requirements, and length of stay: A prospective randomized comparison of two hundred unselected patients undergoing off-pump versus conventional coronary artery bypass grafting. J. Thorac. Cardiovasc. Surg. 2003, 125, 797–808. [Google Scholar] [CrossRef]
- Kobayashi, J.; Tashiro, T.; Ochi, M.; Yaku, H.; Watanabe, G.; Satoh, T.; Tagusari, O.; Nakajima, H.; Kitamura, S.; Japanese Off-Pump Coronary Revascularization Investigation Study Group. Early outcome of a randomized comparison of off-pump and on-pump multiple arterial coronary revascularization. Circulation 2005, 112, I338–I343. [Google Scholar] [CrossRef]
- Probst, C.; Zelle, B.A.; Sittaro, N.A.; Lohse, R.; Krettek, C.; Pape, H.C. Late death after multiple severe trauma: When does it occur and what are the causes? J. Trauma 2009, 66, 1212–1217. [Google Scholar] [CrossRef]
- Martin, G.S.; Mannino, D.M.; Eaton, S.; Moss, M. The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 2003, 348, 1546–1554. [Google Scholar] [CrossRef]
- Paoli, C.J.; Reynolds, M.A.; Sinha, M.; Gitlin, M.; Crouser, E. Epidemiology and Costs of Sepsis in the United States-An Analysis Based on Timing of Diagnosis and Severity Level. Crit. Care Med. 2018, 46, 1889–1897. [Google Scholar] [CrossRef]
- Wall, J.; Naganathar, S.; Praditsuktavorn, B.; Bugg, O.F.; McArthur, S.; Thiemermann, C.; Tremoleda, J.L.; Brohi, K. Modeling Cardiac Dysfunction Following Traumatic Hemorrhage Injury: Impact on Myocardial Integrity. Front. Immunol. 2019, 10, 2774. [Google Scholar] [CrossRef]
- Selvanayagam, J.B.; Petersen, S.E.; Francis, J.M.; Robson, M.D.; Kardos, A.; Neubauer, S.; Taggart, D.P. Effects of off-pump versus on-pump coronary surgery on reversible and irreversible myocardial injury: A randomized trial using cardiovascular magnetic resonance imaging and biochemical markers. Circulation 2004, 109, 345–350. [Google Scholar] [CrossRef]
- Stanley, W.C.; Chandler, M.P. Energy metabolism in the normal and failing heart: Potential for therapeutic interventions. Heart Fail. Rev. 2002, 7, 115–130. [Google Scholar] [CrossRef]
- Suematsu, N.; Tsutsui, H.; Wen, J.; Kang, D.; Ikeuchi, M.; Ide, T.; Hayashidani, S.; Shiomi, T.; Kubota, T.; Hamasaki, N.; et al. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation 2003, 107, 1418–1423. [Google Scholar] [CrossRef]
- Lee, T.I.; Kao, Y.H.; Baigalmaa, L.; Lee, T.W.; Lu, Y.Y.; Chen, Y.C.; Chao, T.F.; Chen, Y.J. Sodium hydrosulphide restores tumour necrosis factor-alpha-induced mitochondrial dysfunction and metabolic dysregulation in HL-1 cells. J. Cell. Mol. Med. 2019, 23, 7641–7650. [Google Scholar] [CrossRef]
- Ayala, A.; Wang, P.; Ba, Z.F.; Perrin, M.M.; Ertel, W.; Chaudry, I.H. Differential alterations in plasma IL-6 and TNF levels after trauma and hemorrhage. Am. J. Physiol. 1991, 260, R167–R171. [Google Scholar] [CrossRef]
- Niederbichler, A.D.; Papst, S.; Claassen, L.; Jokuszies, A.; Steinstraesser, L.; Hirsch, T.; Altintas, M.A.; Ipaktchi, K.R.; Reimers, K.; Kraft, T.; et al. Burn-induced organ dysfunction: Vagus nerve stimulation attenuates organ and serum cytokine levels. Burns 2009, 35, 783–789. [Google Scholar] [CrossRef]
- Bruns, B.; Maass, D.; Barber, R.; Horton, J.; Carlson, D. Alterations in the cardiac inflammatory response to burn trauma in mice lacking a functional Toll-like receptor 4 gene. Shock 2008, 30, 740–746. [Google Scholar] [CrossRef]
- Ballard-Croft, C.; White, D.J.; Maass, D.L.; Hybki, D.P.; Horton, J.W. Role of p38 mitogen-activated protein kinase in cardiac myocyte secretion of the inflammatory cytokine TNF-alpha. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1970–H1981. [Google Scholar] [CrossRef]
- Cain, B.S.; Meldrum, D.R.; Dinarello, C.A.; Meng, X.; Joo, K.S.; Banerjee, A.; Harken, A.H. Tumor necrosis factor-alpha and interleukin-1beta synergistically depress human myocardial function. Crit. Care Med. 1999, 27, 1309–1318. [Google Scholar] [CrossRef]
- Beutler, B.; Milsark, I.W.; Cerami, A.C. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science 1985, 229, 869–871. [Google Scholar] [CrossRef]
- Wang, M.; Sankula, R.; Tsai, B.M.; Meldrum, K.K.; Turrentine, M.; March, K.L.; Brown, J.W.; Dinarello, C.A.; Meldrum, D.R. P38 MAPK mediates myocardial proinflammatory cytokine production and endotoxin-induced contractile suppression. Shock 2004, 21, 170–174. [Google Scholar] [CrossRef]
- Meldrum, D.R. Tumor necrosis factor in the heart. Am. J. Physiol. 1998, 274, R577–R595. [Google Scholar] [CrossRef]
- Kalbitz, M.; Schwarz, S.; Weber, B.; Bosch, B.; Pressmar, J.; Hoenes, F.M.; Braun, C.K.; Horst, K.; Simon, T.P.; Pfeifer, R.; et al. Cardiac Depression in Pigs after Multiple Trauma—Characterization of Posttraumatic Structural and Functional Alterations. Sci. Rep. 2017, 7, 17861. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, C.; Gao, M.; Cao, X.; Ha, T.; Kalbfleisch, J.H.; Williams, D.L.; Li, C.; Kao, R.L. Toll-like receptor 4 plays a central role in cardiac dysfunction during trauma hemorrhage shock. Shock 2014, 42, 31–37. [Google Scholar] [CrossRef]
- Gao, M.; Ha, T.; Zhang, X.; Liu, L.; Wang, X.; Kelley, J.; Singh, K.; Kao, R.; Gao, X.; Williams, D.; et al. Toll-like receptor 3 plays a central role in cardiac dysfunction during polymicrobial sepsis. Crit. Care Med. 2012, 40, 2390–2399. [Google Scholar] [CrossRef]
- Su, X.; Sykes, J.B.; Ao, L.; Raeburn, C.D.; Fullerton, D.A.; Meng, X. Extracellular heat shock cognate protein 70 induces cardiac functional tolerance to endotoxin: Differential effect on TNF-alpha and ICAM-1 levels in heart tissue. Cytokine 2010, 51, 60–66. [Google Scholar] [CrossRef]
- Wang, M.; Baker, L.; Tsai, B.M.; Meldrum, K.K.; Meldrum, D.R. Sex differences in the myocardial inflammatory response to ischemia-reperfusion injury. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E321–E326. [Google Scholar] [CrossRef]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid. Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Offner, P.J.; Moore, E.E.; Biffl, W.L. Male gender is a risk factor for major infections after surgery. Arch. Surg. 1999, 134, 935–938; discussion 938–940. [Google Scholar] [CrossRef]
- Ba, Z.F.; Kuebler, J.F.; Rue, L.W., 3rd; Bland, K.I.; Wang, P.; Chaudry, I.H. Gender dimorphic tissue perfusion response after acute hemorrhage and resuscitation: Role of vascular endothelial cell function. Am. J. Physiol Heart Circ. Physiol. 2003, 284, H2162–H2169. [Google Scholar] [CrossRef][Green Version]
- Gee, A.C.; Sawai, R.S.; Differding, J.; Muller, P.; Underwood, S.; Schreiber, M.A. The influence of sex hormones on coagulation and inflammation in the trauma patient. Shock 2008, 29, 334–341. [Google Scholar] [CrossRef]
- George, R.L.; McGwin, G., Jr.; Windham, S.T.; Melton, S.M.; Metzger, J.; Chaudry, I.H.; Rue, L.W., 3rd. Age-related gender differential in outcome after blunt or penetrating trauma. Shock 2003, 19, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Schroder, J.; Kahlke, V.; Staubach, K.H.; Zabel, P.; Stuber, F. Gender differences in human sepsis. Arch. Surg. 1998, 133, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Frink, M.; Pape, H.C.; van Griensven, M.; Krettek, C.; Chaudry, I.H.; Hildebrand, F. Influence of sex and age on mods and cytokines after multiple injuries. Shock 2007, 27, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Guidry, C.A.; Swenson, B.R.; Davies, S.W.; Dossett, L.A.; Popovsky, K.A.; Bonatti, H.; Evans, H.L.; Metzger, R.; Hedrick, T.L.; Tache-Leon, C.A.; et al. Sex- and diagnosis-dependent differences in mortality and admission cytokine levels among patients admitted for intensive care. Crit. Care Med. 2014, 42, 1110–1120. [Google Scholar] [CrossRef]
- Haider, A.H.; Crompton, J.G.; Oyetunji, T.; Stevens, K.A.; Efron, D.T.; Kieninger, A.N.; Chang, D.C.; Cornwell, E.E., 3rd; Haut, E.R. Females have fewer complications and lower mortality following trauma than similarly injured males: A risk adjusted analysis of adults in the National Trauma Data Bank. Surgery 2009, 146, 308–315. [Google Scholar] [CrossRef]
- Mostafa, G.; Huynh, T.; Sing, R.F.; Miles, W.S.; Norton, H.J.; Thomason, M.H. Gender-related outcomes in trauma. J. Trauma 2002, 53, 430–434; discussion 434–435. [Google Scholar] [CrossRef] [PubMed]
- Deitch, E.A.; Livingston, D.H.; Lavery, R.F.; Monaghan, S.F.; Bongu, A.; Machiedo, G.W. Hormonally active women tolerate shock-trauma better than do men: A prospective study of over 4000 trauma patients. Ann. Surg. 2007, 246, 447–453; discussion 453–455. [Google Scholar] [CrossRef]
- Angus, D.C.; Linde-Zwirble, W.T.; Lidicker, J.; Clermont, G.; Carcillo, J.; Pinsky, M.R. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 2001, 29, 1303–1310. [Google Scholar] [CrossRef]
- Dombrovskiy, V.Y.; Martin, A.A.; Sunderram, J.; Paz, H.L. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: A trend analysis from 1993 to 2003. Crit. Care Med. 2007, 35, 1244–1250. [Google Scholar] [CrossRef]
- Sakr, Y.; Elia, C.; Mascia, L.; Barberis, B.; Cardellino, S.; Livigni, S.; Fiore, G.; Filippini, C.; Ranieri, V.M. The influence of gender on the epidemiology of and outcome from severe sepsis. Crit. Care. 2013, 17, R50. [Google Scholar] [CrossRef]
- Mayr, F.B.; Yende, S.; Angus, D.C. Epidemiology of severe sepsis. Virulence 2014, 5, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tsai, B.M.; Crisostomo, P.R.; Meldrum, D.R. Tumor necrosis factor receptor 1 signaling resistance in the female myocardium during ischemia. Circulation 2006, 114, I282–I289. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Crisostomo, P.R.; Markel, T.A.; Wang, Y.; Meldrum, D.R. Mechanisms of sex differences in TNFR2-mediated cardioprotection. Circulation 2008, 118, S38–S45. [Google Scholar] [CrossRef]
- Wang, M.; Tsai, B.M.; Reiger, K.M.; Brown, J.W.; Meldrum, D.R. 17-beta-Estradiol decreases p38 MAPK-mediated myocardial inflammation and dysfunction following acute ischemia. J. Mol. Cell. Cardiol. 2006, 40, 205–212. [Google Scholar] [CrossRef]
- Wang, M.; Smith, K.; Yu, Q.; Miller, C.; Singh, K.; Sen, C.K. Mitochondrial connexin 43 in sex-dependent myocardial responses and estrogen-mediated cardiac protection following acute ischemia/reperfusion injury. Basic Res. Cardiol. 2019, 115, 1. [Google Scholar] [CrossRef]
- Vijay, V.; Han, T.; Moland, C.L.; Kwekel, J.C.; Fuscoe, J.C.; Desai, V.G. Sexual dimorphism in the expression of mitochondria-related genes in rat heart at different ages. PLoS ONE 2015, 10, e0117047. [Google Scholar] [CrossRef]
- Wang, M.; Markel, T.; Crisostomo, P.; Herring, C.; Meldrum, K.K.; Lillemoe, K.D.; Meldrum, D.R. Deficiency of TNFR1 protects myocardium through SOCS3 and IL-6 but not p38 MAPK or IL-1beta. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1694–H1699. [Google Scholar] [CrossRef]
- Sando, I.C.; Wang, Y.; Crisostomo, P.R.; Markel, T.A.; Sharma, R.; Erwin, G.S.; Guzman, M.J.; Meldrum, D.R.; Wang, M. Females exhibit relative resistance to depressive effects of tumor necrosis factor-alpha on the myocardium. J. Surg. Res. 2008, 150, 92–99. [Google Scholar] [CrossRef]
- Readnower, R.D.; Brainard, R.E.; Hill, B.G.; Jones, S.P. Standardized bioenergetic profiling of adult mouse cardiomyocytes. Physiol. Genom. 2012, 44, 1208–1213. [Google Scholar] [CrossRef]
- Basheer, W.A.; Fu, Y.; Shimura, D.; Xiao, S.; Agvanian, S.; Hernandez, D.M.; Hitzeman, T.C.; Hong, T.; Shaw, R.M. Stress response protein GJA1-20k promotes mitochondrial biogenesis, metabolic quiescence, and cardioprotection against ischemia/reperfusion injury. JCI Insight 2018, 3, e121900. [Google Scholar] [CrossRef]
- Milerova, M.; Drahota, Z.; Chytilova, A.; Tauchmannova, K.; Houstek, J.; Ostadal, B. Sex difference in the sensitivity of cardiac mitochondrial permeability transition pore to calcium load. Mol. Cell. Biochem. 2016, 412, 147–154. [Google Scholar] [CrossRef]
- Wang, M.; Meng, X.; Tsai, B.; Wang, J.F.; Turrentine, M.; Brown, J.W.; Meldrum, D.R. Preconditioning up-regulates the soluble TNF receptor I response to endotoxin. J. Surg. Res. 2004, 121, 20–24. [Google Scholar] [CrossRef]
- Dworatzek, E.; Mahmoodzadeh, S. Targeted basic research to highlight the role of estrogen and estrogen receptors in the cardiovascular system. Pharmacol. Res. 2017, 119, 27–35. [Google Scholar] [CrossRef]
- Blenck, C.L.; Harvey, P.A.; Reckelhoff, J.F.; Leinwand, L.A. The Importance of Biological Sex and Estrogen in Rodent Models of Cardiovascular Health and Disease. Circ. Res. 2016, 118, 1294–1312. [Google Scholar] [CrossRef]
- Wang, M.; Tan, J.; Coffey, A.; Fehrenbacher, J.; Weil, B.R.; Meldrum, D.R. Signal transducer and activator of transcription 3-stimulated hypoxia inducible factor-1alpha mediates estrogen receptor-alpha-induced mesenchymal stem cell vascular endothelial growth factor production. J. Thorac. Cardiovasc. Surg. 2009, 138, 163–171.e1. [Google Scholar] [CrossRef][Green Version]
- Wang, L.; Gu, H.; Turrentine, M.; Wang, M. Estradiol treatment promotes cardiac stem cell (CSC)-derived growth factors, thus improving CSC-mediated cardioprotection after acute ischemia/reperfusion. Surgery 2014, 156, 243–252. [Google Scholar] [CrossRef]
- Marin-Garcia, J.; Goldenthal, M.J. Understanding the impact of mitochondrial defects in cardiovascular disease: A review. J. Card. Fail. 2002, 8, 347–361. [Google Scholar] [CrossRef]
- Ruas, J.S.; Siqueira-Santos, E.S.; Amigo, I.; Rodrigues-Silva, E.; Kowaltowski, A.J.; Castilho, R.F. Underestimation of the Maximal Capacity of the Mitochondrial Electron Transport System in Oligomycin-Treated Cells. PLoS ONE 2016, 11, e0150967. [Google Scholar] [CrossRef]
- Dranka, B.P.; Benavides, G.A.; Diers, A.R.; Giordano, S.; Zelickson, B.R.; Reily, C.; Zou, L.; Chatham, J.C.; Hill, B.G.; Zhang, J.; et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 2011, 51, 1621–1635. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef]
- Lagranha, C.J.; Deschamps, A.; Aponte, A.; Steenbergen, C.; Murphy, E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ. Res. 2010, 106, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Colom, B.; Oliver, J.; Roca, P.; Garcia-Palmer, F.J. Caloric restriction and gender modulate cardiac muscle mitochondrial H2O2 production and oxidative damage. Cardiovasc. Res. 2007, 74, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, A.R.; Abdel-Rahman, E.A.; Mahmoud, A.M.; Ali, M.H.; Noureldin, M.; Saber, S.H.; Mohsen, M.; Ali, S.S. Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiol. Rep. 2017, 5, e13125. [Google Scholar] [CrossRef]
- Ribeiro, R.F., Jr.; Ronconi, K.S.; Morra, E.A.; Do Val Lima, P.R.; Porto, M.L.; Vassallo, D.V.; Figueiredo, S.G.; Stefanon, I. Sex differences in the regulation of spatially distinct cardiac mitochondrial subpopulations. Mol. Cell. Biochem. 2016, 419, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.; Angele, M.K.; Chaudry, I.H. Gender differences in trauma, shock and sepsis. Mil. Med. Res. 2018, 5, 35. [Google Scholar] [CrossRef]
- Hsieh, Y.C.; Frink, M.; Choudhry, M.A.; Bland, K.I.; Chaudry, I.H. Metabolic modulators following trauma sepsis: Sex hormones. Crit. Care Med. 2007, 35, S621–S629. [Google Scholar] [CrossRef]
- Mendelsohn, M.E.; Karas, R.H. Molecular and cellular basis of cardiovascular gender differences. Science 2005, 308, 1583–1587. [Google Scholar] [CrossRef]
- Gilligan, D.M.; Badar, D.M.; Panza, J.A.; Quyyumi, A.A.; Cannon, R.O., 3rd. Acute vascular effects of estrogen in postmenopausal women. Circulation 1994, 90, 786–791. [Google Scholar] [CrossRef]
- Shinohara, T.; Takahashi, N.; Ooie, T.; Ichinose, M.; Hara, M.; Yonemochi, H.; Saikawa, T.; Yoshimatsu, H. Estrogen inhibits hyperthermia-induced expression of heat-shock protein 72 and cardioprotection against ischemia/reperfusion injury in female rat heart. J. Mol. Cell. Cardiol. 2004, 37, 1053–1061. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogenic control of mitochondrial function and biogenesis. J. Cell. Biochem. 2008, 105, 1342–1351. [Google Scholar] [CrossRef]
- Torres, M.J.; Kew, K.A.; Ryan, T.E.; Pennington, E.R.; Lin, C.T.; Buddo, K.A.; Fix, A.M.; Smith, C.A.; Gilliam, L.A.; Karvinen, S.; et al. 17beta-Estradiol Directly Lowers Mitochondrial Membrane Microviscosity and Improves Bioenergetic Function in Skeletal Muscle. Cell Metab. 2018, 27, 167–179.e7. [Google Scholar] [CrossRef]
- Mori, J.; Zhang, L.; Oudit, G.Y.; Lopaschuk, G.D. Impact of the renin-angiotensin system on cardiac energy metabolism in heart failure. J. Mol. Cell. Cardiol. 2013, 63, 98–106. [Google Scholar] [CrossRef]
- Kozlov, A.V.; Duvigneau, J.C.; Hyatt, T.C.; Raju, R.; Behling, T.; Hartl, R.T.; Staniek, K.; Miller, I.; Gregor, W.; Redl, H.; et al. Effect of estrogen on mitochondrial function and intracellular stress markers in rat liver and kidney following trauma-hemorrhagic shock and prolonged hypotension. Mol. Med. 2010, 16, 254–261. [Google Scholar] [CrossRef]
- Hsu, J.T.; Kan, W.H.; Hsieh, C.H.; Choudhry, M.A.; Bland, K.I.; Chaudry, I.H. Mechanism of salutary effects of estrogen on cardiac function following trauma-hemorrhage: Akt-dependent HO-1 up-regulation. Crit. Care Med. 2009, 37, 2338–2344. [Google Scholar] [CrossRef]
- Ba, Z.F.; Hsu, J.T.; Chen, J.; Kan, W.H.; Schwacha, M.G.; Chaudry, I.H. Systematic analysis of the salutary effect of estrogen on cardiac performance after trauma-hemorrhage. Shock 2008, 30, 585–589. [Google Scholar] [CrossRef]
- Messingham, K.A.; Heinrich, S.A.; Kovacs, E.J. Estrogen restores cellular immunity in injured male mice via suppression of interleukin-6 production. J. Leukoc. Biol. 2001, 70, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Wigginton, J.G.; Maass, D.L.; Ma, L.; Carlson, D.; Wolf, S.E.; Minei, J.P.; Zang, Q.S. Estrogen-provided cardiac protection following burn trauma is mediated through a reduction in mitochondria-derived DAMPs. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H882–H894. [Google Scholar] [CrossRef] [PubMed]
- Knoferl, M.W.; Angele, M.K.; Diodato, M.D.; Schwacha, M.G.; Ayala, A.; Cioffi, W.G.; Bland, K.I.; Chaudry, I.H. Female sex hormones regulate macrophage function after trauma-hemorrhage and prevent increased death rate from subsequent sepsis. Ann. Surg. 2002, 235, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Kuebler, J.F.; Jarrar, D.; Toth, B.; Bland, K.I.; Rue, L., 3rd; Wang, P.; Chaudry, I.H. Estradiol administration improves splanchnic perfusion following trauma-hemorrhage and sepsis. Arch. Surg. 2002, 137, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Vornehm, N.D.; Wang, M.; Abarbanell, A.; Herrmann, J.; Weil, B.; Tan, J.; Wang, Y.; Kelly, M.; Meldrum, D.R. Acute postischemic treatment with estrogen receptor-alpha agonist or estrogen receptor-beta agonist improves myocardial recovery. Surgery 2009, 146, 145–154. [Google Scholar] [CrossRef]
- Panov, A.; Orynbayeva, Z. Bioenergetic and antiapoptotic properties of mitochondria from cultured human prostate cancer cell lines PC-3, DU145 and LNCaP. PLoS ONE 2013, 8, e72078. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, A.; Milner, K.; Gupta, A.; Lopaschuk, G.D. Myocardial Energy Substrate Metabolism in Heart Failure: From Pathways to Therapeutic Targets. Curr. Pharm. Des. 2015, 21, 3654–3664. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Piquereau, J.; Garnier, A.; Mericskay, M.; Lemaire, C.; Crozatier, B. Gender issues in cardiovascular diseases. Focus on energy metabolism. Biochim. Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165722. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Jang, S.; Chapa-Dubocq, X.R.; Khuchua, Z.; Camara, A.K. Mitochondrial respiratory supercomplexes in mammalian cells: Structural versus functional role. J. Mol. Med. 2021, 99, 57–73. [Google Scholar] [CrossRef]
- Jang, S.; Lewis, T.S.; Powers, C.; Khuchua, Z.; Baines, C.P.; Wipf, P.; Javadov, S. Elucidating Mitochondrial Electron Transport Chain Supercomplexes in the Heart During Ischemia-Reperfusion. Antioxid. Redox Signal. 2017, 27, 57–69. [Google Scholar] [CrossRef]
- Rosca, M.G.; Vazquez, E.J.; Kerner, J.; Parland, W.; Chandler, M.P.; Stanley, W.; Sabbah, H.N.; Hoppel, C.L. Cardiac mitochondria in heart failure: Decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res. 2008, 80, 30–39. [Google Scholar] [CrossRef]
- Lee, Y.; Singh, J.; Scott, S.R.; Ellis, B.; Zorlutuna, P.; Wang, M. A Recombinant Dimethylarginine Dimethylaminohydrolase-1-Based Biotherapeutics to Pharmacologically Lower Asymmetric Dimethyl Arginine, thus Improving Postischemic Cardiac Function and Cardiomyocyte Mitochondrial Activity. Mol. Pharmacol. 2022, 101, 226–235. [Google Scholar] [CrossRef]
- Redl, H.; Schlag, G.; Adolf, G.R.; Natmessnig, B.; Davies, J. Tumor necrosis factor (TNF)-dependent shedding of the p55 TNF receptor in a baboon model of bacteremia. Infect. Immun. 1995, 63, 297–300. [Google Scholar] [CrossRef]
- Horton, J.W.; White, D.J.; Maass, D.L. Gender-related differences in myocardial inflammatory and contractile responses to major burn trauma. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H202–H213. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scott, S.R.; Singh, K.; Yu, Q.; Sen, C.K.; Wang, M. Sex as Biological Variable in Cardiac Mitochondrial Bioenergetic Responses to Acute Stress. Int. J. Mol. Sci. 2022, 23, 9312. https://doi.org/10.3390/ijms23169312
Scott SR, Singh K, Yu Q, Sen CK, Wang M. Sex as Biological Variable in Cardiac Mitochondrial Bioenergetic Responses to Acute Stress. International Journal of Molecular Sciences. 2022; 23(16):9312. https://doi.org/10.3390/ijms23169312
Chicago/Turabian StyleScott, Susan R., Kanhaiya Singh, Qing Yu, Chandan K. Sen, and Meijing Wang. 2022. "Sex as Biological Variable in Cardiac Mitochondrial Bioenergetic Responses to Acute Stress" International Journal of Molecular Sciences 23, no. 16: 9312. https://doi.org/10.3390/ijms23169312
APA StyleScott, S. R., Singh, K., Yu, Q., Sen, C. K., & Wang, M. (2022). Sex as Biological Variable in Cardiac Mitochondrial Bioenergetic Responses to Acute Stress. International Journal of Molecular Sciences, 23(16), 9312. https://doi.org/10.3390/ijms23169312