
Symptomatic and Disease-Modifying Therapy Pipeline for Alzheimer’s Disease: Towards a Personalized Polypharmacology Patient-Centered Approach


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cholinergic and Dopaminergic System and Ca2+ Signaling
3. Amyloid-β Protein
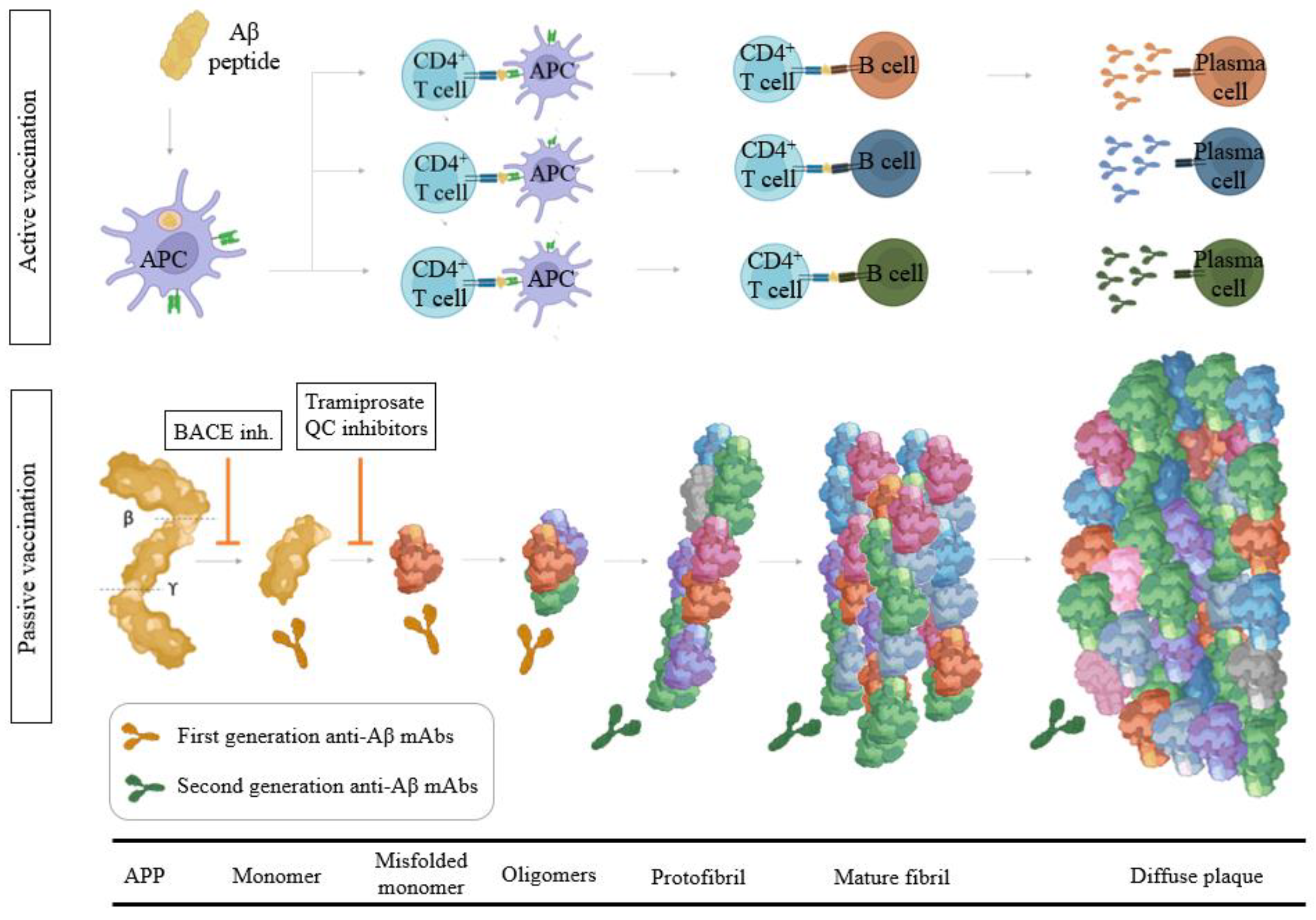
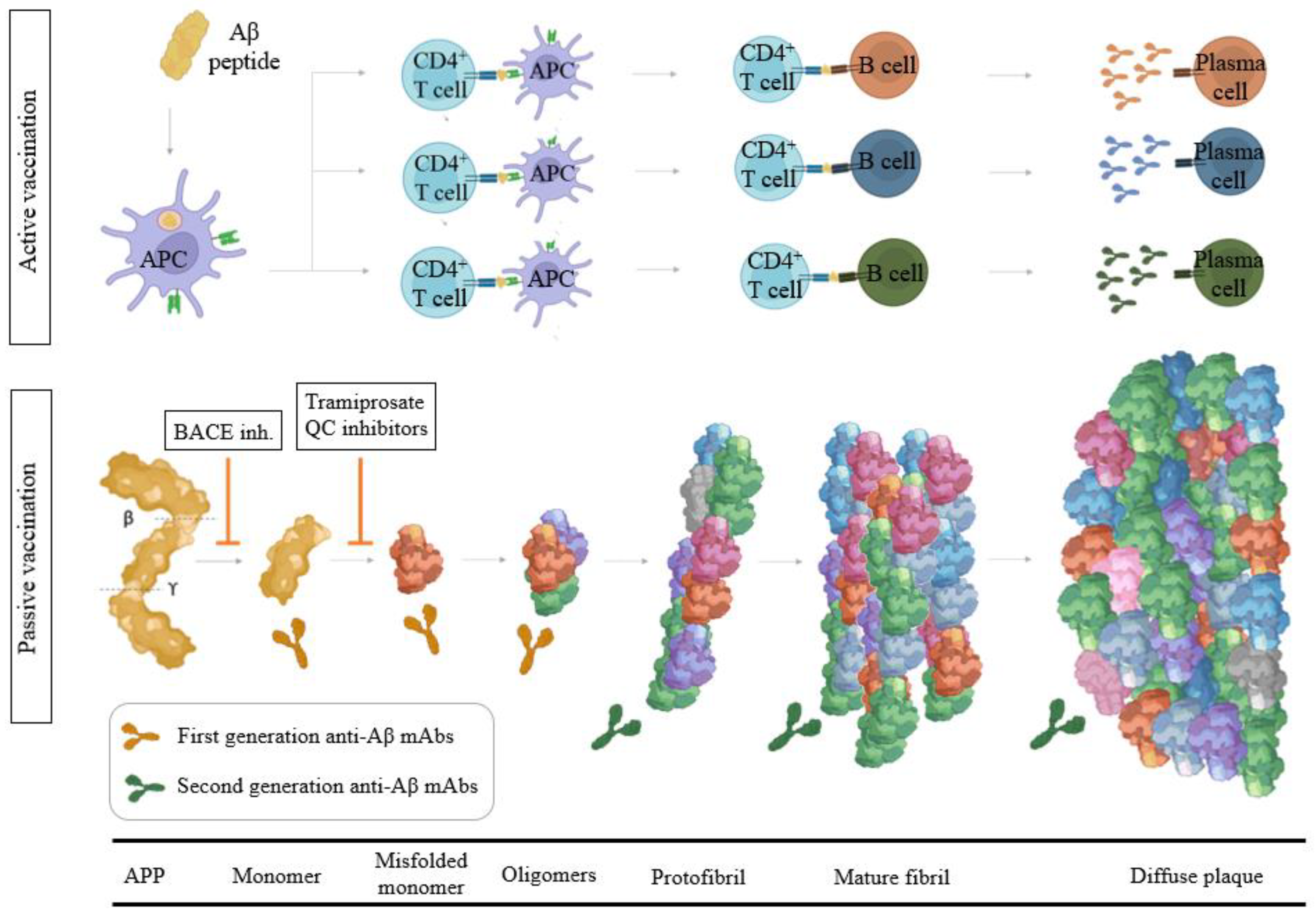
3.1. Modulators of α-, β-, and γ-Secretase Activity
3.2. Aβ Immunotherapy
3.2.1. Aβ Passive Immunotherapy
3.2.2. Aβ Active Immunotherapy
3.3. Modulators of Aβ Toxicity
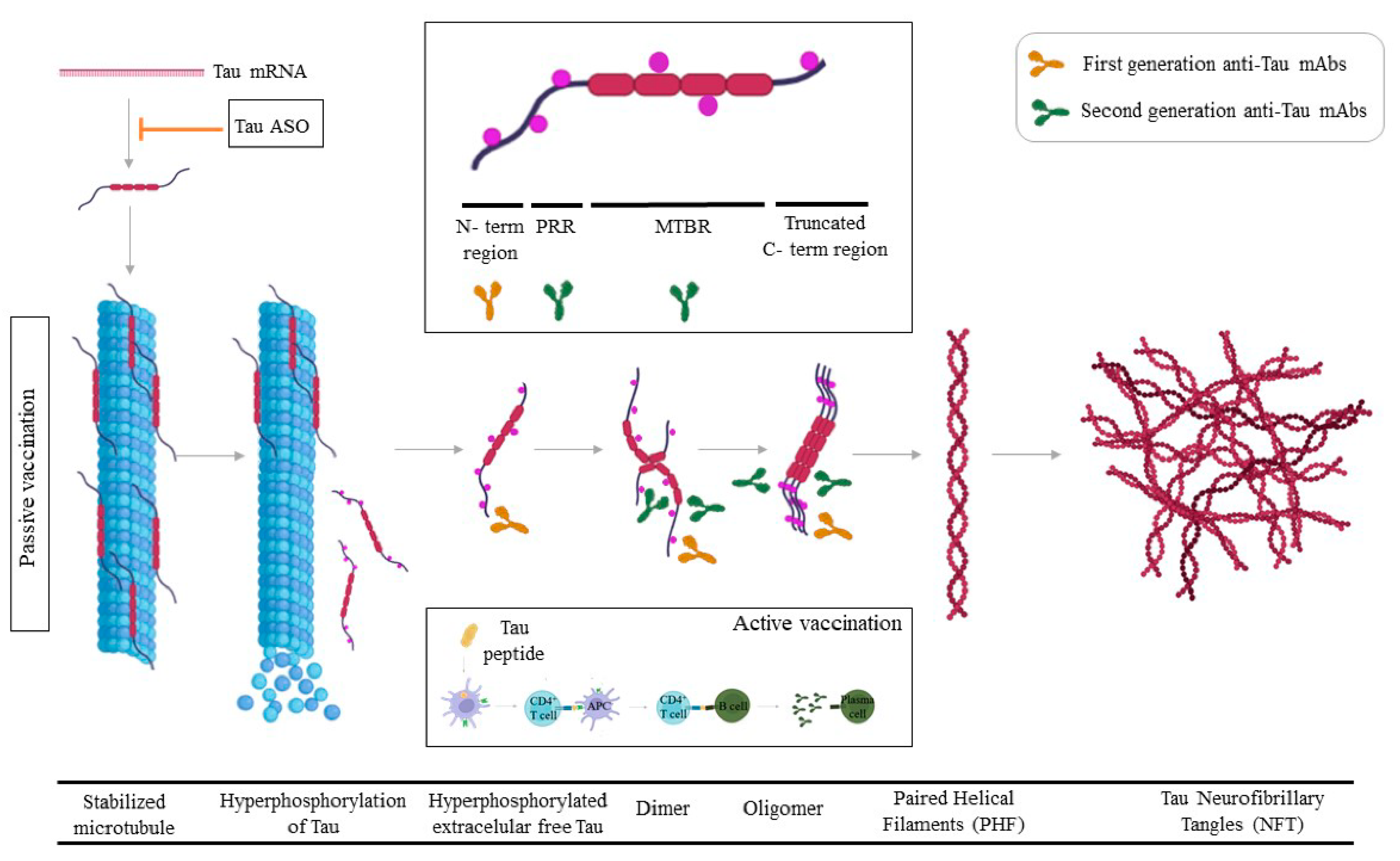
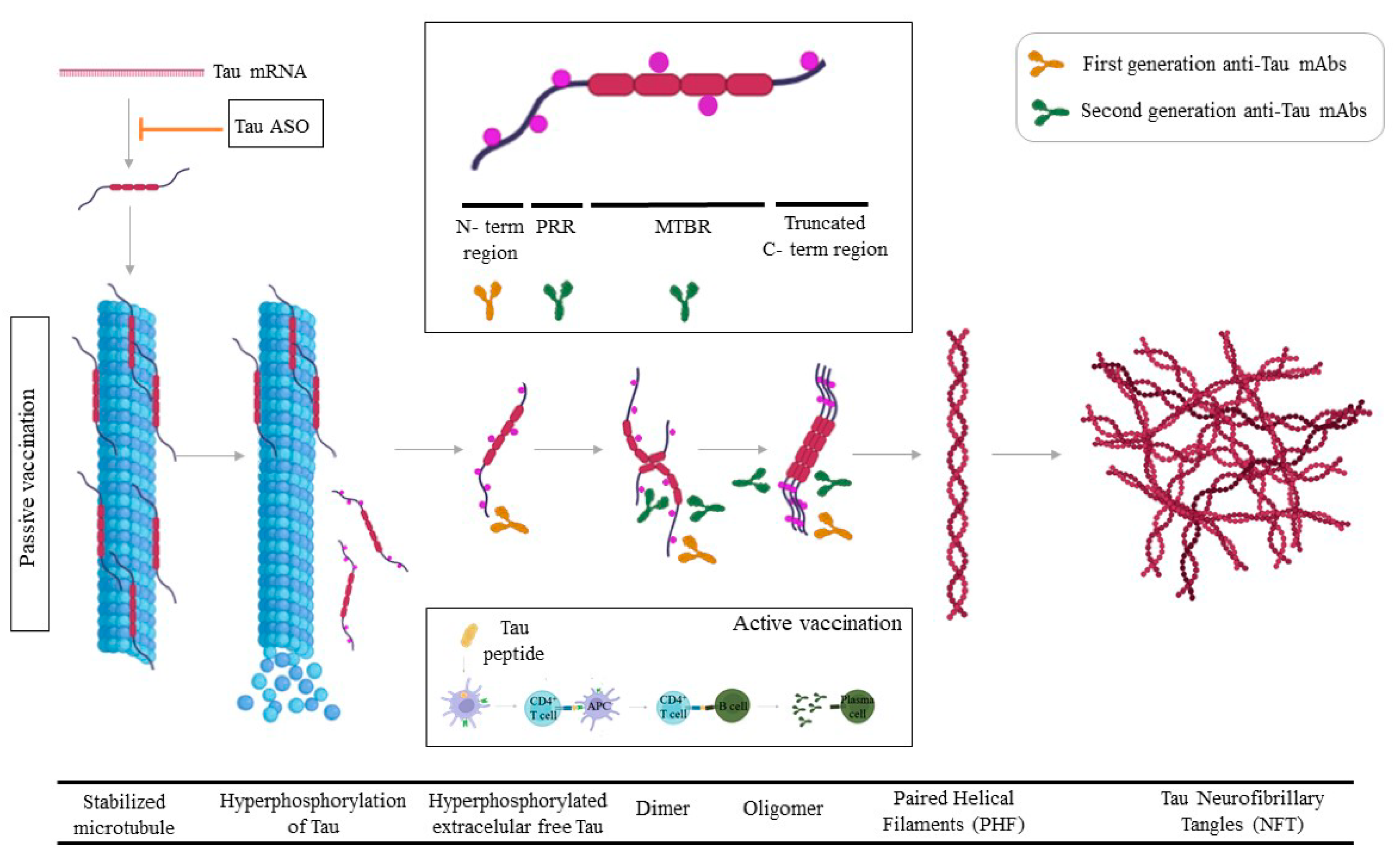
4. Tau
4.1. Tau Passive Immunotherapy
4.2. Active Tau Immunotherapy
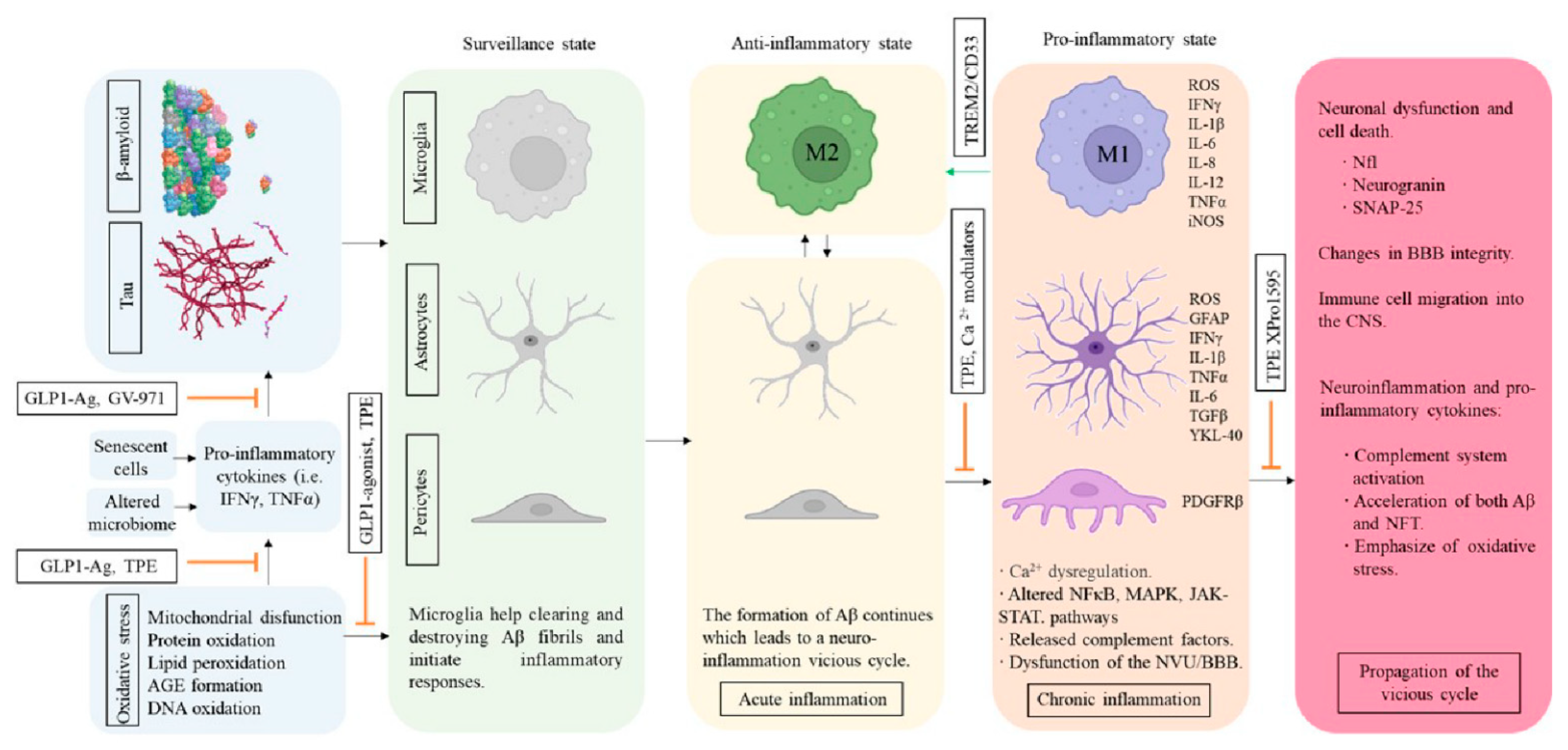
5. Inflammation and AD
5.1. Modulators of Microglial Activity and Neuroinflammation
5.2. Cerebrovascular Structure and Insulin Resistance
6. Lipids and ApoE
7. Plasma Fractions and Therapeutic Plasma Exchange
8. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; Van Der Flier, W.M. Alzheimer’ s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- GBD 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet 2022, 7, 105–125. [Google Scholar] [CrossRef]
- Ávila-Villanueva, M.; Fernández-Blázquez, M.A. Subjective cognitive decline as a preclinical marker for Alzheimer’s disease: The challenge of stability over time. Front. Aging Neurosci. 2017, 9, 377. [Google Scholar] [CrossRef]
- Aisen, P.S.; Cummings, J.; Jack, C.R.; Morris, J.C.; Sperling, R.; Frölich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J.; et al. On the path to 2025: Understanding the Alzheimer’s disease continuum. Alzheimer’s Res. Ther. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.F. Early-onset Alzheimer disease and its variants. Contin. Lifelong Learn. Neurol. 2019, 25, 34–51. [Google Scholar] [CrossRef]
- Herrera-Rivero, M. Late-Onset Alzheimer’s Disease: Risk Factors, Clinical Diagnosis and the Search for Biomarkers. Neurodegener. Dis. 2013. [Google Scholar] [CrossRef]
- Holstege, H.; Hulsman, M.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Grozeva, D.; van Rooij, J.G.J.; Sims, R.; Ahmad, S.; Amin, N.; et al. Exome sequencing identifies rare damaging variants in the ATB8B4 and ABCA1 genes as novel risk factors for Alzheimer’s disease. Alzheimers. Dement. 2021, 17, e055982. [Google Scholar] [CrossRef]
- Griciuc, A.; Tanzi, R.E. The role of innate immune genes in Alzheimer’s disease. Curr. Opin. Neurol. 2021, 34, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Thalamuthu, A.; Mather, K.A.; Crawford, J.; Ulanova, M.; Wong, M.W.K.; Pickford, R.; Sachdev, P.S.; Braidy, N. Plasma lipidome is dysregulated in Alzheimer’s disease and is associated with disease risk genes. Transl. Psychiatry 2021, 11, 344. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef]
- Prokopenko, D.; Hecker, J.; Kirchner, R.; Chapman, B.A.; Hoffman, O.; Mullin, K.; Hide, W.; Bertram, L.; Laird, N.; DeMeo, D.L.; et al. Identification of Novel Alzheimer’s Disease Loci Using Sex-Specific Family-Based Association Analysis of Whole-Genome Sequence Data. Sci. Rep. 2020, 10, 5029. [Google Scholar] [CrossRef]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimery’s disease pathology is associated with early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Ihara, M. Interaction between cerebrovascular disease and Alzheimer pathology. Curr. Opin. Psychiatry 2016, 29, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, D.S. Treatment guidelines for Alzheimer’s disease: Redefining perceptions in primary care. Prim. Care Companion J. Clin. Psychiatry 2007, 9, 113–121. [Google Scholar] [CrossRef]
- Cummings, J.; Fox, N. Defining Disease Modifying Therapy for Alzheimer’S Disease. J. Prev. Alzheimer’s Dis. 2017, 4, 109. [Google Scholar] [CrossRef]
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; Pletnikova, O.; O’Brien, R.; Yang, A.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Neurotransmitter Imbalance in the Brain and Alzheimer’s Disease Pathology. J. Alzheimer’s Dis. 2019, 72, 35–43. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Isabella, M.; Guimaraes, F.R.S. Alzheimer’s Disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Friedli, M.J.; Inestrosa, N.C. Huperzine a and its neuroprotective molecular signaling in alzheimer’s disease. Molecules 2021, 26, 6531. [Google Scholar] [CrossRef]
- Yang, G.; Wang, Y.; Tian, J.; Liu, J.P. Huperzine A for Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. PLoS ONE 2013, 8, e74916. [Google Scholar] [CrossRef]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, H. Role of Glutamate and NMDA in Alzheimer’s desease. J. Alzheimer’s Desese 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Koola, M.M. Galantamine-Memantine combination in the treatment of Alzheimer’s disease and beyond. Psychiatry Res. 2020, 293, 113409. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, Z.; Liu, R.; Huang, Y.; Zhang, N.; Zhang, R. Memantine, Donepezil, or Combination Therapy—What is the best therapy for Alzheimer’s Disease? A Network Meta-Analysis. Brain Behav. 2020, 10, e01831. [Google Scholar] [CrossRef]
- Teipel, S.; Gustafson, D.; Ossenkoppele, R.; Hansson, O.; Babiloni, C.; Wagner, M.; Riedel-Heller, S.; Kilimann, I.; Tang, Y. Alzheimer’s disease—Standard of diagnosis, treatment, care, and prevention. J. Nucl. Med. 2022, 63, 981–985. [Google Scholar] [CrossRef]
- Woods, N.K.; Padmanabhan, J. Neuronal calcium signaling and Alzheimer’s disease. Adv. Exp. Med. Biol. 2012, 740, 1193–1217. [Google Scholar] [CrossRef]
- Ryan, K.C.; Ashkavand, Z.; Norman, K.R. The role of mitochondrial calcium homeostasis in alzheimer’s and related diseases. Int. J. Mol. Sci. 2020, 21, 9153. [Google Scholar] [CrossRef]
- Guan, P.P.; Cao, L.L.; Wang, P. Elevating the levels of calcium ions exacerbate alzheimer’s disease via inducing the production and aggregation of β-amyloid protein and phosphorylated tau. Int. J. Mol. Sci. 2021, 22, 5900. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2021. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12179. [Google Scholar] [CrossRef]
- van der Kall, L.M.; Truong, T.; Burnham, S.C.; Doré, V.; Mulligan, R.S.; Bozinovski, S.; Lamb, F.; Bourgeat, P.; Fripp, J.; Schultz, S.; et al. Association of β-Amyloid Level, Clinical Progression, and Longitudinal Cognitive Change in Normal Older Individuals. Neurology 2021, 96, e662–e670. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.; Rainey-Smith, S.R.; Bird, S.; Bomke, J.; Bourgeat, P.; Brown, B.M.; Burnham, S.C.; Bush, A.I.; Chadunow, C.; Collins, S.; et al. Fifteen Years of the Australian Imaging, Biomarkers and Lifestyle (AIBL) Study: Progress and Observations from 2359 Older Adults Spanning the Spectrum from Cognitive Normality to Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2021, 5, 443–468. [Google Scholar] [CrossRef] [PubMed]
- Kokjohn, T.A.; Roher, A.E. Amyloid precursor protein transgenic mouse models and Alzheimer’s disease: Understanding the paradigms, limitations and contributions. Alzheimers Dement. 2009, 5, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Esquerda-Canals, G.; Montoliu-Gaya, L.; Güell-Bosch, J.; Villegas, S. Mouse Models of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1171–1183. [Google Scholar] [CrossRef]
- Karisetty, B.C.; Bhatnagar, A.; Armour, E.M.; Beaver, M.; Zhang, H.; Elefant, F. Amyloid-β Peptide Impact on Synaptic Function and Neuroepigenetic Gene Control Reveal New Therapeutic Strategies for Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 577622. [Google Scholar] [CrossRef]
- Gosztyla, M.L.; Brothers, H.M.; Robinson, S.R. Alzheimer’s Amyloid-β is an Antimicrobial Peptide: A Review of the Evidence. J. Alzheimer’s Dis. 2018, 62, 1495–1506. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Baumketner, A. Amyloid beta-protein monomer structure: A computational and experimental study. Protein Sci. 2006, 15, 420–428. [Google Scholar] [CrossRef]
- Orellana, A.; García-González, P.; Valero, S.; Montrreal, L.; de Rojas, I.; Hernández, I.; Rosende-Roca, M.; Vargas, L.; Tartari, J.P.; Esteban-De Antonio, E.; et al. Establishing In-House Cutoffs of CSF Alzheimer’s Disease Biomarkers for the AT(N) Stratification of the Alzheimer Center Barcelona Cohort. Int. J. Mol. Sci. 2022, 23, 6891. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Chiu, M.J.; Yang, C.C.; Yang, S.Y.; Scheltens, P.; Zetterberg, H.; Blennow, K. Plasma Amyloid-β (Aβ42) Correlates with Cerebrospinal Fluid Aβ42 in Alzheimer’s Disease. J. Alzheimers. Dis. 2018, 62, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Jäkel, L.; De Kort, A.M.; Klijn, C.J.M.; Schreuder, F.H.B.M.; Verbeek, M.M. Prevalence of cerebral amyloid angiopathy: A systematic review and meta-analysis. Alzheimer’s Dement. 2022, 18, 10–28. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P.; Seeman, N. Alzheimer’s disease: β-amyloid plaque formation in human brain. Synapse 2011, 65, 1289–1297. [Google Scholar] [CrossRef]
- Jongbloed, W.; Bruggink, K.A.; Kester, M.I.; Visser, P.J.; Scheltens, P.; Blankenstein, M.A.; Verbeek, M.M.; Teunissen, C.E.; Veerhuis, R. Amyloid-β oligomers relate to cognitive decline in alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Arbel-Ornath, M.; Hudry, E.; Boivin, J.R.; Hashimoto, T.; Takeda, S.; Kuchibhotla, K.V.; Hou, S.; Lattarulo, C.R.; Belcher, A.M.; Shakerdge, N.; et al. Soluble oligomeric amyloid-β induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol. Neurodegener. 2017, 12, 27. [Google Scholar] [CrossRef]
- Zampar, S.; Klafki, H.W.; Sritharen, K.; Wiltfang, T.A.B.J.; Rostagno, A.; Ghiso, J.; Miles, L.A.; Wirths, O. N-terminal heterogeneity of parenchymal and vascular amyloid-β deposits in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2020, 46, 673–685. [Google Scholar] [CrossRef]
- Gravina, S.A.; Ho, L.; Eckman, C.B.; Long, K.E.; Otvos, L.; Younkin, L.H.; Suzuki, N.; Younkin, S.G. Amyloid β protein (Aβ) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at Aβ40 or Aβ42(43). J. Biol. Chem. 1995, 270, 7013–7016. [Google Scholar] [CrossRef]
- Moro, M.L.; Phillips, A.S.; Gaimster, K.; Paul, C.; Mudher, A.; Nicoll, J.A.R.; Boche, D. Pyroglutamate and Isoaspartate modified Amyloid-Beta in ageing and Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 3. [Google Scholar] [CrossRef]
- Antonyan, A.; Schlenzig, D.; Schilling, S.; Naumann, M.; Sharoyan, S.; Mardanyan, S.; Demuth, H.U. Concerted action of dipeptidyl peptidase IV and glutaminyl cyclase results in formation of pyroglutamate-modified amyloid peptides in vitro. Neurochem. Int. 2018, 113, 112–119. [Google Scholar] [CrossRef]
- Hartlage-Rübsamen, M.; Morawski, M.; Waniek, A.; Jäger, C.; Zeitschel, U.; Koch, B.; Cynis, H.; Schilling, S.; Schliebs, R.; Demuth, H.U.; et al. Glutaminyl cyclase contributes to the formation of focal and diffuse pyroglutamate (pGlu)-Aβ deposits in hippocampus via distinct cellular mechanisms. Acta Neuropathol. 2011, 121, 705–719. [Google Scholar] [CrossRef]
- Morawski, M.; Schilling, S.; Kreuzberger, M.; Waniek, A.; Jäger, C.; Koch, B.; Cynis, H.; Kehlen, A.; Arendt, T.; Hartlage-Rübsamen, M.; et al. Glutaminyl cyclase in human cortex: Correlation with (pGlu)-amyloid-β load and cognitive decline in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 39, 385–400. [Google Scholar] [CrossRef]
- Drummond, E.; Kavanagh, T.; Pires, G.; Marta-Ariza, M.; Kanshin, E.; Nayak, S.; Faustin, A.; Berdah, V.; Ueberheide, B.; Wisniewski, T. The amyloid plaque proteome in early onset Alzheimer’s disease and Down syndrome. Acta Neuropathol. Commun. 2022, 10, 53. [Google Scholar] [CrossRef]
- Neddens, J.; Daurer, M.; Flunkert, S.; Beutl, K.; Loeffler, T.; Walker, L.; Attems, J.; Hutter-Paier, B. Correlation of pyroglutamate amyloid β and ptau Ser202/Thr205 levels in Alzheimer’s disease and related murine models. PLoS ONE 2020, 15, e0235543. [Google Scholar] [CrossRef]
- Gunn, A.P.; Wong, B.X.; McLean, C.; Fowler, C.; Barnard, P.J.; Duce, J.A.; Roberts, B.R. Increased glutaminyl cyclase activity in brains of Alzheimer’s disease individuals. J. Neurochem. 2021, 156, 979–987. [Google Scholar] [CrossRef]
- Sofola-Adesakin, O.; Khericha, M.; Snoeren, I.; Tsuda, L.; Partridge, L. pGluAβ increases accumulation of Aβ and exacerbates toxicity. Acta Neuropathol. Commun. 2016, 4, 109. [Google Scholar] [CrossRef]
- Elder, G.A.; Sosa, M.A.G.; De Gasperi, R.; Dickstein, D.L.; Hof, P.R. Presenilin transgenic mice as models of Alzheimer’s disease. Brain Struct. Funct. 2010, 214, 127–143. [Google Scholar] [CrossRef]
- Grüninger-Leitch, F.; Schlatter, D.; Küng, E.; Nelböck, P.; Döbeli, H. Substrate and inhibitor profile of BACE (β-secretase) and comparison with other mammalian aspartic proteases. J. Biol. Chem. 2002, 277, 4687–4693. [Google Scholar] [CrossRef]
- Luo, Y.; Bolon, B.; Damore, M.A.; Fitzpatrick, D.; Liu, H.; Zhang, J.; Yan, Q.; Vassar, R.; Citron, M. BACE1 (β-secretase) knockout mice do not acquire compensatory gene expression changes or develop neural lesions over time. Neurobiol. Dis. 2003, 14, 81–88. [Google Scholar] [CrossRef]
- Yan, R.; Robert Vassar, P. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014, 13, 319–329. [Google Scholar] [CrossRef]
- Vassar, R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 89. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Maloney, B.; Long, J.M.; Greig, N.H. Lessons from a BACE1 inhibitor trial: Off-site but not off base. Alzheimer’s Dement. 2014, 10, S411–S419. [Google Scholar] [CrossRef]
- Willis, B.A.; Lowe, S.L.; Monk, S.A.; Cocke, P.J.; Aluise, C.D.; Boggs, L.N.; Borders, A.R.; Brier, R.A.; Dean, R.A.; Green, S.J.; et al. Robust Pharmacodynamic Effect of LY3202626, a Central Nervous System Penetrant, Low Dose BACE1 Inhibitor, in Humans and Nonclinical Species. J. Alzheimer’s Dis. Reports 2022, 6, 1–15. [Google Scholar] [CrossRef]
- Xia, Q.; Yang, X.Y.; Shi, J.B.; Liu, Z.J.; Peng, Y.H.; Wang, W.J.; Li, B.W.; Zhao, Y.; Xiao, J.Y.; Huang, L.; et al. The Protective A673T Mutation of Amyloid Precursor Protein (APP) in Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 4038–4050. [Google Scholar] [CrossRef]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guldo, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; et al. Immunization with amyloid-β attenuates Alzheimer disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef]
- Menendez-Gonzalez, M.; Perez-Pinera, P.; Martinez-Rivera, M.; Lopez Muniz, A.; Vega, J.A. Immunotherapy for Alzheimer’s Disease: Rational Basis in Ongoing Clinical Trials. Curr. Pharm. Des. 2011, 17, 508–520. [Google Scholar] [CrossRef]
- Relkin, N.R.; Thomas, R.G.; Rissman, R.A.; Brewer, J.B.; Rafii, M.S.; Van Dyck, C.H.; Jack, C.R.; Sano, M.; Knopman, D.S.; Raman, R.; et al. A phase 3 trial of IV immunoglobulin for Alzheimer disease. Neurology 2017, 88, 1768–1775. [Google Scholar] [CrossRef]
- Kile, S.; Au, W.; Parise, C.; Rose, K.; Donnel, T.; Hankins, A.; Chan, M.; Ghassemi, A. IVIG treatment of mild cognitive impairment due to Alzheimer’s disease: A randomised double-blinded exploratory study of the effect on brain atrophy, cognition and conversion to dementia. J. Neurol. Neurosurg. Psychiatry 2017, 88, 106–112. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two Phase 3 Trials of Bapineuzumab in Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef]
- Crespi, G.A.N.; Hermans, S.J.; Parker, M.W.; Miles, L.A. Molecular basis for mid-region amyloid-β capture by leading Alzheimer’s disease immunotherapies. Sci. Rep. 2015, 5, 2–6. [Google Scholar] [CrossRef]
- Willis, B.A.; Sundell, K.; Lachno, D.R.; Ferguson-Sells, L.R.; Case, M.G.; Holdridge, K.; DeMattos, R.B.; Raskin, J.; Siemers, E.R.; Dean, R.A. Central pharmacodynamic activity of solanezumab in mild Alzheimer’s disease dementia. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 652–660. [Google Scholar] [CrossRef]
- Yoshida, K.; Moein, A.; Bittner, T.; Ostrowitzki, S.; Lin, H.; Honigberg, L.; Jin, J.Y.; Quartino, A. Pharmacokinetics and pharmacodynamic effect of crenezumab on plasma and cerebrospinal fluid beta-amyloid in patients with mild-to-moderate Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 16. [Google Scholar] [CrossRef]
- Yang, T.; Dang, Y.; Ostaszewski, B.; Mengel, D.; Steffen, V.; Rabe, C.; Bittner, T.; Walsh, D.M.; Selkoe, D.J. Target engagement in an alzheimer trial: Crenezumab lowers amyloid β oligomers in cerebrospinal fluid. Ann. Neurol. 2019, 86, 215–224. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801—The first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimer’s Res. Ther. 2020, 12, 95. [Google Scholar] [CrossRef]
- Yang, P.; Sun, F. Aducanumab: The first targeted Alzheimer’s therapy. Drug Discov. Ther. 2021, 15, 166–168. [Google Scholar] [CrossRef]
- Cummings, J.; Rabinovici, G.D.; Atri, A.; Aisen, P.; Apostolova, L.G.; Hendrix, S.; Sabbagh, M.; Selkoe, D.; Weiner, M.; Salloway, S. Aducanumab: Appropriate Use Recommendations Update. J. Prev. Alzheimer’s Dis. 2022, 9, 221–230. [Google Scholar] [CrossRef]
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimer’s Dement. 2021, 17, 696–701. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.; Lemere, C.; Atri, A.; Sabbagh, M.; Salloway, S. Aducanumab produced a clinically meaningful benefit in association with amyloid lowering. Alzheimer’s Res. Ther. 2021, 13, 10–12. [Google Scholar] [CrossRef]
- VandeVrede, L.; Gibbs, D.M.; Koestler, M.; La Joie, R.; Ljubenkov, P.A.; Provost, K.; Soleimani-Meigooni, D.; Strom, A.; Tsoy, E.; Rabinovici, G.D.; et al. Symptomatic amyloid-related imaging abnormalities in an APOE ε4/ε4 patient treated with aducanumab. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12101. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- Lowe, S.L.; Duggan Evans, C.; Shcherbinin, S.; Cheng, Y.J.; Willis, B.A.; Gueorguieva, I.; Lo, A.C.; Fleisher, A.S.; Dage, J.L.; Ardayfio, P.; et al. Donanemab (LY3002813) Phase 1b Study in Alzheimer’s Disease: Rapid and Sustained Reduction of Brain Amyloid Measured by Florbetapir F18 Imaging. J. Prev. Alzheimer’s Dis. 2021, 8, 414–424. [Google Scholar] [CrossRef]
- Hultqvist, G.; Syvänen, S.; Fang, X.T.; Lannfelt, L.; Sehlin, D. Bivalent brain shuttle increases antibody uptake by monovalent binding to the transferrin receptor. Theranostics 2017, 7, 308–318. [Google Scholar] [CrossRef]
- Weber, F.; Bohrmann, B.; Niewoehner, J.; Fischer, J.A.A.; Rueger, P.; Tiefenthaler, G.; Moelleken, J.; Bujotzek, A.; Brady, K.; Singer, T.; et al. Brain Shuttle Antibody for Alzheimer’s Disease with Attenuated Peripheral Effector Function due to an Inverted Binding Mode. Cell Rep. 2018, 22, 149–162. [Google Scholar] [CrossRef]
- Tian Hui Kwan, A.; Arfaie, S.; Therriault, J.; Rosa-Neto, P.; Gauthier, S. Lessons Learnt from the Second Generation of Anti-Amyloid Monoclonal Antibodies Clinical Trials. Dement. Geriatr. Cogn. Disord. 2021, 49, 334–348. [Google Scholar] [CrossRef]
- Withington, C.G.; Turner, R.S. Amyloid-Related Imaging Abnormalities With Anti-amyloid Antibodies for the Treatment of Dementia Due to Alzheimer’s Disease. Front. Neurol. 2022, 13, 862369. [Google Scholar] [CrossRef]
- Sperling, R.A.; Jack, C.R.; Black, S.E.; Frosch, M.P.; Greenberg, S.M.; Hyman, B.T.; Scheltens, P.; Carrillo, M.C.; Thies, W.; Bednar, M.M.; et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimer’s Dement. 2011, 7, 367–385. [Google Scholar] [CrossRef]
- Chouhan, N.B.; Siegel, G.J. Intracerebroventricular passive immunization in transgenic mouse models of Alzheimer’s disease. Expert Rev. Vaccines 2004, 3, 717–725. [Google Scholar] [CrossRef]
- Cacabelos, R. How plausible is an Alzheimer’s disease vaccine? Expert Opin. Drug Discov. 2020, 15, 1–6. [Google Scholar] [CrossRef]
- Asuni, A.A.; Boutajangout, A.; Scholtzova, H.; Knudsen, E.; Li, Y.S.; Quartermain, D.; Frangione, B.; Wisniewski, T.; Sigurdsson, E.M. Vaccination of Alzheimer’s model mice with Aβ derivative in alum adjuvant reduces Aβ burden without microhemorrhages. Eur. J. Neurosci. 2006, 24, 2530–2542. [Google Scholar] [CrossRef]
- Gilman, S.; Koller, M.; Black, R.S.; Jenkins, L.; Griffith, S.G.; Fox, N.C.; Eisner, L.; Kirby, L.; Rovira, M.B.; Forette, F.; et al. Clinical Effects of Abeta immunization(AN1792) in patients with AD in an interrupted trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef]
- Fox, N.C.; Black, R.S.; Gilman, S.; Rossor, M.N.; Griffith, S.G.; Jenkins, L.; Koller, M. Effects of Aβ immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 2005, 64, 1563–1572. [Google Scholar] [CrossRef]
- Vellas, B.; Black, R.; Thal, L.; Fox, N.; Daniels, M.; McLennan, G.; Tompkins, C.; Leibman, C.; Pomfret, M.; Grundman, M. Long-Term Follow-Up of Patients Immunized with AN1792: Reduced Functional Decline in Antibody Responders. Curr. Alzheimer Res. 2009, 6, 144–151. [Google Scholar] [CrossRef]
- Hull, M.; Sadowsky, C.; Arai, H.; Leterme, G.L.P.; Holstein, A.; Booth, K.; Peng, Y.; Yoshiyama, T.; Suzuki, H.; Ketter, N.; et al. Long-Term Extensions of Randomized Vaccination Trials of ACC-001 and QS-21 in Mild to Moderate Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 696–708. [Google Scholar] [CrossRef]
- Mandler, M.; Santic, R.; Gruber, P.; Cinar, Y.; Pichler, D.; Funke, S.A.; Willbold, D.; Schneeberger, A.; Schmidt, W.; Mattner, F. Tailoring the antibody response to aggregated aβ using novel Alzheimer-vaccines. PLoS ONE 2015, 10, e0115237. [Google Scholar] [CrossRef]
- Vassilakopoulou, V.; Karachaliou, C.E.; Evangelou, A.; Zikos, C.; Livaniou, E. Peptide-based vaccines for neurodegenerative diseases: Recent endeavors and future perspectives. Vaccines 2021, 9, 1278. [Google Scholar] [CrossRef]
- Winblad, B.; Andreasen, N.; Minthon, L.; Floesser, A.; Imbert, G.; Dumortier, T.; Maguire, R.P.; Blennow, K.; Lundmark, J.; Staufenbiel, M.; et al. Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: Randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012, 11, 597–604. [Google Scholar] [CrossRef]
- Vandenberghe, R.; Riviere, M.E.; Caputo, A.; Sovago, J.; Maguire, R.P.; Farlow, M.; Marotta, G.; Sanchez-Valle, R.; Scheltens, P.; Ryan, J.M.; et al. Active Aβ immunotherapy CAD106 in Alzheimer’s disease: A phase 2b study. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 10–22. [Google Scholar] [CrossRef]
- Muhs, A.; Hickman, D.T.; Pihlgren, M.; Chuard, N.; Giriens, V.; Meerschman, C.; Van Der Auwera, I.; Van Leuven, F.; Sugawara, M.; Weingertner, M.C.; et al. Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proc. Natl. Acad. Sci. USA 2007, 104, 9810–9815. [Google Scholar] [CrossRef]
- Wang, C.Y.; Wang, P.N.; Chiu, M.J.; Finstad, C.L.; Lin, F.; Lynn, S.; Tai, Y.H.; De Fang, X.; Zhao, K.; Hung, C.H.; et al. UB-311, a novel UBITh® amyloid β peptide vaccine for mild Alzheimer’s disease. Alzheimer’s Dement. 2017, 3, 262–272. [Google Scholar] [CrossRef]
- Lacosta, A.M.; Pascual-Lucas, M.; Pesini, P.; Casabona, D.; Pérez-Grijalba, V.; Marcos-Campos, I.; Sarasa, L.; Canudas, J.; Badi, H.; Monleón, I.; et al. Safety, tolerability and immunogenicity of an active anti-Aβ 40 vaccine (ABvac40) in patients with Alzheimer’s disease: A randomised, double-blind, placebo-controlled, phase i trial. Alzheimer’s Res. Ther. 2018, 10, 12. [Google Scholar] [CrossRef]
- Grundman, M.; Morgan, R.; Lickliter, J.D.; Schneider, L.S.; DeKosky, S.; Izzo, N.J.; Guttendorf, R.; Higgin, M.; Pribyl, J.; Mozzoni, K.; et al. A phase 1 clinical trial of the sigma-2 receptor complex allosteric antagonist CT1812, a novel therapeutic candidate for Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Izzo, N.J.; Yuede, C.M.; LaBarbera, K.M.; Limegrover, C.S.; Rehak, C.; Yurko, R.; Waybright, L.; Look, G.; Rishton, G.; Safferstein, H.; et al. Preclinical and clinical biomarker studies of CT1812: A novel approach to Alzheimer’s disease modification. Alzheimer’s Dement. 2021, 17, 1365–1382. [Google Scholar] [CrossRef] [PubMed]
- Hey, J.A.; Kocis, P.; Hort, J.; Abushakra, S.; Power, A.; Vyhnálek, M.; Yu, J.Y.; Tolar, M. Discovery and Identification of an Endogenous Metabolite of Tramiprosate and Its Prodrug ALZ-801 that Inhibits Beta Amyloid Oligomer Formation in the Human Brain. CNS Drugs 2018, 32, 849–861. [Google Scholar] [CrossRef]
- Manzano, S.; Agüera, L.; Aguilar, M.; Olazarán, J. A Review on Tramiprosate (Homotaurine) in Alzheimer’s Disease and Other Neurocognitive Disorders. Front. Neurol. 2020, 11, 614. [Google Scholar] [CrossRef]
- Hey, J.A.; Yu, J.Y.; Versavel, M.; Abushakra, S.; Kocis, P.; Power, A.; Kaplan, P.L.; Amedio, J.; Tolar, M. Clinical Pharmacokinetics and Safety of ALZ-801, a Novel Prodrug of Tramiprosate in Development for the Treatment of Alzheimer’s Disease. Clin. Pharmacokinet. 2018, 57, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Hallikainen, M.; Grimmer, T.; Duning, T.; Gouw, A.A.; Teunissen, C.E.; Wink, A.M.; Maruff, P.; Harrison, J.; Van Baal, C.M.; et al. Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer’s disease: Results of a randomized, double-blind, placebo-controlled phase 2a study. Alzheimer’s Res. Ther. 2018, 10, 107. [Google Scholar] [CrossRef]
- Vijverberg, E.G.B.; Axelsen, T.M.; Bihlet, A.R.; Henriksen, K.; Weber, F.; Fuchs, K.; Harrison, J.E.; Kühn-Wache, K.; Alexandersen, P.; Prins, N.D.; et al. Rationale and study design of a randomized, placebo-controlled, double-blind phase 2b trial to evaluate efficacy, safety, and tolerability of an oral glutaminyl cyclase inhibitor varoglutamstat (PQ912) in study participants with MCI and mild AD—VIVIAD. Alzheimer’s Res. Ther. 2021, 13, 142. [Google Scholar] [CrossRef]
- Cynis, H.; Scheel, E.; Saido, T.C.; Schilling, S.; Demuth, H.U. Amyloidogenic processing of amyloid precursor protein: Evidence of a pivotal role of glutaminyl cyclase in generation of pyroglutamate-modified amyloid-β. Biochemistry 2008, 47, 7405–7413. [Google Scholar] [CrossRef]
- Brooksa, A.F.; Jacksona, I.M.; Shaoa, X.; Kropoga, G.W.; Shermana, P.; Quesadaa, C.A.; Scotta, P.J.H. Synthesis and evaluation of [11C]PBD150, a radiolabeled glutaminyl cyclase inhibitor for the potential detection of Alzheimer’s disease prior to amyloid β aggregation. Medchemcomm 2015, 1, 1065–1068. [Google Scholar] [CrossRef]
- Pereira, J.B.; Janelidze, S.; Ossenkoppele, R.; Kvartsberg, H.; Brinkmalm, A.; Mattsson-Carlgren, N.; Stomrud, E.; Smith, R.; Zetterberg, H.; Blennow, K.; et al. Untangling the association of amyloid-β and tau with synaptic and axonal loss in Alzheimer’s disease. Brain 2021, 144, 310–324. [Google Scholar] [CrossRef]
- Stancu, I.; Vasconcelos, B.; Terwel, D.; Dewachter, I. Models of beta-amyloid induced Tau-pathology: The long and folded road to understand the mechanism. Mol. Neurodegener. 2014, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimers. Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.H.; Kumar, S.; Pinotsi, D.; Tunnacliffe, A.; George-Hyslop, P.S.; Mandelkow, E.; Mandelkow, E.M.; Kaminski, C.F.; Schierle, G.S.K. Extracellular monomeric tau protein is sufficient to initiate the spread of tau protein pathology. J. Biol. Chem. 2014, 289, 956–967. [Google Scholar] [CrossRef]
- Sebastián-Serrano, Á.; De Diego-García, L.; Díaz-Hernández, M. The neurotoxic role of extracellular tau protein. Int. J. Mol. Sci. 2018, 19, 998. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Simón, D.; Díaz-Hernández, M.; Pintor, J.; Hernández, F. Sources of extracellular tau and its signaling. J. Alzheimer’s Dis. 2014, 40, 7–15. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, M.; Wang, D. The propagation mechanisms of extracellular tau in Alzheimer’s disease. J. Neurol. 2022, 269, 1164–1181. [Google Scholar] [CrossRef]
- Zeng, Y.; Yang, J.; Zhang, B.; Gao, M.; Su, Z.; Huang, Y. The structure and phase of tau: From monomer to amyloid filament. Cell. Mol. Life Sci. 2021, 78, 1873–1886. [Google Scholar] [CrossRef]
- Sopko, R.; Golonzhka, O.; Arndt, J.; Quan, C.; Czerkowicz, J.; Cameron, A.; Smith, B.; Murugesan, Y.; Gibbons, G.; Kim, S.J.; et al. Characterization of tau binding by gosuranemab. Neurobiol. Dis. 2020, 146, 105120. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Litvan, I.; Mendonca, N.; Wang, D.; Zheng, H.; Rendenbach-Mueller, B.; Lon, H.K.; Jin, Z.; Fisseha, N.; Budur, K.; et al. Safety and efficacy of tilavonemab in progressive supranuclear palsy: A phase 2, randomised, placebo-controlled trial. Lancet Neurol. 2021, 20, 182–192. [Google Scholar] [CrossRef]
- West, T.; Hu, Y.; Verghese, P.B.; Bateman, R.J.; Braunstein, J.B.; Fogelman, I.; Budur, K.; Florian, H.; Mendonca, N.; Holtzman, D.M. Preclinical and Clinical Development of ABBV-8E12, a Humanized Anti-Tau Antibody, for Treatment of Alzheimer’s Disease and Other Tauopathies. J. Prev. Alzheimer’s Dis. 2017, 4, 236–241. [Google Scholar] [CrossRef]
- Teng, E.; Manser, P.T.; Pickthorn, K.; Brunstein, F.; Blendstrup, M.; Bohorquez, S.S. Safety and Efficacy of Semorinemab in individuals with prodromal to MIld Alzheimer disease. A Randomized Clinical Trial. JAMA Neurol. 2022, 79, 758–767. [Google Scholar] [CrossRef]
- Barthélemy, N.R.; Horie, K.; Sato, C.; Bateman, R.J. Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. J. Exp. Med. 2020, 217, 1–12. [Google Scholar] [CrossRef]
- Palmqvist, S.; Tideman, P.; Cullen, N.; Zetterberg, H.; Blennow, K.; Dage, J.L.; Stomrud, E.; Janelidze, S.; Mattsson-Carlgren, N.; Hansson, O. Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nat. Med. 2021, 27, 1034–1042. [Google Scholar] [CrossRef]
- Kondo, A.; Shahpasand, K.; Mannix, R.; Qiu, J.; Moncaster, J.; Chen, C.; Yao, Y.; Lin, Y.; Driver, J.A.; Sun, Y.; et al. Cis P-Tau: Early Driver of Brain Injury and Tauopathy Blocked By Antibody. Nature 2015, 523, 431–436. [Google Scholar] [CrossRef]
- Lu, K.P.; Kondo, A.; Albayram, O.; Herbert, M.K.; Liu, H.; Zhou, X.Z. Potential of the antibody against cis-phosphorylated tau in the early diagnosis, treatment, and prevention of Alzheimer disease and brain injury. JAMA Neurol. 2016, 73, 1356–1362. [Google Scholar] [CrossRef]
- Courade, J.P.; Angers, R.; Mairet-Coello, G.; Pacico, N.; Tyson, K.; Lightwood, D.; Munro, R.; McMillan, D.; Griffin, R.; Baker, T.; et al. Epitope determines efficacy of therapeutic anti-Tau antibodies in a functional assay with human Alzheimer Tau. Acta Neuropathol. 2018, 136, 729–745. [Google Scholar] [CrossRef]
- Theunis, C.; Crespo-Biel, N.; Gafner, V.; Pihlgren, M.; López-Deber, M.P.; Reis, P.; Hickman, D.T.; Adolfsson, O.; Chuard, N.; Ndao, D.M.; et al. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in Tau.P301L mice that model tauopathy. PLoS ONE 2013, 8, e72301. [Google Scholar] [CrossRef]
- Novak, P.; Zilka, N.; Zilkova, M.; Kovacech, B.; Skrabana, R.; Ondrus, M.; Fialova, L.; Kontsekova, E.; Otto, M.; Novak, M. AADvac1, an Active Immunotherapy for Alzheimer’s Disease and Non Alzheimer Tauopathies: An Overview of Preclinical and Clinical Development. J. Prev. Alzheimer’s Dis. 2019, 6, 63–69. [Google Scholar] [CrossRef]
- Novak, P.; Schmidt, R.; Kontsekova, E.; Kovacech, B.; Smolek, T.; Katina, S.; Fialova, L.; Prcina, M.; Parrak, V.; Dal-Bianco, P.; et al. FUNDAMANT: An interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 108. [Google Scholar] [CrossRef]
- Eikelenboom, P.; Van Exel, E.; Hoozemans, J.J.M.; Veerhuis, R.; Rozemuller, A.J.M.; Van Gool, W.A. Neuroinflammation—An early event in both the history and pathogenesis of Alzheimer’s disease. Neurodegener. Dis. 2010, 7, 38–41. [Google Scholar] [CrossRef]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Pan, Y.T.; Zhang, Z.Y.; Yang, H.; Yu, S.Y.; Zheng, Y.; Ma, J.H.; Wang, X.M. Systemic activation of NLRP3 inflammasome and plasma α-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J. Neuroinflamm. 2020, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Zhang, Q. Activation of NLRP3 Inflammasome and Onset of Alzheimer’s Disease. Front. Immunol. 2021, 12, 2998. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Sun, Y.; Peng, G. Neuroinflammation as a Potential Therapeutic Target in Alzheimer’s Disease. Clin. Interv. Aging 2022, 17, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 2017, 140, 792–803. [Google Scholar] [CrossRef]
- Wang, M.-M.; Miao, D.; Cao, X.-P.; Tan, L.; Tan, L. Innate immune activation in Alzheimer’s disease. Ann. Transl. Med. 2018, 6, 177. [Google Scholar] [CrossRef]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 179. [Google Scholar] [CrossRef]
- Llorens, F.; Thüne, K.; Tahir, W.; Kanata, E.; Diaz-Lucena, D.; Xanthopoulos, K.; Kovatsi, E.; Pleschka, C.; Garcia-Esparcia, P.; Schmitz, M.; et al. YKL-40 in the brain and cerebrospinal fluid of neurodegenerative dementias. Mol. Neurodegener. 2017, 12, 83. [Google Scholar] [CrossRef]
- Wennström, M.; Surova, Y.; Hall, S.; Nilsson, C.; Minthon, L.; Hansson, O.; Nielsen, H.M. The inflammatory marker YKL-40 is elevated in cerebrospinal fluid from patients with Alzheimer’s but not Parkinson’s disease or dementia with Lewy bodies. PLoS ONE 2015, 10, e0135458. [Google Scholar] [CrossRef]
- Li, J.; Shui, X.; Sun, R.; Wan, L.; Zhang, B.; Xiao, B.; Luo, Z. Microglial Phenotypic Transition: Signaling Pathways and Influencing Modulators Involved in Regulation in Central Nervous System Diseases. Front. Cell. Neurosci. 2021, 15, 736310. [Google Scholar] [CrossRef]
- Sobue, A.; Komine, O.; Hara, Y.; Endo, F.; Mizoguchi, H.; Watanabe, S.; Murayama, S.; Saito, T.; Saido, T.C.; Sahara, N.; et al. Microglial gene signature reveals loss of homeostatic microglia associated with neurodegeneration of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, Y.; Wang, D.; Zhang, J.; Zhang, F. NSAID exposure and risk of Alzheimer’s disease: An updated meta-analysis from cohort studies. Front. Aging Neurosci. 2018, 10, 83. [Google Scholar] [CrossRef]
- Ettcheto, M.; Sánchez-Lopez, E.; Cano, A.; Carrasco, M.; Herrera, K.; Manzine, P.R.; Espinosa-Jimenez, T.; Busquets, O.; Verdaguer, E.; Olloquequi, J.; et al. Dexibuprofen ameliorates peripheral and central risk factors associated with Alzheimer’s disease in metabolically stressed APPswe/PS1dE9 mice. Cell Biosci. 2021, 11, 141. [Google Scholar] [CrossRef] [PubMed]
- Ettcheto, M.; Sánchez-López, E.; Pons, L.; Busquets, O.; Olloquequi, J.; Beas-Zarate, C.; Pallas, M.; García, M.L.; Auladell, C.; Folch, J.; et al. Dexibuprofen prevents neurodegeneration and cognitive decline in APPswe/PS1dE9 through multiple signaling pathways. Redox Biol. 2017, 13, 345–352. [Google Scholar] [CrossRef]
- Meyer, P.-F.; Tremblay-Mercier, J.; Leoutsakos, J.; Madjar, C.; Lafaille-Maignan, M.-É.; Savard, M.; Rosa-Neto, P.; Poirier, J.; Etienne, P.; Breitner, J. Intrepad. Neurology 2019, 92, e2070–e2080. [Google Scholar] [CrossRef]
- Aisen, P.S.; Schafer, K.A.; Grundman, M.; Pfeiffer, E.; Sano, M.; Davis, K.L.; Farlow, M.R.; Jin, S.; Thomas, R.G.; Thal, L.J. Effects of Rofecoxib or Naproxen vs Placebo on Alzheimer Disease Progression: A Randomized Controlled Trial. J. Am. Med. Assoc. 2003, 289, 2819–2826. [Google Scholar] [CrossRef]
- Hori, Y.; Takeda, S.; Cho, H.; Wegmann, S.; Shoup, T.M.; Takahashi, K.; Irimia, D.; Elmaleh, D.R.; Hyman, B.T.; Hudry, E. A Food and Drug Administration-approved asthma therapeutic agent impacts amyloid β in the brain in a transgenic model of Alzheimer disease. J. Biol. Chem. 2015, 290, 1966–1978. [Google Scholar] [CrossRef]
- Zhang, C.; Griciuc, A.; Hudry, E.; Wan, Y.; Quinti, L.; Ward, J.; Forte, A.M.; Shen, X.; Ran, C.Z.; Elmaleh, D.R.; et al. Cromolyn Reduces Levels of the Alzheimer’s Disease-Associated Amyloid β-Protein by Promoting Microglial Phagocytosis. Sci. Rep. 2018, 8, 1144. [Google Scholar] [CrossRef]
- Wang, Y.J.; Monteagudo, A.; Downey, M.A.; Ashton-Rickardt, P.G.; Elmaleh, D.R. Cromolyn inhibits the secretion of inflammatory cytokines by human microglia (HMC3). Sci. Rep. 2021, 11, 8054. [Google Scholar] [CrossRef]
- Steeland, S.; Gorlé, N.; Vandendriessche, C.; Balusu, S.; Brkic, M.; Van Cauwenberghe, C.; Van Imschoot, G.; Van Wonterghem, E.; De Rycke, R.; Kremer, A.; et al. Counteracting the effects of TNF receptor-1 has therapeutic potential in Alzheimer’s disease. EMBO Mol. Med. 2018, 10, e8300. [Google Scholar] [CrossRef]
- De Sousa Rodrigues, M.E.; Houser, M.C.; Walker, D.I.; Jones, D.P.; Chang, J.; Barnum, C.J.; Tansey, M.G. Targeting soluble tumor necrosis factor as a potential intervention to lower risk for late-onset Alzheimer’s disease associated with obesity, metabolic syndrome, and type 2 diabetes. Alzheimer’s Res. Ther. 2019, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- MacPhersona, K.P.; Sompolb, P.; Kannarkata, G.T.; Changa, J.; Sniffena, L.; Wildnera, M.E.; Norrisb, C.M. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 2017, 102, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Tufan, A.N.; Tufan, F. Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 2015, 85, 2083–2084. [Google Scholar] [CrossRef] [PubMed]
- Renee, M.; Elmore, P.; Najafi, A.R.; Koike, M.A.; Nazih, N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Nguyen, H.; West, B.L.; et al. CSF1 receptor signaling is necessary for microglia viability, which unmasks a cell that rapidly repopulates the microglia-depleted adult brain. Neuron 2015, 82, 380–397. [Google Scholar] [CrossRef]
- Pons, V.; Lévesque, P.; Plante, M.M.; Rivest, S. Conditional genetic deletion of CSF1 receptor in microglia ameliorates the physiopathology of Alzheimer’s disease. Alzheimer’s Res. Ther. 2021, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef]
- Lei, F.; Cui, N.; Zhou, C.; Chodosh, J.; Vavvas, D.G.; Paschalis, E.I. CSF1R inhibition by a small-molecule inhibitor is not microglia specific; Affecting hematopoiesis and the function of macrophages. Proc. Natl. Acad. Sci. USA 2020, 117, 23336–23338. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Teng, Z.; Liu, C.; Li, Q.; Yin, Y.; Tang, Y. TREM2, microglia, and Alzheimer’s disease. Mech. Ageing Dev. 2021, 195, 111438. [Google Scholar] [CrossRef]
- Zhong, L.; Zhang, Z.L.; Li, X.; Liao, C.; Mou, P.; Wang, T.; Wang, Z.; Wang, Z.; Wei, M.; Xu, H.; et al. TREM2/DAP12 complex regulates inflammatory responses in Microglia via the JNK signaling pathway. Front. Aging Neurosci. 2017, 9, 204. [Google Scholar] [CrossRef]
- Cheng-Hathaway, P.J.; Reed-Geaghan, E.G.; Jay, T.R.; Casali, B.T.; Bemiller, S.M.; Puntambekar, S.S.; Von Saucken, V.E.; Williams, R.Y.; Karlo, J.C.; Moutinho, M.; et al. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 29. [Google Scholar] [CrossRef] [PubMed]
- Fassler, M.; Rappaport, M.S.; Cuño, C.B.; George, J. Engagement of TREM2 by a novel monoclonal antibody induces activation of microglia and improves cognitive function in Alzheimer’s disease models. J. Neuroinflamm. 2021, 18, 19. [Google Scholar] [CrossRef] [PubMed]
- Price, B.R.; Sudduth, T.L.; Weekman, E.M.; Johnson, S.; Hawthorne, D.; Woolums, A.; Wilcock, D.M. Therapeutic Trem2 activation ameliorates amyloid-beta deposition and improves cognition in the 5XFAD model of amyloid deposition. J. Neuroinflamm. 2020, 17, 238. [Google Scholar] [CrossRef]
- Schlepckow, K.; Monroe, K.M.; Kleinberger, G.; Cantuti-Castelvetri, L.; Parhizkar, S.; Xia, D.; Willem, M.; Werner, G.; Pettkus, N.; Brunner, B.; et al. Enhancing protective microglial activities with a dual function TREM 2 antibody to the stalk region. EMBO Mol. Med. 2020, 12, e11227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L. CD33 in Alzheimer’s disease—Biology, pathogenesis, and therapeutics: A mini-review. Gerontology 2019, 65, 323–331. [Google Scholar] [CrossRef]
- Wißfeld, J.; Nozaki, I.; Mathews, M.; Raschka, T.; Ebeling, C.; Hornung, V.; Brüstle, O.; Neumann, H. Deletion of Alzheimer’s disease-associated CD33 results in an inflammatory human microglia phenotype. Glia 2021, 69, 1393–1412. [Google Scholar] [CrossRef]
- Miles, L.A.; Hermans, S.J.; Crespi, G.A.N.; Gooi, J.H.; Doughty, L.; Nero, T.L.; Markulić, J.; Ebneth, A.; Wroblowski, B.; Oehlrich, D.; et al. Small Molecule Binding to Alzheimer Risk Factor CD33 Promotes Aβ Phagocytosis. iScience 2019, 19, 110–118. [Google Scholar] [CrossRef]
- Siddiqui, S.S.; Springer, S.A.; Verhagen, A.; Sundaramurthy, V.; Alisson-Silva, F.; Jiang, W.; Ghosh, P.; Varki, A. The Alzheimer’s Disease–protective CD33 splice variant mediates adaptive loss of function via diversion to an intracellular pool. J. Biol. Chem. 2017, 292, 15312–15320. [Google Scholar] [CrossRef]
- Gbadamosi, M.; Meshinchi, S.; Lamba, J.K. Gemtuzumab ozogamicin for treatment of newly diagnosed CD33-positive acute myeloid leukemia. Futur. Oncol. 2018, 14, 3199–3213. [Google Scholar] [CrossRef]
- Zhang, M.; Schmitt-Ulms, G.; Sato, C.; Xi, Z.; Zhang, Y.; Zhou, Y.; George-Hyslop, P.S.; Rogaeva, E. Drug repositioning for Alzheimer’s disease based on systematic “omics” data mining. PLoS ONE 2016, 11, e0168812. [Google Scholar] [CrossRef]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Chan, P.; Wang, T.; Hong, Z.; Wang, S.; Kuang, W.; He, J. Group, phase 3 clinical trial of sodium oligomannate for mild-to-moderate. Alzheimer‘s Dement. 2021, 7, 1–11. [Google Scholar]
- Sochocka, M.; Ochnik, M.; Sobczyński, M.; Gębura, K.; Zambrowicz, A.; Naporowski, P.; Leszek, J. Ginkgo Biloba Leaf Extract Improves an Innate Immune Response of Peripheral Blood Leukocytes of Alzheimer’s Disease Patients. Nutrients 2022, 14, 2022. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Ai, D.; Sun, L.; Xu, X.; Cao, X. EGb 761 inhibits Aβ 1-42-induced neuroinflammatory response by suppressing P38 MAPK signaling pathway in BV-2 microglial cells. Neuroreport 2019, 30, 434–440. [Google Scholar] [CrossRef]
- Bowman, G.L.; Dayon, L.; Kirkland, R.; Wojcik, J.; Peyratout, G.; Severin, I.C.; Henry, H.; Oikonomidi, A.; Migliavacca, E.; Bacher, M.; et al. Blood-brain barrier breakdown, neuroinflammation, and cognitive decline in older adults. Alzheimer’s Dement. 2018, 14, 1640–1650. [Google Scholar] [CrossRef]
- Muszyski, P.; Kulczyska-Przybik, A.; Borawska, R.; Litman-Zawadzka, A.; Sowik, A.; Klimkowicz-Mrowiec, A.; Pera, J.; Dziedzic, T.; Mroczko, B. The Relationship between Markers of Inflammation and Degeneration in the Central Nervous System and the Blood-Brain Barrier Impairment in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 59, 903–912. [Google Scholar] [CrossRef]
- Rahman, M.M.; Lendel, C. Extracellular protein components of amyloid plaques and their roles in Alzheimer’s disease pathology. Mol. Neurodegener. 2021, 16, 59. [Google Scholar] [CrossRef]
- Korte, N.; Nortley, R.; Attwell, D. Cerebral blood flow decrease as an early pathological mechanism in Alzheimer’s disease. Acta Neuropathol. 2020, 140, 793–810. [Google Scholar] [CrossRef]
- Su, X.; Huang, L.; Qu, Y.; Xiao, D.; Mu, D. Pericytes in Cerebrovascular Diseases: An Emerging Therapeutic Target. Front. Cell. Neurosci. 2019, 13, 519. [Google Scholar] [CrossRef]
- Laredo, F.; Plebanski, J.; Tedeschi, A. Pericytes: Problems and Promises for CNS Repair. Front. Cell. Neurosci. 2019, 13, 546. [Google Scholar] [CrossRef]
- Miners, J.S.; Kehoe, P.G.; Love, S.; Zetterberg, H.; Blennow, K. CSF evidence of pericyte damage in Alzheimer’s disease is associated with markers of blood-brain barrier dysfunction and disease pathology. Alzheimer’s Res. Ther. 2019, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid b oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, aav9518. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fan, D.Y.; Li, H.Y.; He, C.Y.; Shen, Y.Y.; Zeng, G.H.; Chen, D.W.; Yi, X.; Ma, Y.H.; Yu, J.T.; et al. Dynamic changes of CSF sPDGFRβ during ageing and AD progression and associations with CSF ATN biomarkers. Mol. Neurodegener. 2022, 17, 9. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Korte, N.; Nortley, R.; Sethi, H.; Tang, Y.; Attwell, D. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018, 136, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Korte, N.; Ilkan, Z.; Pearson, C.L.; Pfeiffer, T.; Singhal, P.; Rock, J.R.; Sethi, H.; Gill, D.; Attwell, D.; Tammaro, P. The Ca2+-gated channel TMEM16A amplifies capillary pericyte contraction and reduces cerebral blood flow after ischemia. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Smyth, L.C.D.; Highet, B.; Jansson, D.; Wu, J.; Rustenhoven, J.; Aalderink, M.; Tan, A.; Li, S.; Johnson, R.; Coppieters, N.; et al. Characterisation of PDGF-BB:PDGFRβ signalling pathways in human brain pericytes: Evidence of disruption in Alzheimer’s disease. Commun. Biol. 2022, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- Kubis-Kubiak, A.; Dyba, A.; Piwowar, A. The interplay between diabetes and alzheimer’s disease—In the hunt for biomarkers. Int. J. Mol. Sci. 2020, 21, 2744. [Google Scholar] [CrossRef]
- Cheng, G.; Huang, C.; Deng, H.; Wang, H. Diabetes as a risk factor for dementia and mild cognitive impairment: A meta-analysis of longitudinal studies. Intern. Med. J. 2012, 42, 484–491. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Giau, V. Van Type 3 diabetes and its role implications in alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 3165. [Google Scholar] [CrossRef]
- Kleinridders, A. Deciphering Brain Insulin Receptor and Insulin-Like Growth Factor 1 Receptor Signalling. J. Neuroendocrinol. 2016, 28, 1–13. [Google Scholar] [CrossRef]
- Gonçalves, R.A.; Wijesekara, N.; Fraser, P.E.; De Felice, F.G. The link between tau and insulin signaling: Implications for alzheimer’s disease and other tauopathies. Front. Cell. Neurosci. 2019, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; et al. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, C. Brain insulin resistance: Role in neurodegenerative disease and potential for targeting. Expert Opin. Investig. Drugs 2020, 29, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Rachdaoui, N.; Polo-Parada, L.; Ismail-Beigi, F. Prolonged exposure to insulin inactivates Akt and ERK1/2 and increases pancreatic islet and InS1e B-cell apoptosis. J. Endocr. Soc. 2019, 3, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Akindehin, S.E.; Orsso, C.E.; Waldner, R.C.; DiMarchi, R.D.; Müller, T.D.; Haqq, A.M. Recent Advances in Incretin-Based Pharmacotherapies for the Treatment of Obesity and Diabetes. Front. Endocrinol. 2022, 13, 838410. [Google Scholar] [CrossRef]
- Ballard, C.; Nørgaard, C.H.; Friedrich, S.; Mørch, L.S.; Gerds, T.; Møller, D.V.; Knudsen, L.B.; Kvist, K.; Zinman, B.; Holm, E.; et al. Liraglutide and semaglutide: Pooled post hoc analysis to evaluate risk of dementia in patients with type 2 diabetes. Alzheimer’s Dement. 2020, 16, e042909. [Google Scholar] [CrossRef]
- Hayes, M.R.; Borner, T.; De Jonghe, B.C. The Role of GIP in the Regulation of GLP-1 Satiety and Nausea. Diabetes 2021, 70, 1956–1961. [Google Scholar] [CrossRef]
- Zhao, T.C. Glucagon-like peptide-1 (GLP-1) and protective effects in cardiovascular disease: A new therapeutic approach for myocardial protection. Cardiovasc. Diabetol. 2013, 12, 90. [Google Scholar] [CrossRef]
- Erbil, D.; Eren, C.Y.; Demirel, C.; Küçüker, M.U.; Solaroğlu, I.; Eser, H.Y. GLP-1′s role in neuroprotection: A systematic review. Brain Inj. 2019, 33, 734–819. [Google Scholar] [CrossRef]
- Fu, Z.; Gong, L.; Liu, J.; Wu, J.; Barrett, E.J.; Aylor, K.W.; Liu, Z. Brain Endothelial Cells Regulate Glucagon-Like Peptide 1 Entry Into the Brain via a Receptor-Mediated Process. Front. Physiol. 2020, 11, 555. [Google Scholar] [CrossRef]
- Robinson, E.; Tate, M.; Lockhart, S.; McPeake, C.; O’Neill, K.M.; Edgar, K.S.; Calderwood, D.; Green, B.D.; McDermott, B.J.; Grieve, D.J. Metabolically-inactive glucagon-like peptide-1(9-36)amide confers selective protective actions against post-myocardial infarction remodelling. Cardiovasc. Diabetol. 2016, 15, 65. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Frangou, E.; Love, S.B.; Busza, G.; Holmes, C.; Ritchie, C.; Lawrence, R.; McFarlane, B.; Tadros, G.; Ridha, B.H.; et al. Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer’s disease: Study protocol for a randomised controlled trial (ELAD study). Trials 2019, 20, 191. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.M.; Nuffer, W.; Smith, B.A. GLP-1 receptor agonists: An updated review of head-to-head clinical studies. Ther. Adv. Endocrinol. Metab. 2021, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Q.; Hölscher, C. GIP has neuroprotective effects in Alzheimer and Parkinson’s disease models. Peptides 2020, 125, 170184. [Google Scholar] [CrossRef]
- Hölscher, C. Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology 2018, 136, 251–259. [Google Scholar] [CrossRef]
- Lumsden, A.L.; Mulugeta, A.; Zhou, A.; Hyppönen, E. Apolipoprotein E (APOE) genotype-associated disease risks: A phenome-wide, registry-based, case-control study utilising the UK Biobank. EBioMedicine 2020, 59, 102954. [Google Scholar] [CrossRef]
- Husain, M.A.; Laurent, B.; Plourde, M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front. Neurosci. 2021, 15, 630502. [Google Scholar] [CrossRef]
- Chew, H.; Solomon, V.A.; Fonteh, A.N. Involvement of Lipids in Alzheimer’s Disease Pathology and Potential Therapies. Front. Physiol. 2020, 11, 598. [Google Scholar] [CrossRef] [PubMed]
- Ramkumar, S.; Raghunath, A.; Raghunath, S. Statin therapy: Review of safety and potential side effects. Acta Cardiol. Sin. 2016, 32, 631–639. [Google Scholar] [CrossRef]
- Davignon, J. Beneficial cardiovascular pleiotropic effects of statins. Circulation 2004, 109, 39–43. [Google Scholar] [CrossRef]
- Dalla, Y.; Singh, N.; Jaggi, A.S.; Singh, D. Memory restorative role of statins in experimental dementia: An evidence of their cholesterol dependent and independent actions. Pharmacol. Reports 2010, 62, 784–796. [Google Scholar] [CrossRef]
- Langness, V.F.; Van der Kant, R.; Das, U.; Wang, L.; Dos Santos Chaves, R.; Goldstein, L.S.B. Cholesterol-lowering drugs reduce APP processing to Aβ by inducing APP dimerization. Mol. Biol. Cell 2021, 32, 247–259. [Google Scholar] [CrossRef]
- Parvathy, S.; Ehrlich, M.; Pedrini, S.; Diaz, N.; Refolo, L.; Buxbaum, J.D.; Bogush, A.; Petanceska, S.; Gandy, S. Atorvastatin-induced activation of Alzheimer’s α secretase is resistant to standard inhibitors of protein phosphorylation-regulated ectodomain shedding. J. Neurochem. 2004, 90, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Drummond, E. APOE-amyloid interaction: Therapeutic targets. Neurobiol. Dis. 2021, 138, 104784. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kantor, B.; Chiba-Falek, O. Apoe: The new frontier in the development of a therapeutic target towards precision medicine in late-onset alzheimer’s. Int. J. Mol. Sci. 2021, 22, 1244. [Google Scholar] [CrossRef] [PubMed]
- Luz, I.; Liraz, O.; Michaelson, D.M. An Anti-apoE4 Specific Monoclonal Antibody Counteracts the Pathological Effects of apoE4 In Vivo. Curr. Alzheimer Res. 2016, 13, 918–929. [Google Scholar] [CrossRef]
- Xiong, M.; Jiang, H.; Serrano, J.R.; Gonzales, E.R.; Gratuze, M.; Hoyle, R.; Bien-ly, N.; Silverman, A.P.; Patrick, M.; Watts, R.J.; et al. APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci. Transl. Med. 2022, 13, eabd7522. [Google Scholar] [CrossRef]
- Zhao, L.; Gottesdiener, A.J.; Parmar, M.; Li, M.; Kaminsky, S.M.; Chiuchiolo, M.J.; Sondhi, D.; Sullivan, P.M.; Holtzman, D.M.; Crystal, R.G.; et al. Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer’s disease mouse models. Neurobiol. Aging 2016, 44, 159–172. [Google Scholar] [CrossRef]
- Rosenberg, J.B.; Kaplitt, M.G.; De, B.P.; Chen, A.; Flagiello, T.; Salami, C.; Pey, E.; Zhao, L.; Ricart Arbona, R.J.; Monette, S.; et al. AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer’s Disease. Hum. Gene Ther. Clin. Dev. 2018, 29, 24–47. [Google Scholar] [CrossRef]
- Johnson, A.A.; Shokhirev, M.N.; Lehallier, B. The protein inputs of an ultra-predictive aging clock represent viable anti-aging drug targets. Ageing Res. Rev. 2021, 70, 101404. [Google Scholar] [CrossRef]
- Shin, J.; Noh, J.R.; Choe, D.; Lee, N.; Song, Y.; Cho, S.; Kang, E.J.; Go, M.J.; Ha, S.K.; Chang, D.H.; et al. Ageing and rejuvenation models reveal changes in key microbial communities associated with healthy ageing. Microbiome 2021, 9, 240. [Google Scholar] [CrossRef] [PubMed]
- Rebo, J.; Mehdipour, M.; Gathwala, R.; Causey, K.; Liu, Y.; Conboy, M.J.; Conboy, I.M. A single heterochronic blood exchange reveals rapid inhibition of multiple tissues by old blood. Nat. Commun. 2016, 7, 13363. [Google Scholar] [CrossRef] [PubMed]
- Middeldorp, J.; Lehallier, B.; Villeda, S.A.; Miedema, S.S.M.; Evans, E.; Czirr, E.; Zhang, H.; Luo, J.; Stan, T.; Mosher, K.I.; et al. Preclinical Assessment of Young Blood Plasma for Alzheimer Disease. JAMA Neurol. 2016, 73, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Das, M.M.; Godoy, M.; Chen, S.; Moser, V.A.; Avalos, P.; Roxas, K.M.; Dang, I.; Yáñez, A.; Zhang, W.; Bresee, C.; et al. Young bone marrow transplantation preserves learning and memory in old mice. Commun. Biol. 2019, 2, 73. [Google Scholar] [CrossRef] [PubMed]
- Seyhanli, A.; Yavuz, B.; Selimoglu, I.; Sengun, I.S.; Aslan, A.T.; Ozsan, G.H.; Alacacioglu, I.; Demirkan, F. Therapeutic plasma exchange in neurological diseases: Eleven years experience at a tertiary care center in Turkey. Ther. Apher. Dial. 2022, 26, 465–470. [Google Scholar] [CrossRef]
- Picón-Pagès, P.; Bonet, J.; García-García, J.; Garcia-Buendia, J.; Gutierrez, D.; Valle, J.; Gómez-Casuso, C.E.S.; Sidelkivska, V.; Alvarez, A.; Perálvarez-Marín, A.; et al. Human Albumin Impairs Amyloid β-peptide Fibrillation Through its C-terminus: From docking Modeling to Protection Against Neurotoxicity in Alzheimer’s disease. Comput. Struct. Biotechnol. J. 2019, 17, 963–971. [Google Scholar] [CrossRef]
- Costa, M.; Páez, A. Emerging insights into the role of albumin with plasma exchange in Alzheimer’s disease management. Transfus. Apher. Sci. 2021, 60, 103164. [Google Scholar] [CrossRef]
- Mehdipour, M.; Skinner, C.; Wong, N.; Lieb, M.; Liu, C.; Etienne, J.; Kato, C.; Kiprov, D.; Conboy, M.J.; Conboy, I.M. Rejuvenation of three germ layers tissues by exchanging old blood plasma with saline-albumin. Aging 2020, 12, 8790–8819. [Google Scholar] [CrossRef]
- Boada, M.; Anaya, F.; Ortiz, P.; Olazarán, J.; Shua-Haim, J.R.; Obisesan, T.O.; Hernández, I.; Muñoz, J.; Buendia, M.; Alegret, M.; et al. Efficacy and safety of plasma exchange with 5% albumin to modify cerebrospinal fluid and plasma amyloid-β concentrations and cognition outcomes in Alzheimer’s disease patients: A multicenter, randomized, controlled clinical trial. J. Alzheimer’s Dis. 2017, 56, 129–143. [Google Scholar] [CrossRef]
- Boada, M.; López, O.L.; Olazarán, J.; Núñez, L.; Pfeffer, M.; Paricio, M.; Lorites, J.; Piñol-Ripoll, G.; Gámez, J.E.; Anaya, F.; et al. A randomized, controlled clinical trial of plasma exchange with albumin replacement for Alzheimer’s disease: Primary results of the AMBAR Study. Alzheimer’s Dement. 2020, 16, 1412–1425. [Google Scholar] [CrossRef]
- Ortiz, A.M.; Minguet, C.; Gonzalo, R.; Núñez, L.; Ruiz, A.; Lopez, O.L.; Boada, M.; Páez, A.; Costa, M. Inflammatory biomarkers in patients undergoing therapeutic plasma exchange with albumin replacement as a treatment for Alzheimer’s disease. Alzheimer’s Dement. 2021, 17, e057735. [Google Scholar] [CrossRef]
- Cuberas-Borrós, G.; Roca, I.; Castell-Conesa, J.; Núñez, L.; Boada, M.; López, O.L.; Grifols, C.; Barceló, M.; Pareto, D.; Páez, A. Neuroimaging analyses from a randomized, controlled study to evaluate plasma exchange with albumin replacement in mild-to-moderate Alzheimer’s disease: Additional results from the AMBAR study. Eur. J. Nucl. Med. Mol. Imaging 2022. [Google Scholar] [CrossRef] [PubMed]
- Sha, S.J.; Deutsch, G.K.; Tian, L.; Richardson, K.; Coburn, M.; Gaudioso, J.L.; Marcal, T.; Solomon, E.; Boumis, A.; Bet, A.; et al. Safety, Tolerability, and Feasibility of Young Plasma Infusion in the Plasma for Alzheimer Symptom Amelioration Study: A Randomized Clinical Trial. JAMA Neurol. 2019, 76, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Hannestad, J.; Koborsi, K.; Klutzaritz, V.; Chao, W.; Ray, R.; Páez, A.; Jackson, S.; Lohr, S.; Cummings, J.L.; Kay, G.; et al. Safety and tolerability of GRF6019 in mild-to-moderate Alzheimer’s disease dementia. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, e12115. [Google Scholar] [CrossRef]
- Hannestad, J.; Duclos, T.; Chao, W.; Koborsi, K.; Klutzaritz, V.; Beck, B.; Patel, A.K.; Scott, J.; Thein, S.G.; Cummings, J.L.; et al. Safety and Tolerability of GRF6019 Infusions in Severe Alzheimer’s Disease: A Phase II Double-Blind Placebo-Controlled Trial. J. Alzheimer’s Dis. 2021, 81, 1649–1662. [Google Scholar] [CrossRef]
- Struble, R.G.; Ala, T.; Patrylo, P.R.; Brewer, G.J.; Yan, X.X. Is brain amyloid production a cause or a result of dementia of the Alzheimer’s type? J. Alzheimer’s Dis. 2010, 22, 393–399. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennettb, D.A.; Blennowc, K.; Carrillod, M.C.; Dunne, B.; Haeberleinf, S.B.; Holtzmang, D.M.; Jagusth, W.; Jesseni, F.; Karlawishj, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Higashi, S.; Iseki, E.; Yamamoto, R.; Minegishi, M.; Hino, H.; Fujisawa, K.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Concurrence of TDP-43, tau and α-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 2007, 1184, 284–294. [Google Scholar] [CrossRef]
- Bouter, Y.; Liekefeld, H.; Pichlo, S.; Westhoff, A.C.; Fenn, L.; Bakrania, P.; Bayer, T.A. Donanemab detects a minor fraction of amyloid-β plaques in post-mortem brain tissue of patients with Alzheimer’s disease and Down syndrome. Acta Neuropathol. 2022, 143, 601–603. [Google Scholar] [CrossRef]
- Hoffmann, T.; Rahfeld, J.U.; Schenk, M.; Ponath, F.; Makioka, K.; Hutter-Paier, B.; Lues, I.; Lemere, C.A.; Schilling, S. Combination of the glutaminyl cyclase inhibitor PQ912 (Varoglutamstat) and the murine monoclonal antibody PBD-C06 (m6) shows additive effects on brain Aβ pathology in transgenic mice. Int. J. Mol. Sci. 2021, 22, 11791. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front. Aging Neurosci. 2020, 11, 373. [Google Scholar] [CrossRef] [PubMed]
- Whittington, M.D.; Campbell, J.D.; Rind, D.; Fluetsch, N.; Lin, G.A.; Pearson, S.D. Cost-Effectiveness and Value-Based Pricing of Aducanumab for Patients With Early Alzheimer Disease. Neurology 2022, 98, 968–977. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morató, X.; Pytel, V.; Jofresa, S.; Ruiz, A.; Boada, M. Symptomatic and Disease-Modifying Therapy Pipeline for Alzheimer’s Disease: Towards a Personalized Polypharmacology Patient-Centered Approach. Int. J. Mol. Sci. 2022, 23, 9305. https://doi.org/10.3390/ijms23169305
Morató X, Pytel V, Jofresa S, Ruiz A, Boada M. Symptomatic and Disease-Modifying Therapy Pipeline for Alzheimer’s Disease: Towards a Personalized Polypharmacology Patient-Centered Approach. International Journal of Molecular Sciences. 2022; 23(16):9305. https://doi.org/10.3390/ijms23169305
Chicago/Turabian StyleMorató, Xavier, Vanesa Pytel, Sara Jofresa, Agustín Ruiz, and Mercè Boada. 2022. "Symptomatic and Disease-Modifying Therapy Pipeline for Alzheimer’s Disease: Towards a Personalized Polypharmacology Patient-Centered Approach" International Journal of Molecular Sciences 23, no. 16: 9305. https://doi.org/10.3390/ijms23169305
APA StyleMorató, X., Pytel, V., Jofresa, S., Ruiz, A., & Boada, M. (2022). Symptomatic and Disease-Modifying Therapy Pipeline for Alzheimer’s Disease: Towards a Personalized Polypharmacology Patient-Centered Approach. International Journal of Molecular Sciences, 23(16), 9305. https://doi.org/10.3390/ijms23169305