
Transcriptomic Analysis of Human Fragile X Syndrome Neurons Reveals Neurite Outgrowth Modulation by the TGFβ/BMP Pathway

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
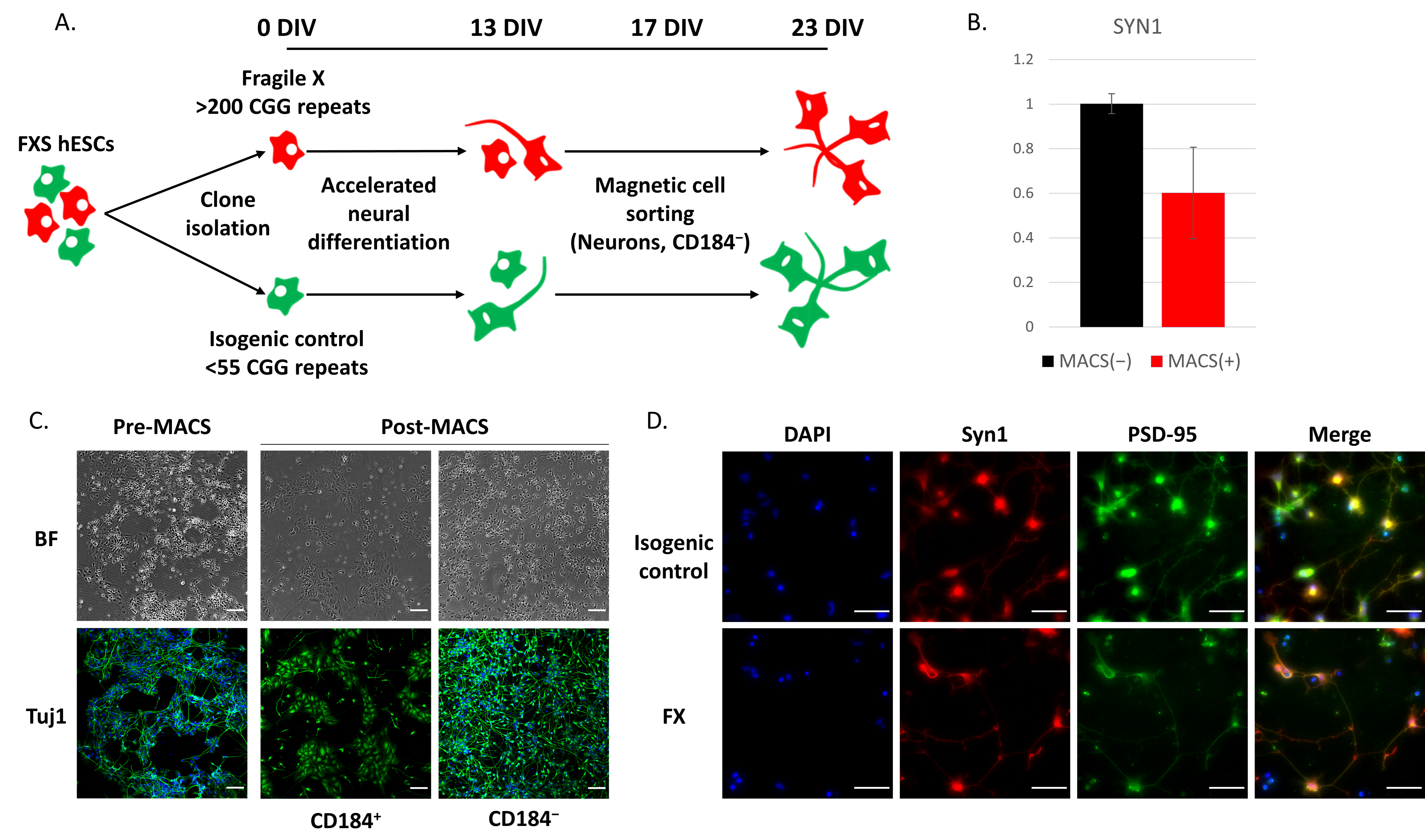
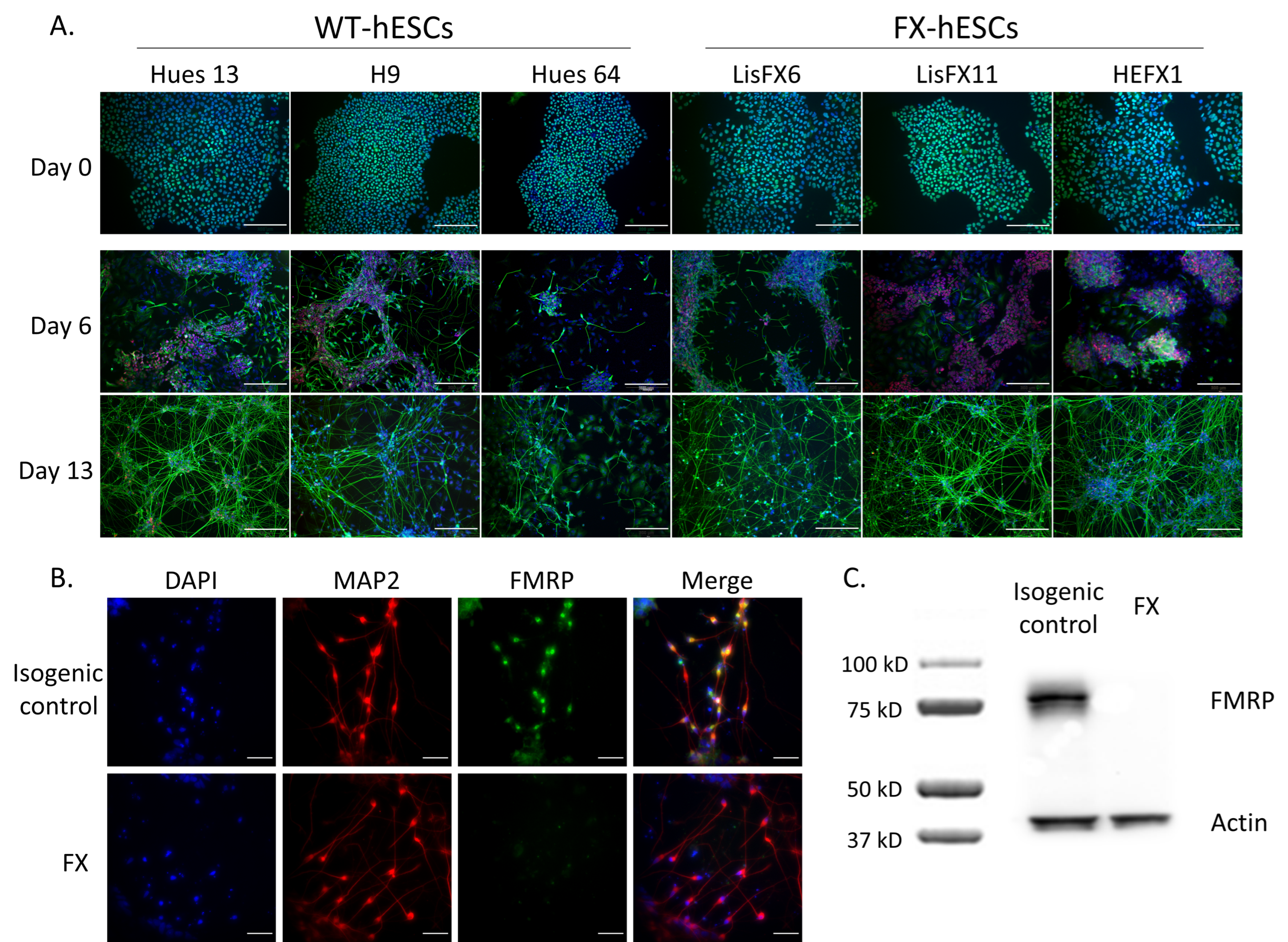
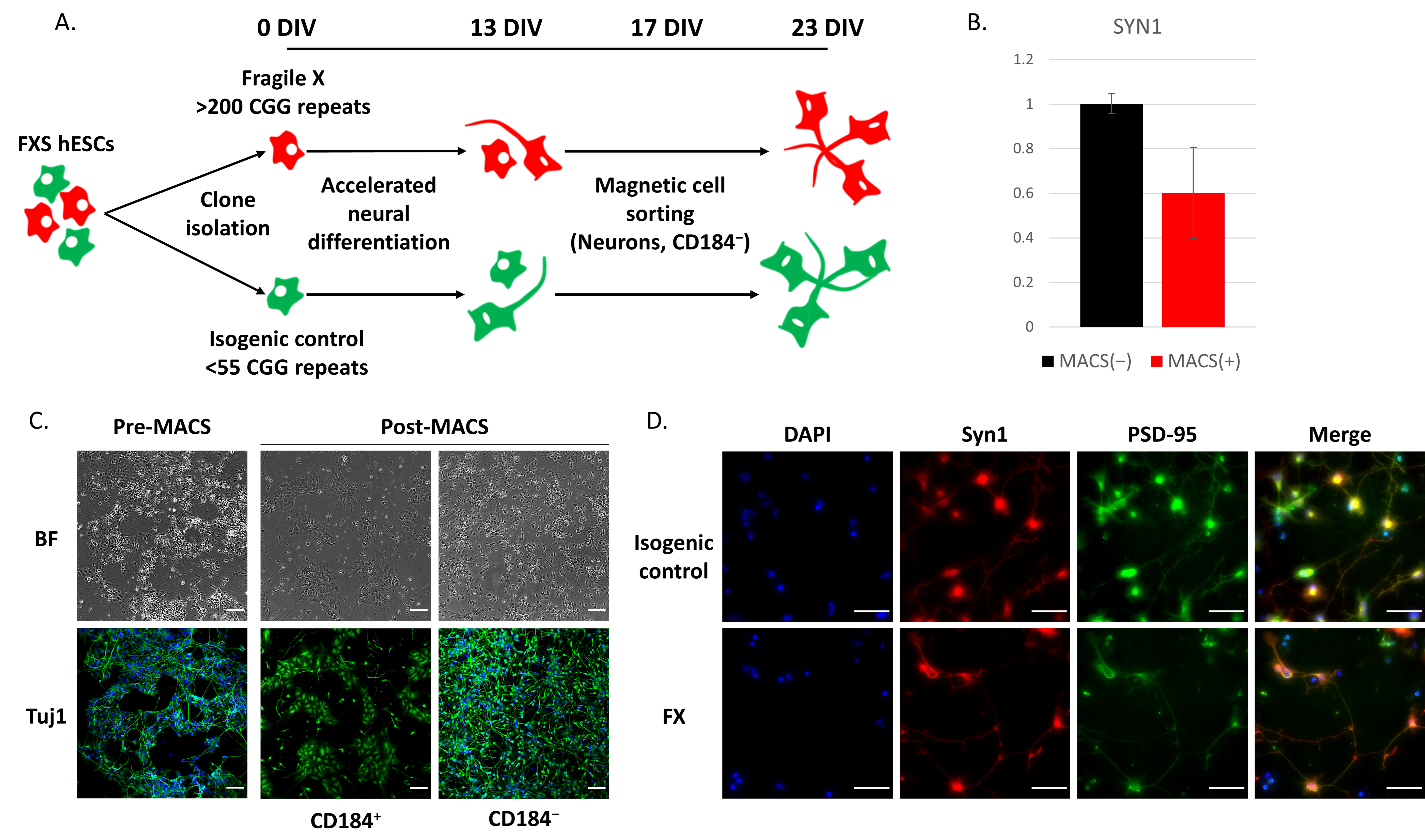
2.1. Delayed Neurodevelopment of FX-hESCs
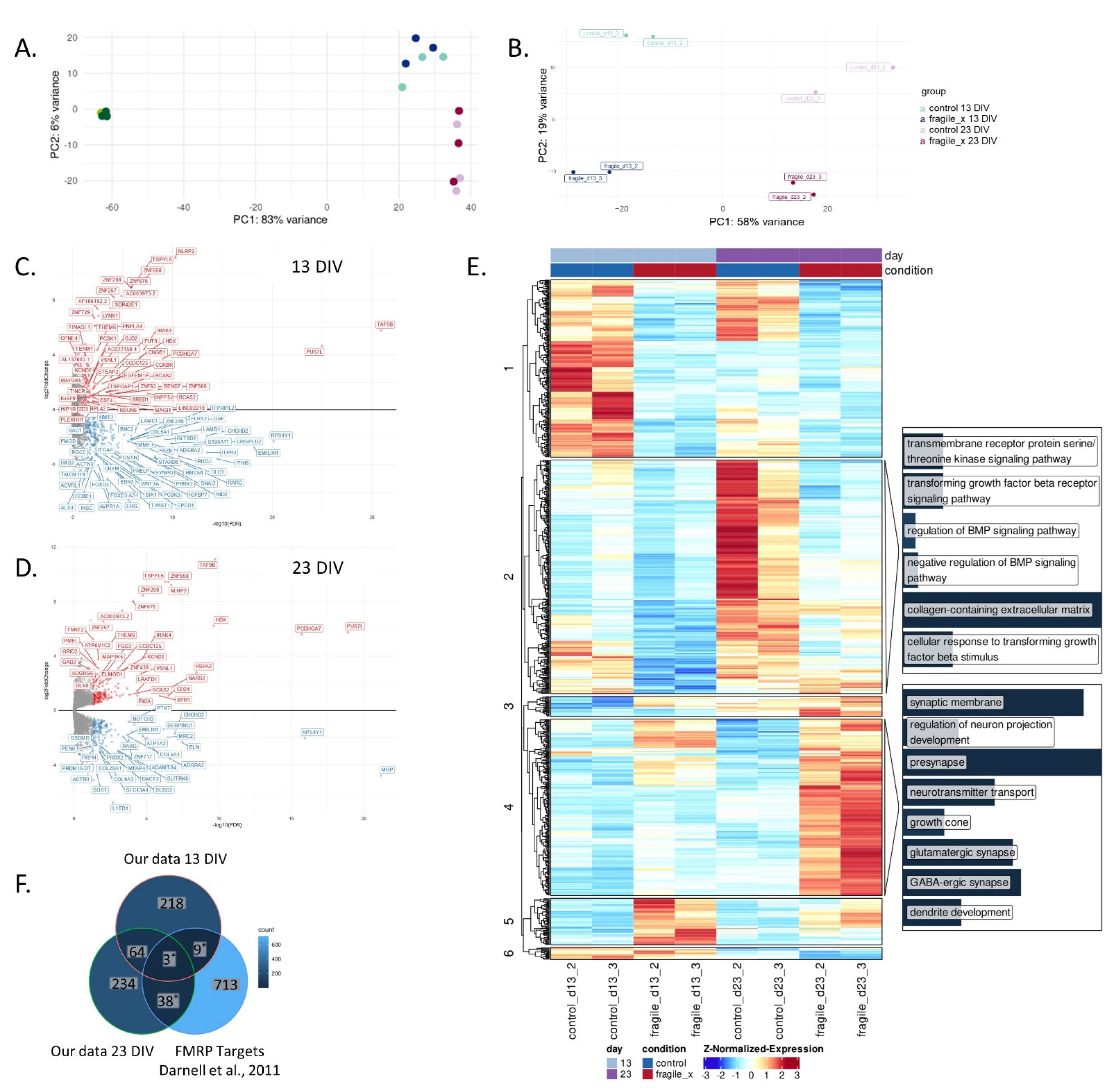
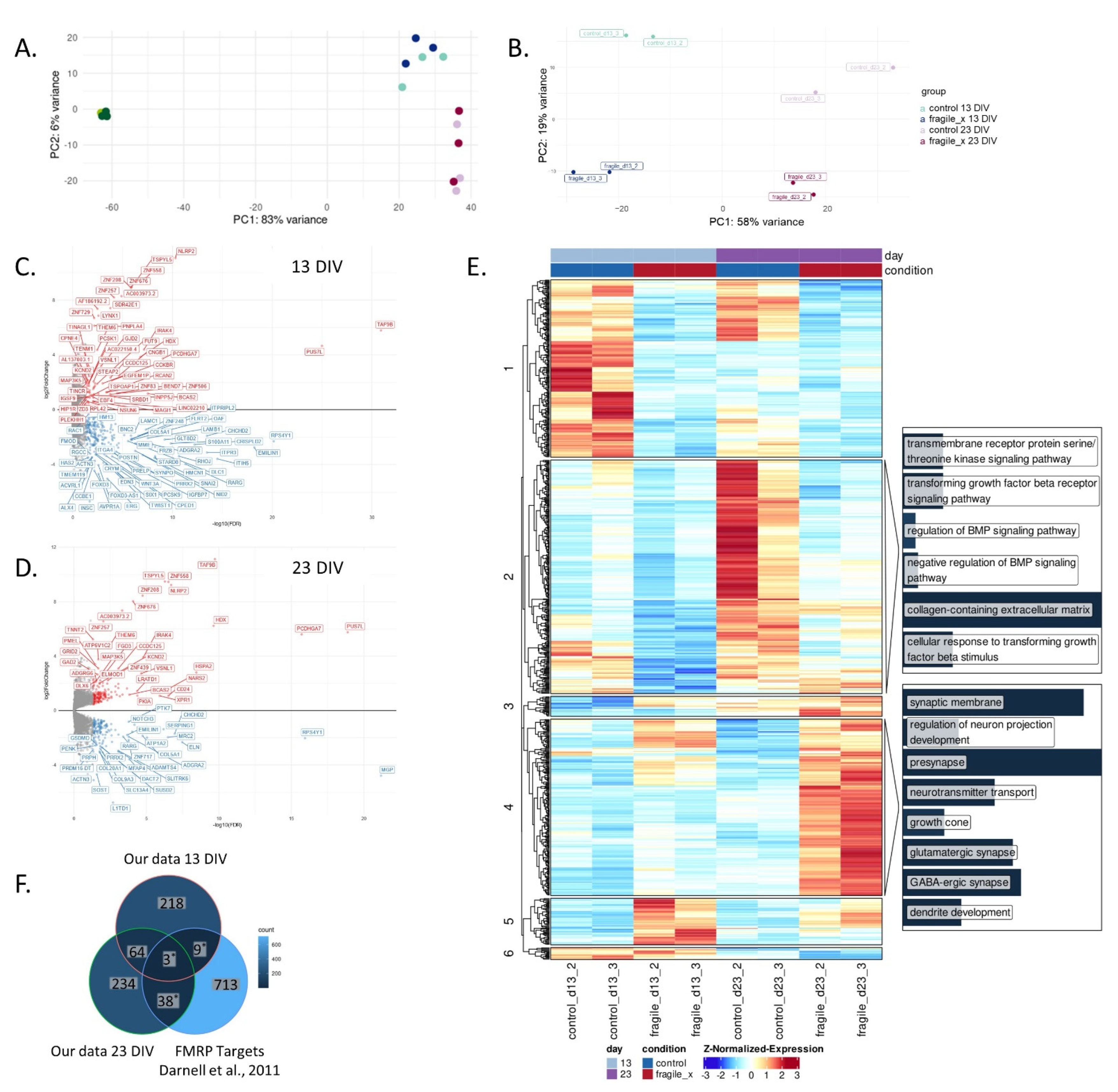
2.2. Loss of FMRP Leads to Altered Gene Expression throughout Neuronal Differentiation
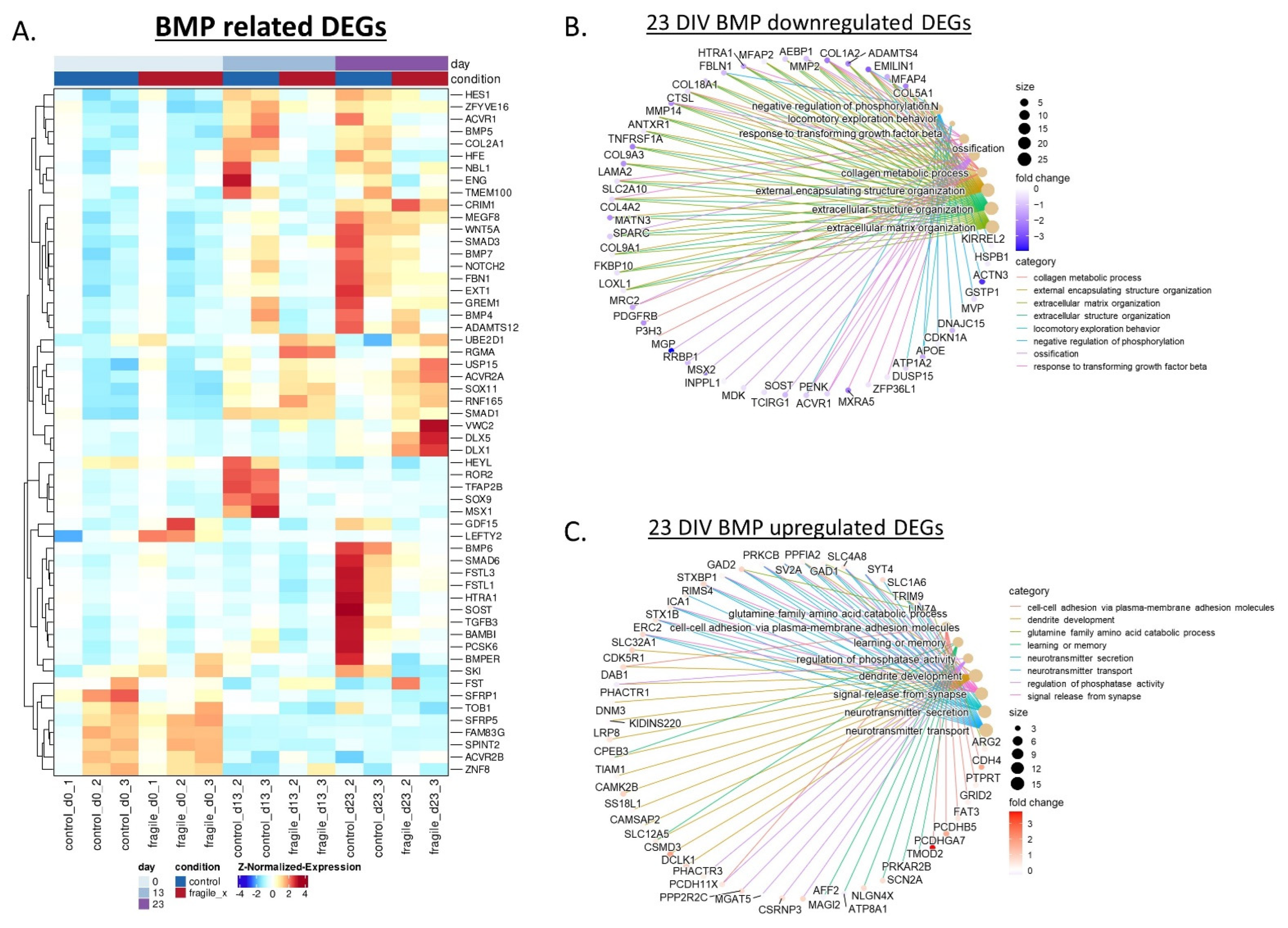
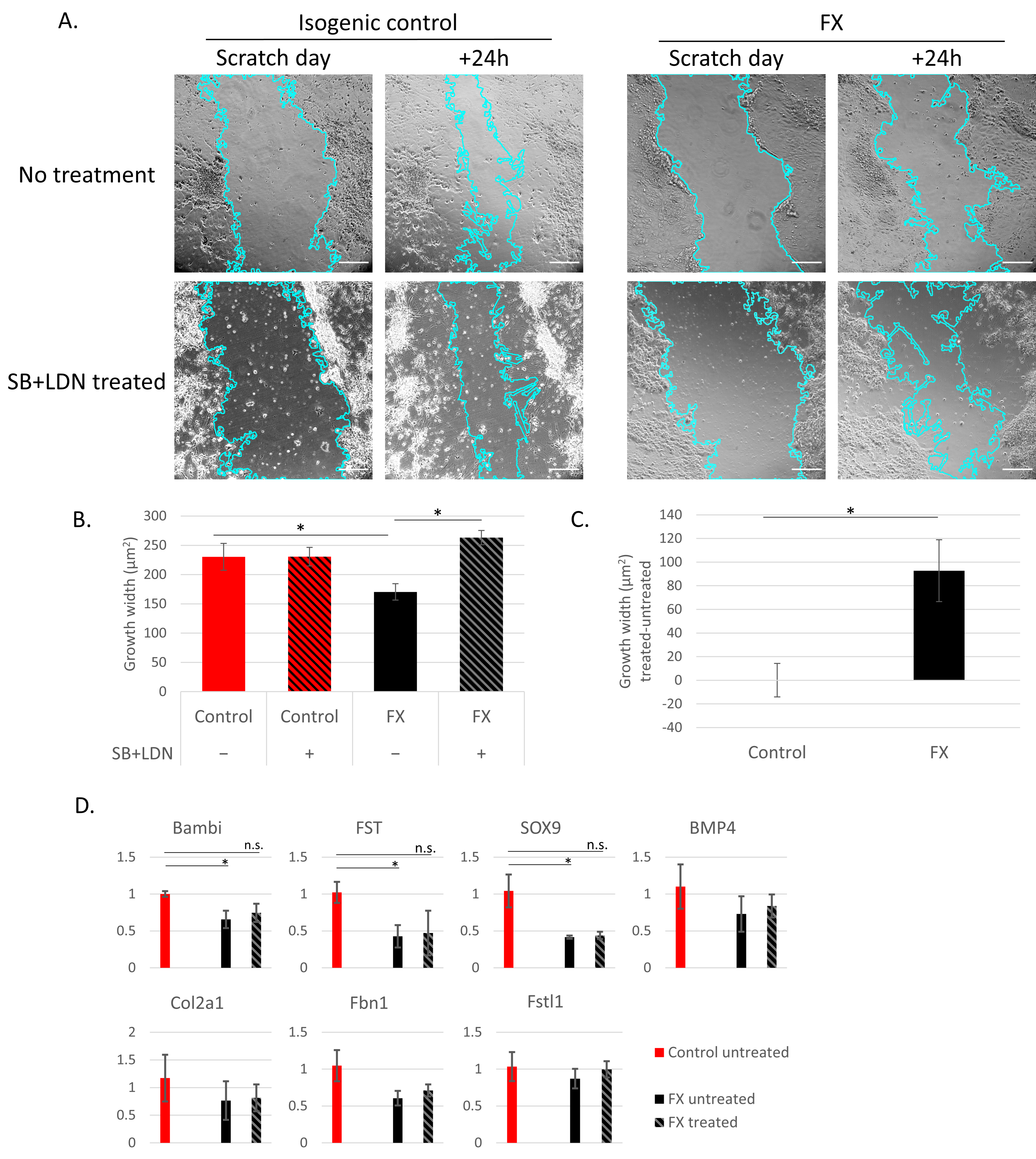
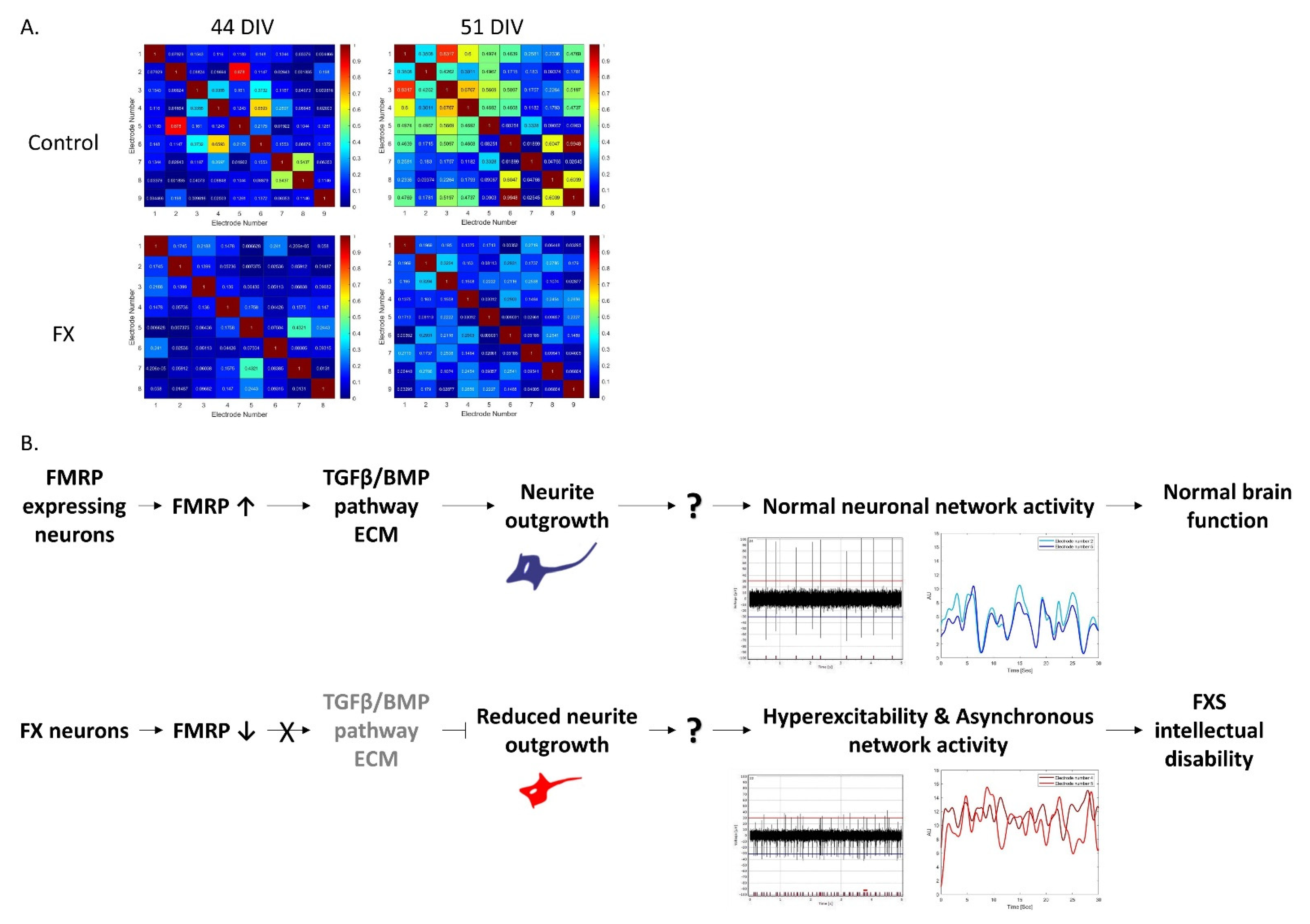
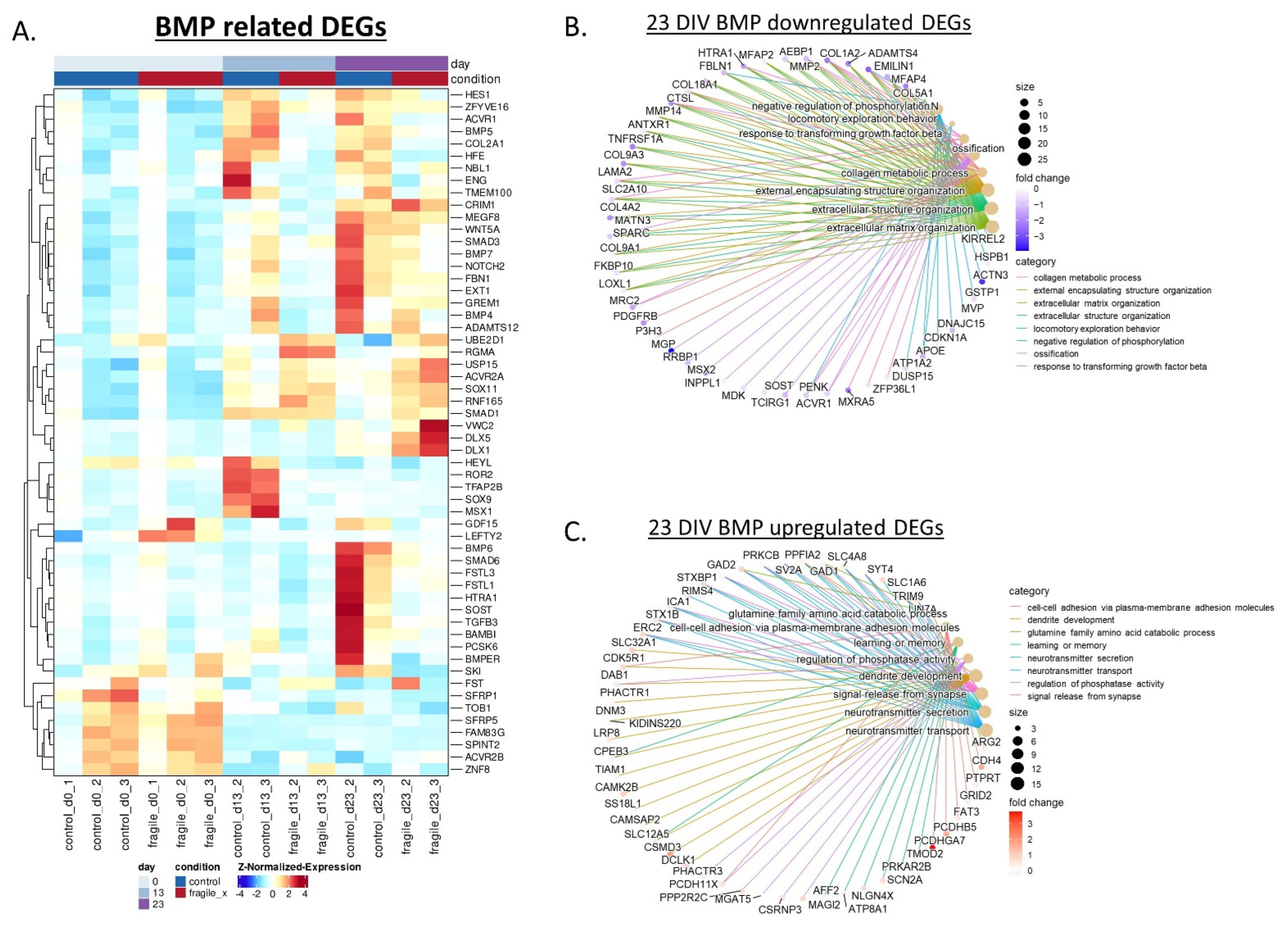
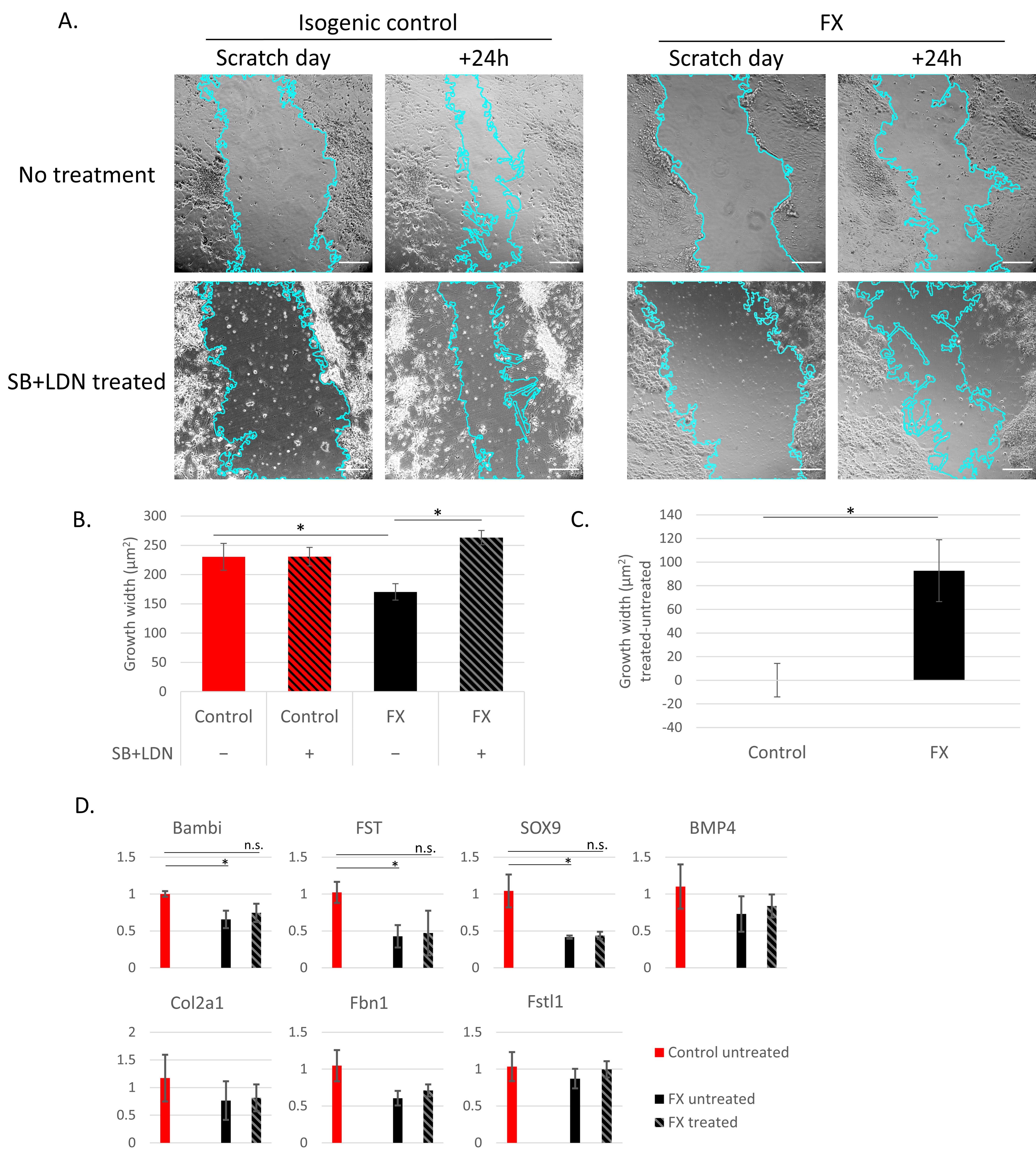
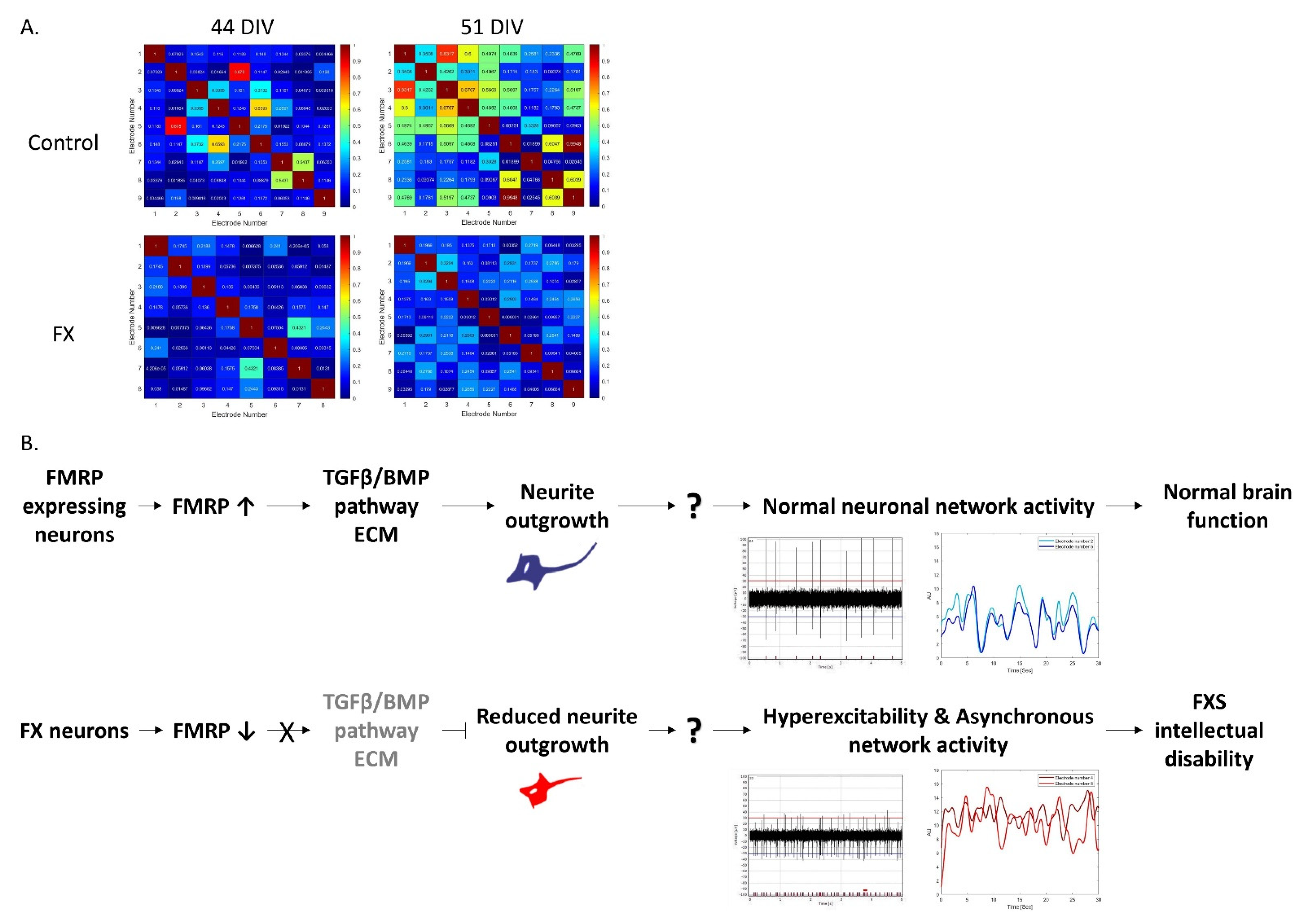
2.3. Fragile X-Derived Neurons Display a Neurite Outgrowth Defect Manifested by the BMP Pathway
3. Discussion
4. Materials and Methods
4.1. Human Embryonic Stem Cell Culture
4.2. In Vitro Neural Differentiation
4.3. Immunofluorescence
4.4. Western Blot Analysis
4.5. RNA Extraction and Quantitative Real-Time PCR
4.6. Magnetic-Activated Cell Sorting (MACS)
4.7. RNA Sequencing and Bioinformatic Analysis
4.8. Scratch Assay
4.9. Statistical Analysis and Experimental Design
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verkerk, A.J.M.H.; Pieretti, Y.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.A.; Pixxuti, A.; Refner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Richter, J.D.; Zhao, X. The molecular biology of FMRP: New insights into fragile X syndrome. Nat. Rev. Neurosci. 2021, 22, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; van Driesche, S.J.; Zhang, C.; Hung, K.Y.S.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on MRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Ascano, M.J.; Mukherjee, N.; Bandaru, P.; Miller, J.B.; Nusbaum, J.D.; Corcoran, D.L.; Langlois, C.; Munschauer, M.; Dewell, S.; Hafner, M.; et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 2012, 492, 382–386. [Google Scholar] [CrossRef]
- Utami, K.H.; Yusof, A.; Kwa, J.E.; Peteri, U.-K.; Castrén, M.; Pouladi, M.A. Elevated de novo protein synthesis in FMRP-deficient human neurons and its correction by metformin treatment. Mol. Autism 2020, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Utami, K.H.; Skotte, N.H.; Colaço, A.R.; Yusof, N.A.B.M.; Sim, B.; Yeo, X.Y.; Bae, H.G.; Garcia-Miralles, M.; Radulescu, C.I.; Chen, Q.; et al. Integrative analysis identifies key molecular signatures underlying neurodevelopmental deficits in fragile X syndrome. Biol. Psychiatry 2020, 88, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Castrén, M.; Tervonen, T.; Kärkkäinen, V.; Heinonen, S.; Castré, E.; Larsson, K.; Bakker, C.E.; Oostra, B.A.; Åkerman, K. Altered differentiation of neural stem cells in fragile X syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 17834–17839. [Google Scholar] [CrossRef]
- Gross, C.; Berry-Kravis, E.M.; Bassell, G.J. Therapeutic strategies in fragile X syndrome: Dysregulated MGluR signaling and beyond. Neuropsychopharmacology 2012, 37, 178–195. [Google Scholar] [CrossRef]
- Dahlhaus, R. Of men and mice: Modeling the fragile X syndrome. Front. Mol. Neurosci. 2018, 11, 41. [Google Scholar] [CrossRef]
- Kazdoba, T.M.; Leach, P.T.; Silverman, J.L.; Crawley, J.N. Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare Dis. Res. 2014, 3, 118–133. [Google Scholar] [CrossRef]
- Drozd, M.; Bardoni, B.; Capovilla, M. Modeling fragile X syndrome in drosophila. Front. Mol. Neurosci. 2018, 11, 124. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.C.; Yang, Y.L.; Lu, K.T. Behavioral and synaptic circuit features in a zebrafish model of fragile X syndrome. PLoS ONE 2013, 8, e51456. [Google Scholar] [CrossRef] [PubMed]
- Irwin, S.A.; Patel, B.; Idupulapati, M.; Harris, J.B.; Crisostomo, R.A.; Larsen, B.P.; Kooy, F.; Willems, P.J.; Cras, P.; Kozlowski, P.B.; et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: A quantitative examination. Am. J. Med. Genet. 2001, 98, 161–167. [Google Scholar] [CrossRef]
- Schwartz, P.H.; Tassone, F.; Greco, C.M.; Nethercott, H.E.; Ziaeian, B.; Hagerman, R.J.; Hagerman, P.J. Neural progenitor cells from an adult patient with fragile X syndrome. BMC Med. Genet. 2005, 6, 2. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; McMillan, E.; Wallace, K.; Tubon, T.C.; Capowski, E.E.; Svendsen, C.N. Normal neurogenesis but abnormal gene expression in human fragile X cortical progenitor cells. Stem Cells Dev. 2008, 17, 107–117. [Google Scholar] [CrossRef]
- Sheridan, S.D.; Theriault, K.M.; Reis, S.A.; Zhou, F.; Madison, J.M.; Daheron, L.; Loring, J.F.; Haggarty, S.J. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS ONE 2011, 6, e26203. [Google Scholar] [CrossRef]
- Halevy, T.; Czech, C.; Benvenisty, N. Molecular mechanisms regulating the defects in fragile x syndrome neurons derived from human pluripotent stem cells. Stem Cell Rep. 2015, 4, 37–46. [Google Scholar] [CrossRef]
- Telias, M.; Mayshar, Y.; Amit, A.; Ben-Yosef, D. Molecular mechanisms regulating impaired neurogenesis of fragile x syndrome human embryonic stem cells. Stem Cells Dev. 2015, 24, 2353–2365. [Google Scholar] [CrossRef]
- Handel, A.E.; Chintawar, S.; Lalic, T.; Whiteley, E.; Vowles, J.; Giustacchini, A.; Argoud, K.; Sopp, P.; Nakanishi, M.; Bowden, R.; et al. Assessing similarity to primary tissue and cortical layer identity in induced pluripotent stem cell-derived cortical neurons through single-cell transcriptomics. Hum. Mol. Genet. 2016, 25, 989–1000. [Google Scholar] [CrossRef]
- Eiges, R.; Urbach, A.; Malcov, M.; Frumkin, T.; Schwartz, T.; Amit, A.; Yaron, Y.; Eden, A.; Yanuka, O.; Benvenisty, N.; et al. Developmental study of fragile x syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 2007, 1, 568–577. [Google Scholar] [CrossRef]
- Telias, M.; Segal, M.; Ben-Yosef, D. Neural differentiation of fragile x human embryonic stem cells reveals abnormal patterns of development despite successful neurogenesis. Dev. Biol. 2013, 374, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Telias, M.; Segal, M.; Telias, M. Immature responses to GABA in fragile x neurons derived from human embryonic stem cells. Front. Cell. Neurosci. 2016, 10, 121. [Google Scholar] [CrossRef] [PubMed]
- Telias, M.; Kuznitsov-Yanovsky, L.; Sega, M.; Ben-Yosef, D. Functional deficiencies in fragile x neurons derived from human embryonic stem cells. J. Neurosci. 2015, 35, 15295–15306. [Google Scholar] [CrossRef] [PubMed]
- Gildin, L.; Rauti, R.; Vardi, O.; Kuznitsov-Yanovsky, L.; Maoz, B.M.; Segal, M.; Ben-Yosef, D. Impaired functional connectivity underlies fragile x syndrome. Int. J. Mol. Sci. 2022, 23, 2048. [Google Scholar] [CrossRef]
- Boland, M.J.; Nazor, K.L.; Tran, H.T.; Szücs, A.; Lynch, C.L.; Paredes, R.; Tassone, F.; Sanna, P.P.; Hagerman, R.J.; Loring, J.F. Molecular analyses of neurogenic defects in a human pluripotent stem cell model of fragile x syndrome. Brain 2017, 140, 582–598. [Google Scholar] [CrossRef]
- Doers, M.E.; Musser, M.T.; Nichol, R.; Berndt, E.R.; Baker, M.; Gomez, T.M.; Zhang, S.C.; Abbeduto, L.; Bhattacharyya, A. IPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev. 2014, 23, 1777–1787. [Google Scholar] [CrossRef]
- Li, M.; Shin, J.; Risgaard, R.D.; Parries, M.J.; Wang, J.; Chasman, D.; Liu, S.; Roy, S.; Bhattacharyya, A.; Zhao, X. Identification of FMR1-regulated molecular networks in human neurodevelopment. Genome Res. 2020, 30, 361–374. [Google Scholar] [CrossRef]
- Urbach, A.; Bar-Nur, O.; Daley, G.Q.; Benvenisty, N. Differential modeling of fragile x syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 2010, 6, 407–411. [Google Scholar] [CrossRef]
- Casingal, C.R.; Kikkawa, T.; Inada, H.; Sasaki, Y.; Osumi, N. Identification of FMRP target MRNAs in the developmental brain: FMRP might coordinate ras/MAPK, wnt/β-catenin, and MTOR signaling during corticogenesis. Mol. Brain 2020, 13, 167. [Google Scholar] [CrossRef]
- Hinds, H.L.; Ashley, C.T.; Sutcliffe, J.S.; Nelson, D.L.; Warren, S.T.; Housman, D.E.; Schalling, M. Tissue specific expression of FMR–1 provides evidence for a functional role in fragile x syndrome. Nat. Genet. 1993, 3, 36–43. [Google Scholar] [CrossRef]
- Beroun, A.; Mitra, S.; Michaluk, P.; Pijet, B.; Stefaniuk, M.; Kaczmarek, L. MMPs in learning and memory and neuropsychiatric disorders. Cell. Mol. Life Sci. 2019, 76, 3207–3228. [Google Scholar] [CrossRef] [PubMed]
- Melrose, J.; Hayes, A.J.; Bix, G. The CNS/PNS extracellular matrix provides instructive guidance cues to neural cells and neuroregulatory proteins in neural development and repair. Int. J. Mol. Sci. 2021, 22, 5583. [Google Scholar] [CrossRef] [PubMed]
- Kashima, R.; Hata, A. The role of TGF-β superfamily signaling in neurological disorders. Acta Biochim. Biophys. Sin. 2018, 50, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Meyers, E.A.; Kessler, J.A. TGF-β family signaling in neural and neuronal differentiation, development, and function. Cold Spring Harb. Perspect. Biol. 2017, 9, a022244. [Google Scholar] [CrossRef]
- Ebendal, T.; Bengtsson, H.; Söderström, S. Bone morphogenetic proteins and their receptors: Potential functions in the brain. J. Neurosci. Res. 1998, 51, 139–146. [Google Scholar] [CrossRef]
- Fukushima, T.; Liu, R.Y.; Byrne, J.H. Transforming growth factor-Β2 modulates synaptic efficacy and plasticity and induces phosphorylation of CREB in hippocampal neurons. Hippocampus 2007, 17, 5–9. [Google Scholar] [CrossRef]
- Kashima, R.; Roy, S.; Ascano, M.; Martinez-Cerdeno, V.; Ariza-Torres, J.; Kim, S.; Louie, J.; Lu, Y.; Leyton, P.; Bloch, K.D.; et al. Augmented noncanonical BMP type II receptor signaling mediates the synaptic abnormality of fragile x syndrome. Sci. Signal. 2016, 9, ra58. [Google Scholar] [CrossRef] [PubMed]
- Kashima, R.; Redmond, P.L.; Ghatpande, P.; Roy, S.; Kornberg, T.B.; Hanke, T.; Knapp, S.; Lagna, G.; Hata, A. Hyperactive locomotion in a drosophila model is a functional readout for the synaptic abnormalities underlying fragile x syndrome. Sci. Signal. 2017, 10, eaai8133. [Google Scholar] [CrossRef]
- Comery, T.A.; Harris, J.B.; Willems, P.J.; Oostra, B.A.; Irwin, S.A.; Jeanne Weiler, I.; Greenough, W.T. Abnormal dendritic spines in fragile x knockout mice: Maturation and pruning deficits. Proc. Natl. Acad. Sci. USA 1997, 94, 5401–5404. [Google Scholar] [CrossRef]
- Irwin, S.A.; Galvez, R.; Greenough, W.T. Dendritic spine structural anomalies in fragile-x mental retardation syndrome. Cereb. Cortex 2000, 10, 1038–1044. [Google Scholar] [CrossRef]
- Lee-Hoeflich, S.T.; Causing, C.G.; Podkowa, M.; Zhao, X.; Wrana, J.L.; Attisano, L. Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J. 2004, 23, 4792–4801. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.M.; Bhalala, O.G.; Kessler, J.A. The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Dev. Neurobiol. 2012, 72, 1068–1084. [Google Scholar] [CrossRef] [PubMed]
- Georges, P.C.; Hadzimichalis, N.M.; Sweet, E.S.; Firestein, B.L. The yin-yang of dendrite morphology: Unity of actin and microtubules. Mol. Neurobiol. 2008, 38, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Podkowa, M.; Zhao, X.; Chow, C.-W.; Coffey, E.T.; Davis, R.J.; Attisano, L. Microtubule stabilization by bone morphogenetic protein receptor-mediated scaffolding of c-jun n-terminal kinase promotes dendrite formation. Mol. Cell. Biol. 2010, 30, 2241–2250. [Google Scholar] [CrossRef]
- Siegenthaler, J.A.; Miller, M.W. Transforming growth factor Β1 modulates cell migration in rat cortex: Effects of ethanol. Cereb. Cortex 2004, 14, 791–802. [Google Scholar] [CrossRef]
- Avitzour, M.; Mor-Shaked, H.; Yanovsky-Dagan, S.; Aharoni, S.; Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Schonberger, O.; Levy-Lahad, E.; Epsztejn-Litman, S.; et al. FMR1 epigenetic silencing commonly occurs in undifferentiated fragile x-affected embryonic stem cells. Stem Cell Rep. 2014, 3, 699–706. [Google Scholar] [CrossRef]
- Frumkin, T.; Malcov, M.; Telias, M.; Gold, V.; Schwartz, T.; Azem, F.; Amit, A.; Yaron, Y.; Ben-Yosef, D. Human embryonic stem cells carrying mutations for severe genetic disorders. Vitr. Cell. Dev. Biol.-Anim. 2010, 46, 327–336. [Google Scholar] [CrossRef]
- Cowan, C.A.; Klimanskaya, I.; McMahon, J.; Atienza, J.; Witmyer, J.; Zucker, J.P.; Wang, S.; Morton, C.C.; McMahon, A.P.; Powers, D.; et al. Derivation of embryonic stem-cell lines from human blastocysts. N. Engl. J. Med. 2004, 350, 1353–1356. [Google Scholar] [CrossRef]
- Chen, A.E.; Egli, D.; Niakan, K.; Deng, J.; Akutsu, H.; Yamaki, M.; Cowan, C.; Fitz-Gerald, C.; Zhang, K.; Melton, D.A.; et al. Optimal timing of inner cell mass isolation increases the efficiency of human embryonic stem cell derivation and allows generation of sibling cell lines. Cell Stem Cell 2009, 4, 103–106. [Google Scholar] [CrossRef]
- Thomson, J.A. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Elkabetz, Y.; Panagiotakos, G.; al Shamy, G.; Socci, N.D.; Tabar, V.; Studer, L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008, 22, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Zhang, X.J.; Renier, N.; Wu, Z.; Atkin, T.; Sun, Z.; Ozair, M.Z.; Tchieu, J.; Zimmer, B.; Fattahi, F.; et al. Combined small-molecule inhibition accelerates the derivation of functional cortical neurons from human pluripotent stem cells. Nat. Biotechnol. 2017, 35, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Kuznitsov-Yanovsky, L.; Mayshar, Y.; Ben-Yosef, D. Modeling FXS: Human pluripotent stem cells and in vitro neural differentiation. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2019; Volume 1942. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Suarez-Arnedo, A.; Figueroa, F.T.; Clavijo, C.; Arbeláez, P.; Cruz, J.C.; Muñoz-Camargo, C. An image j plugin for the high throughput image analysis of in vitro scratch wound healing assays. PLoS ONE 2020, 15, e0232565. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuznitsov-Yanovsky, L.; Shapira, G.; Gildin, L.; Shomron, N.; Ben-Yosef, D. Transcriptomic Analysis of Human Fragile X Syndrome Neurons Reveals Neurite Outgrowth Modulation by the TGFβ/BMP Pathway. Int. J. Mol. Sci. 2022, 23, 9278. https://doi.org/10.3390/ijms23169278
Kuznitsov-Yanovsky L, Shapira G, Gildin L, Shomron N, Ben-Yosef D. Transcriptomic Analysis of Human Fragile X Syndrome Neurons Reveals Neurite Outgrowth Modulation by the TGFβ/BMP Pathway. International Journal of Molecular Sciences. 2022; 23(16):9278. https://doi.org/10.3390/ijms23169278
Chicago/Turabian StyleKuznitsov-Yanovsky, Liron, Guy Shapira, Lital Gildin, Noam Shomron, and Dalit Ben-Yosef. 2022. "Transcriptomic Analysis of Human Fragile X Syndrome Neurons Reveals Neurite Outgrowth Modulation by the TGFβ/BMP Pathway" International Journal of Molecular Sciences 23, no. 16: 9278. https://doi.org/10.3390/ijms23169278
APA StyleKuznitsov-Yanovsky, L., Shapira, G., Gildin, L., Shomron, N., & Ben-Yosef, D. (2022). Transcriptomic Analysis of Human Fragile X Syndrome Neurons Reveals Neurite Outgrowth Modulation by the TGFβ/BMP Pathway. International Journal of Molecular Sciences, 23(16), 9278. https://doi.org/10.3390/ijms23169278