
Targeting ABCC6 in Mesenchymal Stem Cells: Impairment of Mature Adipocyte Lipid Homeostasis

 ,
,
Abstract
:1. Introduction
2. Results
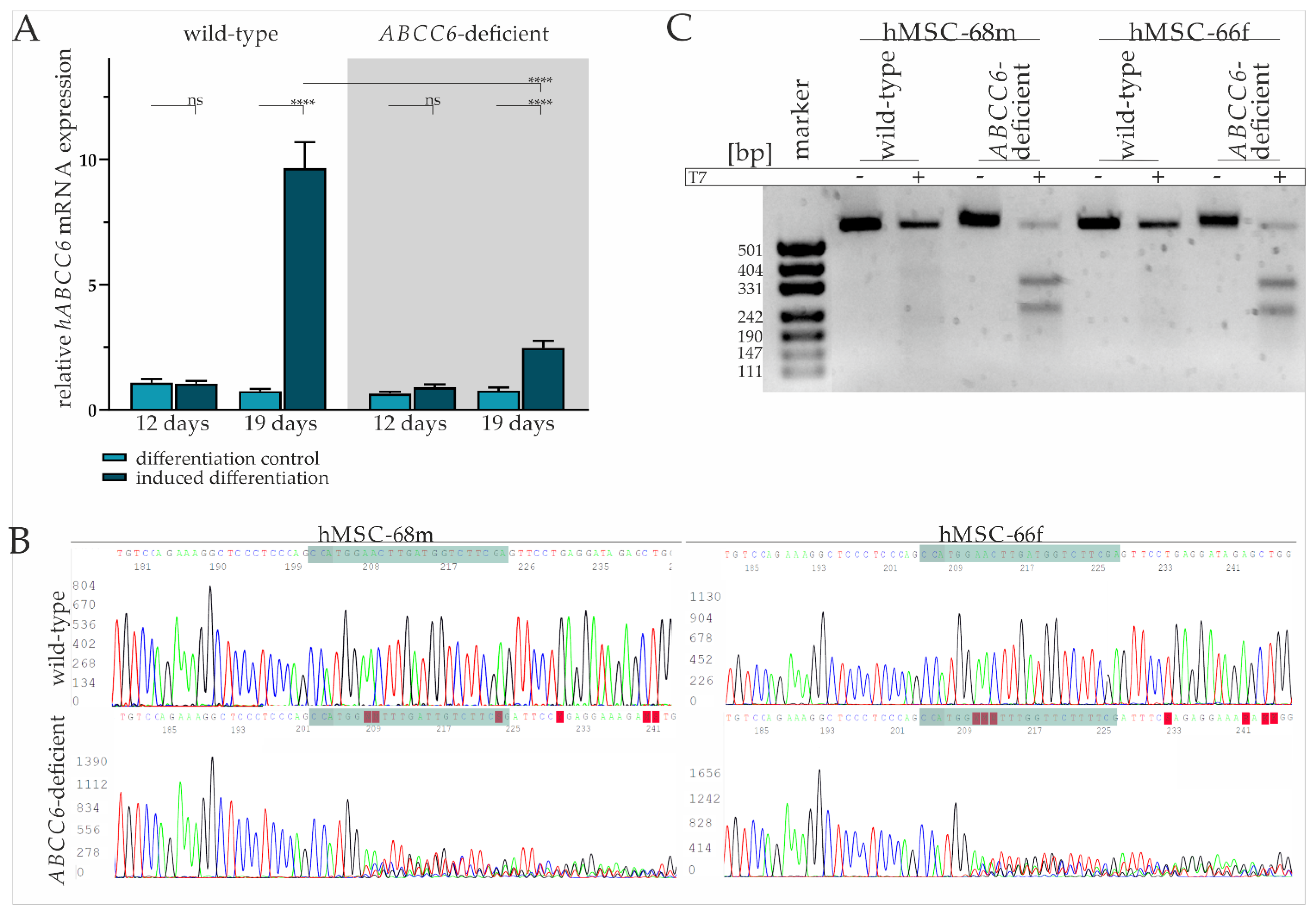
2.1. Diminished ABCC6 Expression in Differentiated hMSCs via CRISPR–Cas9 Genome Editing
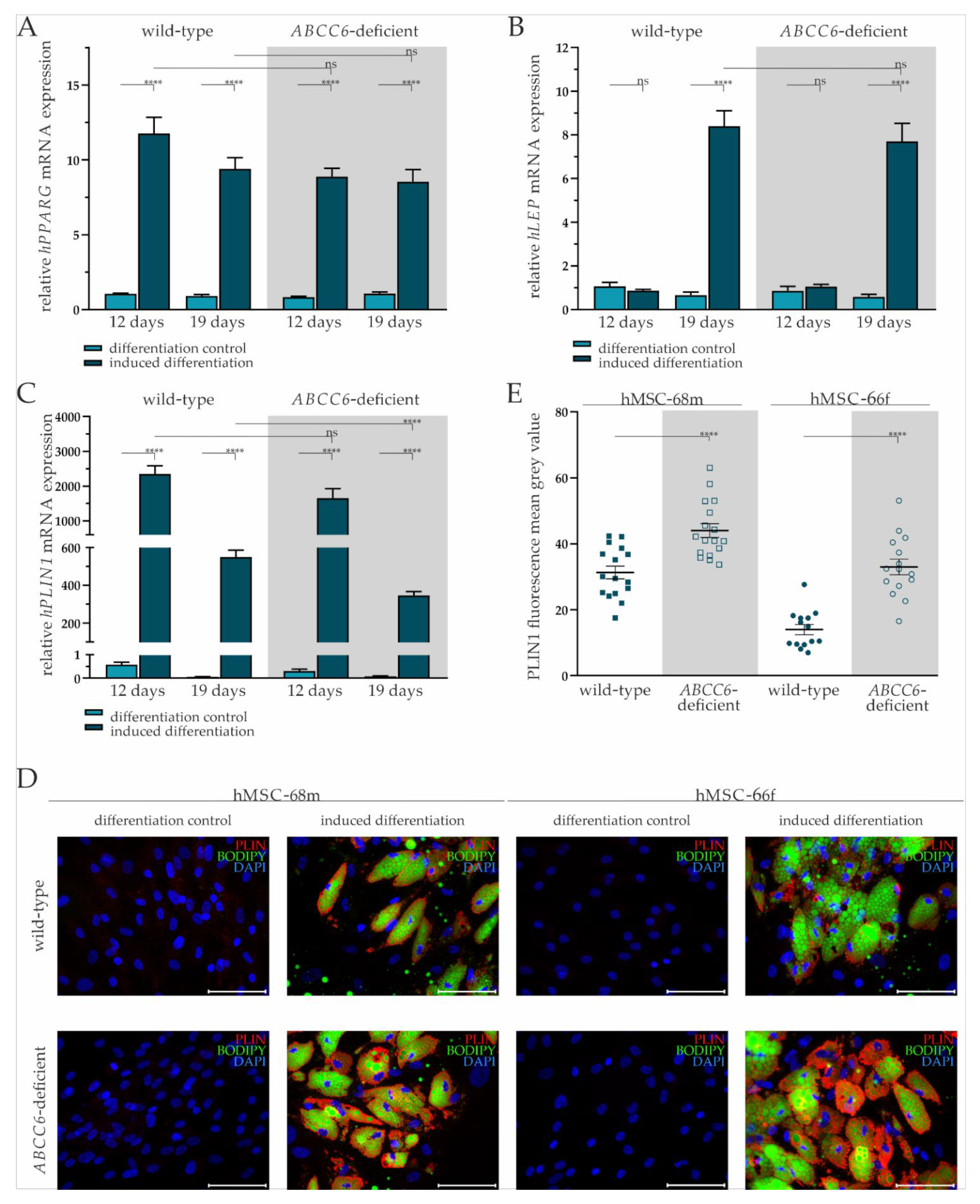
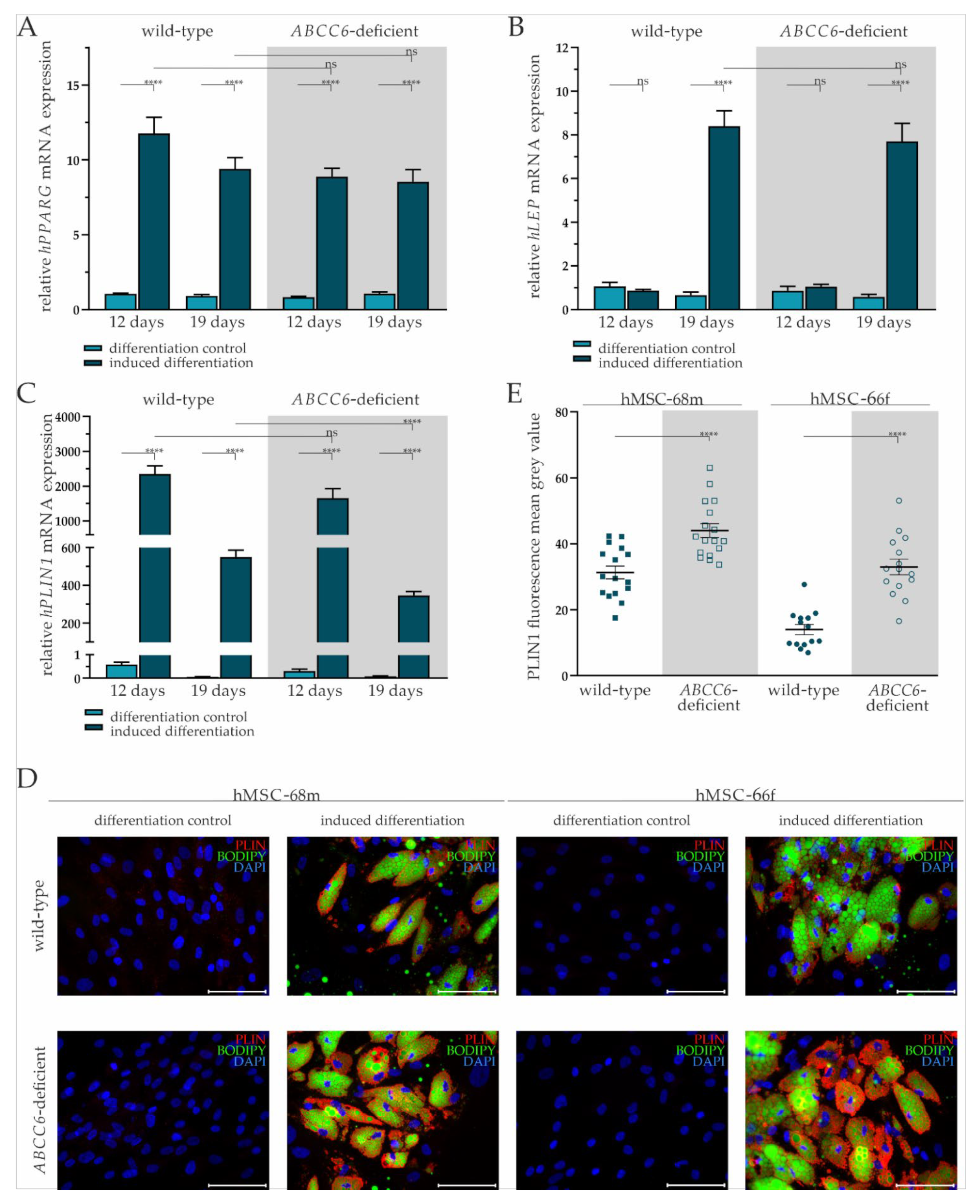
2.2. Successful Differentiation of hMSCs into Adipocytes Is Largely Independent of ABCC6
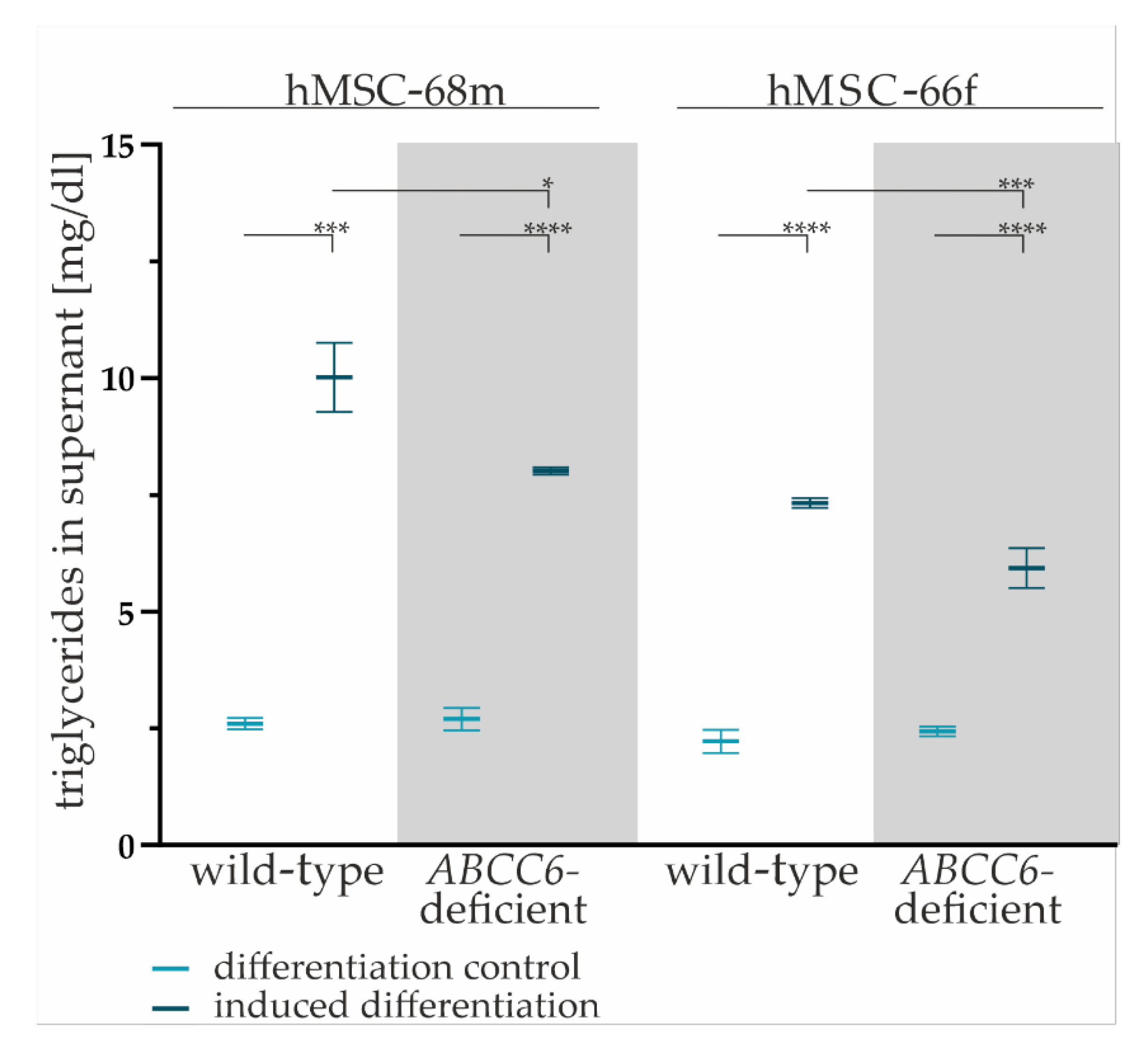
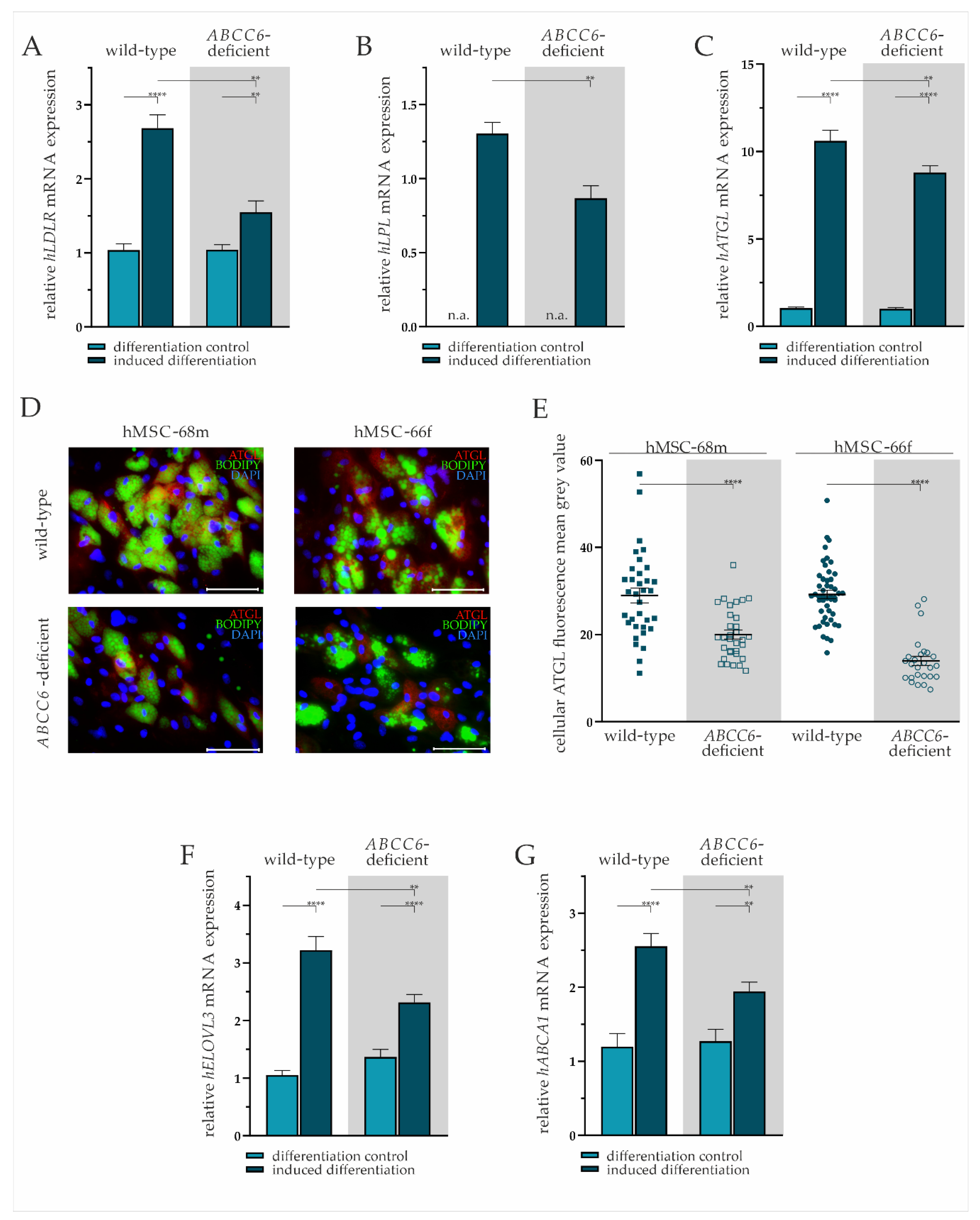
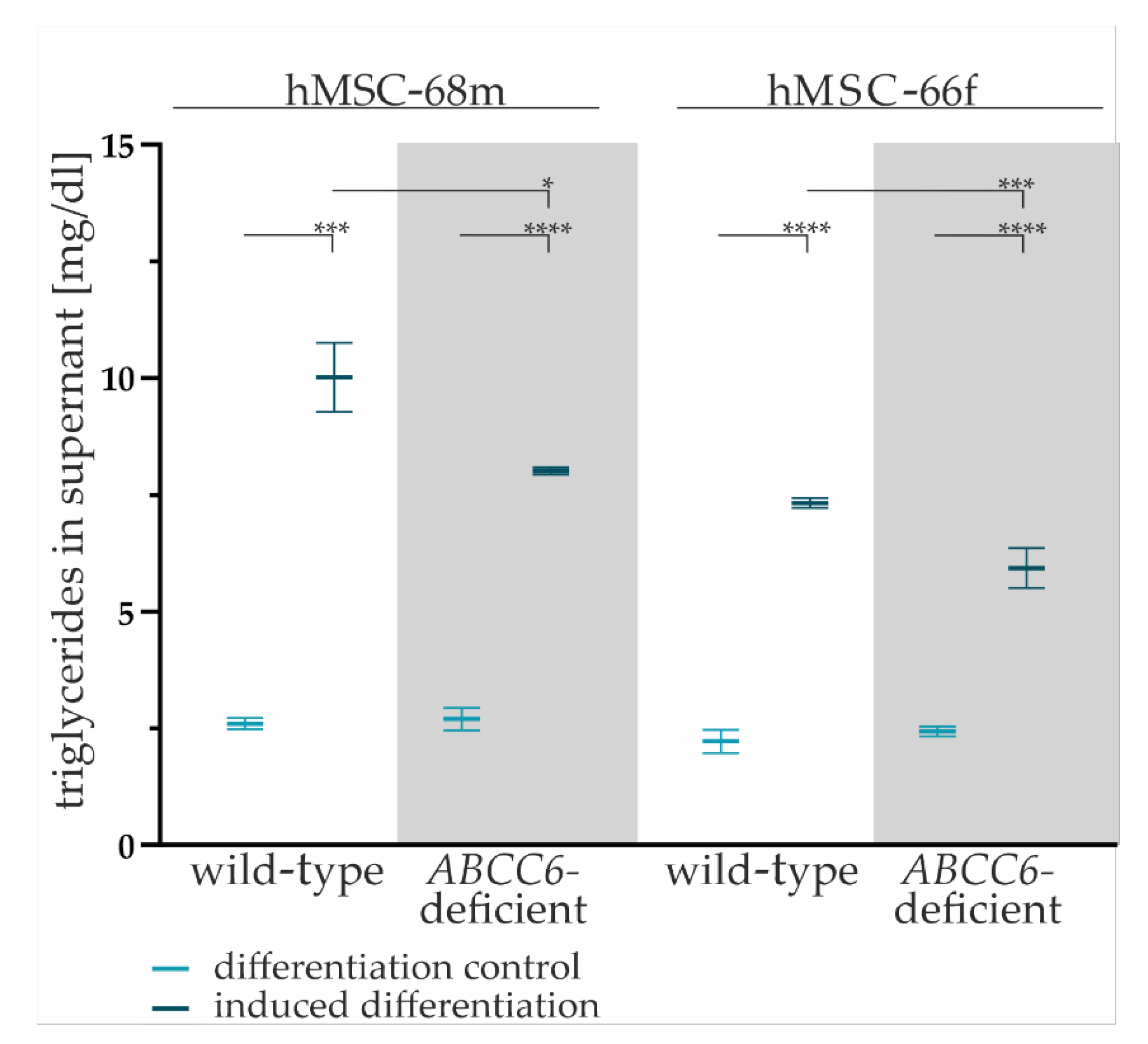
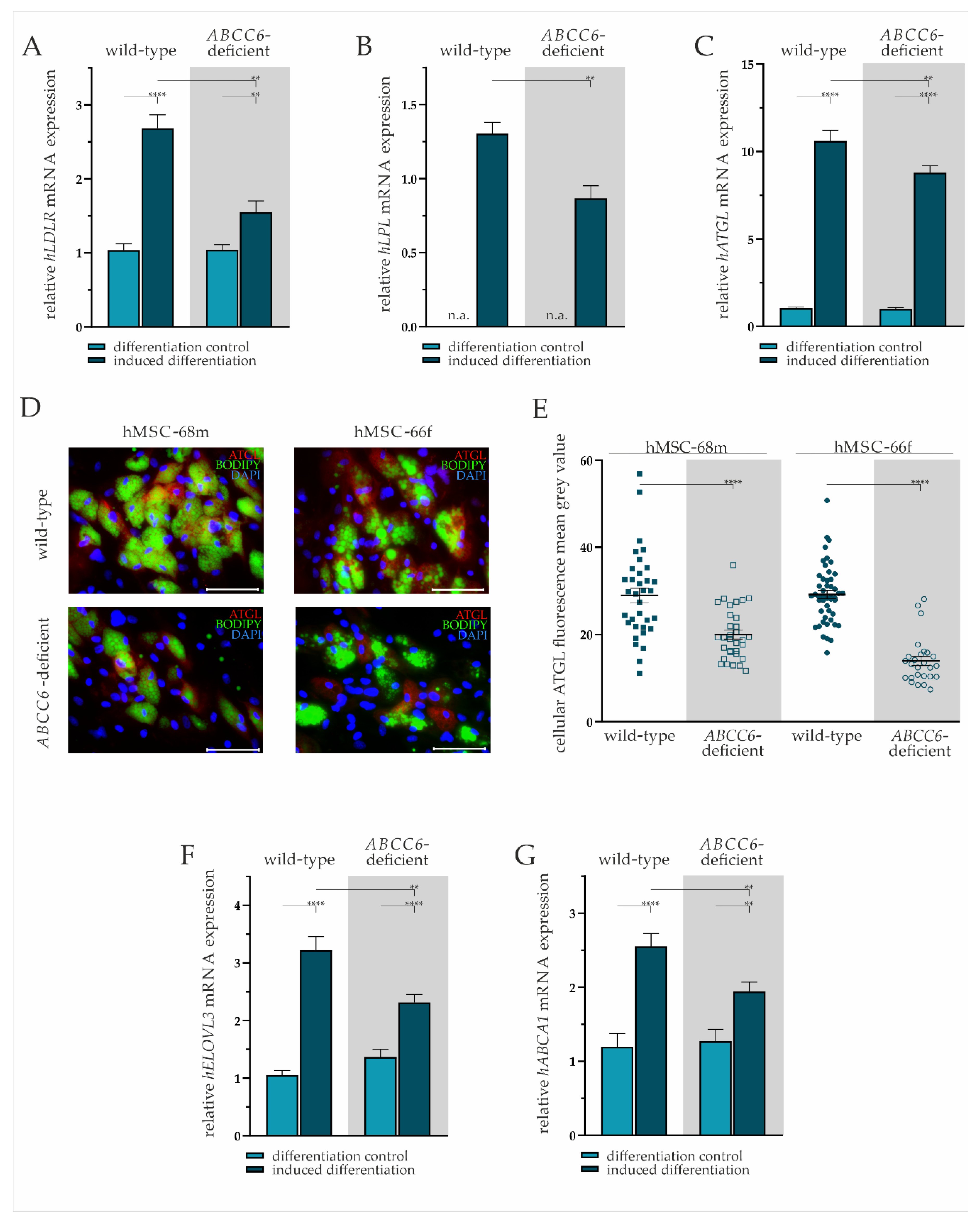
2.3. ABCC6 Deficiency Impairs Lipid Homeostasis in Adipocytes
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Adipogenic Differentiation
4.2. CRISPR–Cas9 Mediated ABCC6 Knock-Out
4.3. Nucleic Acid Isolation
4.4. Gene Expression Analysis
4.5. Genomic DNA Amplification
4.6. T7 Endonuclease I Mutagenesis Assay
4.7. TA cloning for Single Allele Sequencing
4.8. Sanger Sequencing
4.9. Immunofluorescent Staining, Bodipy Staining, and Fluorescence Microscopy
4.10. Triglyceride Quantification
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Contri, M.B.; Boraldi, F.; Taparelli, F.; De Paepe, A.; Ronchetti, I.P. Matrix proteins with high affinity for calcium ions are associated with mineralization within the elastic fibers of pseudoxanthoma elasticum dermis. Am. J. Pathol. 1996, 148, 569–577. [Google Scholar] [PubMed]
- Hosen, M.J.; Lamoen, A.; De Paepe, A.; Vanakker, O.M. Histopathology of Pseudoxanthoma Elasticum and Related Disorders: Histological Hallmarks and Diagnostic Clues. Scientifica 2012, 2012, e598262. [Google Scholar] [CrossRef] [PubMed]
- Neldner, K.H. Pseudoxanthoma Elasticum. Int. J. Dermatol. 1988, 27, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Plomp, A.S.; van Soest, S.; Wijnholds, J.; de Jong, P.T.V.M.; Bergen, A.A.B. Pseudoxanthoma elasticum: A clinical, histopathological, and molecular update. Surv. Ophthalmol. 2003, 48, 424–438. [Google Scholar] [CrossRef]
- Jensen, O.A. Bruch’s membrane in pseudoxanthoma elasticum. Albrecht Graefes Arch. Klin. Exp. Ophthalmol. 1977, 203, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Connor, P.J.; Juergens, J.L.; Perry, H.O.; Hollenhorst, R.W.; Edwards, J.E. Pseudoxanthoma elasticum and angioid streaks: A review of 106 cases. Am. J. Med. 1961, 30, 537–543. [Google Scholar] [CrossRef]
- Laube, S.; Moss, C. Pseudoxanthoma elasticum. Arch. Dis. Child. 2005, 90, 754–756. [Google Scholar] [CrossRef]
- Campens, L.; Vanakker, O.M.; Trachet, B.; Segers, P.; Leroy, B.P.; De Zaeytijd, J.; Voet, D.; De Paepe, A.; De Backer, T.; De Backer, J. Characterization of Cardiovascular Involvement in Pseudoxanthoma Elasticum Families. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2646–2652. [Google Scholar] [CrossRef]
- Bergen, A.A.B.; Plomp, A.S.; Schuurman, E.J.; Terry, S.; Breuning, M.; Dauwerse, H.; Swart, J.; Kool, M.; van Soest, S.; Baas, F.; et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 228–231. [Google Scholar] [CrossRef]
- Pseudoxanthoma Elasticum and ABCC6—ClinVar—NCBI. Available online: https://www.ncbi.nlm.nih.gov/clinvar/?term=pseudoxanthoma+elasticum+abcc6 (accessed on 23 March 2022).
- Beck, K.; Hayashi, K.; Dang, K.; Hayashi, M.; Boyd, C.D. Analysis of ABCC6 (MRP6) in normal human tissues. Histochem. Cell Biol. 2005, 123, 517–528. [Google Scholar] [CrossRef]
- Jiang, Q.; Endo, M.; Dibra, F.; Wang, K.; Uitto, J. Pseudoxanthoma Elasticum Is a Metabolic Disease. J. Investig. Dermatol. 2009, 129, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Hosen, M.J.; Zubaer, A.; Thapa, S.; Khadka, B.; Paepe, A.D.; Vanakker, O.M. Molecular Docking Simulations Provide Insights in the Substrate Binding Sites and Possible Substrates of the ABCC6 Transporter. PLoS ONE 2014, 9, e102779. [Google Scholar] [CrossRef] [PubMed]
- Kuzaj, P.; Kuhn, J.; Dabisch-Ruthe, M.; Faust, I.; Götting, C.; Knabbe, C.; Hendig, D. ABCC6- a new player in cellular cholesterol and lipoprotein metabolism? Lipids Health Dis. 2014, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Tarling, E.J.; de Aguiar Vallim, T.Q.; Edwards, P.A. Role of ABC transporters in lipid transport and human disease. Trends Endocrinol. Metab. 2013, 24, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Van Greevenbroek, M.M.; de Bruin, T.W. Chylomicron synthesis by intestinal cells in vitro and in vivo. Atherosclerosis 1998, 141, S9–S16. [Google Scholar] [CrossRef]
- Nilsson-Ehle, P.; Garfinkel, A.S.; Schotz, M.C. Lipolytic Enzymes and Plasma Lipoprotein Metabolism. Annu. Rev. Biochem. 1980, 49, 667–693. [Google Scholar] [CrossRef]
- Dashti, M.; Kulik, W.; Hoek, F.; Veerman, E.C.; Peppelenbosch, M.P.; Rezaee, F. A Phospholipidomic Analysis of All Defined Human Plasma Lipoproteins. Sci. Rep. 2011, 1, 139. [Google Scholar] [CrossRef]
- Merkel, M.; Eckel, R.H.; Goldberg, I.J. Lipoprotein lipase. J. Lipid Res. 2002, 43, 1997–2006. [Google Scholar] [CrossRef]
- Nomura, D.K.; Casida, J.E. Lipases and their inhibitors in health and disease. Chem. Biol. Interact. 2016, 259, 211–222. [Google Scholar] [CrossRef]
- Wang, S.; Smith, J.D. ABCA1 and nascent HDL biogenesis. BioFactors 2014, 40, 547–554. [Google Scholar] [CrossRef]
- Schweiger, M.; Lass, A.; Zimmermann, R.; Eichmann, T.O.; Zechner, R. Neutral lipid storage disease: Genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, E289–E296. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat Mobilization in Adipose Tissue Is Promoted by Adipose Triglyceride Lipase. Science 2004, 306, 1383–1386. [Google Scholar] [CrossRef]
- Kobayashi, T.; Fujimori, K. Very long-chain-fatty acids enhance adipogenesis through coregulation of Elovl3 and PPARγ in 3T3-L1 cells. Am. J. Physiol.-Endocrinol. Metab. 2012, 302, E1461–E1471. [Google Scholar] [CrossRef] [PubMed]
- Alexandersson, I.; Harms, M.J.; Boucher, J. Isolation and Culture of Human Mature Adipocytes Using Membrane Mature Adipocyte Aggregate Cultures (MAAC). JoVE J. Vis. Exp. 2020, 156, e60485. [Google Scholar] [CrossRef] [PubMed]
- Janderová, L.; McNeil, M.; Murrell, A.N.; Mynatt, R.L.; Smith, S.R. Human Mesenchymal Stem Cells as an in Vitro Model for Human Adipogenesis. Obes. Res. 2003, 11, 65–74. [Google Scholar] [CrossRef]
- Yin, Y.; Yuan, H.; Wang, C.; Pattabiraman, N.; Rao, M.; Pestell, R.G.; Glazer, R.I. 3-Phosphoinositide-Dependent Protein Kinase-1 Activates the Peroxisome Proliferator-Activated Receptor-γ and Promotes Adipocyte Differentiation. Mol. Endocrinol. 2006, 20, 268–278. [Google Scholar] [CrossRef]
- Yi, X.; Wu, P.; Liu, J.; Gong, Y.; Xu, X.; Li, W. Identification of the potential key genes for adipogenesis from human mesenchymal stem cells by RNA-Seq. J. Cell. Physiol. 2019, 234, 20217–20227. [Google Scholar] [CrossRef]
- Guo, H.; Li, Q.; Chou, D.W.; Uitto, J. Atorvastatin counteracts aberrant soft tissue mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6−/−). J. Mol. Med. 2013, 91, 1177–1184. [Google Scholar] [CrossRef]
- Stumpf, M.J.; Schahab, N.; Nickenig, G.; Skowasch, D.; Schaefer, C.A. Therapy of Pseudoxanthoma Elasticum: Current Knowledge and Future Perspectives. Biomedicines 2021, 9, 1895. [Google Scholar] [CrossRef]
- Tiemann, J.; Lindenkamp, C.; Plümers, R.; Faust, I.; Knabbe, C.; Hendig, D. Statins as a Therapeutic Approach for the Treatment of Pseudoxanthoma Elasticum Patients: Evaluation of the Spectrum Efficacy of Atorvastatin In Vitro. Cells 2021, 10, 442. [Google Scholar] [CrossRef]
- Liu, S.; Lorenzen, E.D.; Fumagalli, M.; Li, B.; Harris, K.; Xiong, Z.; Zhou, L.; Korneliussen, T.S.; Somel, M.; Babbitt, C.; et al. Population Genomics Reveal Recent Speciation and Rapid Evolutionary Adaptation in Polar Bears. Cell 2014, 157, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, L.; Rabasa-Lhoret, R.; Faraj, M.; Lavoie, M.-È.; Mill, J.; Pérusse, L.; Vohl, M.-C. Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction. Am. J. Clin. Nutr. 2010, 91, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Poitou, C.; Perret, C.; Mathieu, F.; Truong, V.; Blum, Y.; Durand, H.; Alili, R.; Chelghoum, N.; Pelloux, V.; Aron-Wisnewsky, J.; et al. Bariatric Surgery Induces Disruption in Inflammatory Signaling Pathways Mediated by Immune Cells in Adipose Tissue: A RNA-Seq Study. PLoS ONE 2015, 10, e0125718. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Barak, Y.; Hevener, A.; Olson, P.; Liao, D.; Le, J.; Nelson, M.; Ong, E.; Olefsky, J.M.; Evans, R.M. Adipose-specific peroxisome proliferator-activated receptor γ knockout causes insulin resistance in fat and liver but not in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 15712–15717. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, Y.; Gong, Z.; Sheng, X.; Li, Z.; Zhang, W.; Qin, Y. Berberine inhibits 3T3-L1 adipocyte differentiation through the PPARγ pathway. Biochem. Biophys. Res. Commun. 2006, 348, 571–578. [Google Scholar] [CrossRef]
- Arimura, N.; Horiba, T.; Imagawa, M.; Shimizu, M.; Sato, R. The Peroxisome Proliferator-activated Receptor γ Regulates Expression of the Perilipin Gene in Adipocytes. J. Biol. Chem. 2004, 279, 10070–10076. [Google Scholar] [CrossRef]
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2017, 1862, 1221–1232. [Google Scholar] [CrossRef]
- Shijun, L.; Khan, R.; Raza, S.H.A.; Jieyun, H.; Chugang, M.; Kaster, N.; Gong, C.; Chunping, Z.; Schreurs, N.M.; Linsen, Z. Function and characterization of the promoter region of perilipin 1 (PLIN1): Roles of E2F1, PLAG1, C/EBPβ, and SMAD3 in bovine adipocytes. Genomics 2020, 112, 2400–2409. [Google Scholar] [CrossRef]
- Xu, G.; Sztalryd, C.; Londos, C. Degradation of perilipin is mediated through ubiquitination-proteasome pathway. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2006, 1761, 83–90. [Google Scholar] [CrossRef]
- Nielsen, T.S.; Jessen, N.; Jørgensen, J.O.L.; Møller, N.; Lund, S. Dissecting adipose tissue lipolysis: Molecular regulation and implications for metabolic disease. J. Mol. Endocrinol. 2014, 52, R199–R222. [Google Scholar] [CrossRef]
- Zhang, X.; Xie, X.; Heckmann, B.L.; Saarinen, A.M.; Czyzyk, T.A.; Liu, J. Targeted Disruption of G0/G1 Switch Gene 2 Enhances Adipose Lipolysis, Alters Hepatic Energy Balance, and Alleviates High-Fat Diet–Induced Liver Steatosis. Diabetes 2014, 63, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Brampton, C.; Pomozi, V.; Chen, L.-H.; Apana, A.; McCurdy, S.; Zoll, J.; Boisvert, W.A.; Lambert, G.; Henrion, D.; Blanchard, S.; et al. ABCC6 deficiency promotes dyslipidemia and atherosclerosis. Sci. Rep. 2021, 11, 3881. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, B.; Heeren, J.; Blaeser, M.; Radner, H.; Kayser, D.; Aydin, B.; Merkel, M. Lipoprotein lipase-facilitated uptake of LDL is mediated by the LDL receptor. J. Lipid Res. 2007, 48, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, V.W.; Cremer, P.; Eberle, E.; Manke, A.; Schulze, F.; Wieland, H.; Kreuzer, H.; Seidel, D. The association between serum Lp(a) concentrations and angiographically assessed coronary atherosclerosis: Dependence on serum LDL levels. Atherosclerosis 1986, 62, 249–257. [Google Scholar] [CrossRef]
- Pisciotta, L.; Tarugi, P.; Borrini, C.; Bellocchio, A.; Fresa, R.; Guerra, D.; Quaglino, D.; Ronchetti, I.; Calandra, S.; Bertolini, S. Pseudoxanthoma elasticum and familial hypercholesterolemia: A deleterious combination of cardiovascular risk factors. Atherosclerosis 2010, 210, 173–176. [Google Scholar] [CrossRef]
- Getz, G.S.; Reardon, C.A. Animal Models of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1104–1115. [Google Scholar] [CrossRef]
- Jakobsson, A.; Westerberg, R.; Jacobsson, A. Fatty acid elongases in mammals: Their regulation and roles in metabolism. Prog. Lipid Res. 2006, 45, 237–249. [Google Scholar] [CrossRef]
- Brooks-Wilson, A.; Marcil, M.; Clee, S.M.; Zhang, L.-H.; Roomp, K.; van Dam, M.; Yu, L.; Brewer, C.; Collins, J.A.; Molhuizen, H.O.F.; et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 1999, 22, 336–345. [Google Scholar] [CrossRef]
- Ibold, B.; Tiemann, J.; Faust, I.; Ceglarek, U.; Dittrich, J.; Gorgels, T.G.M.F.; Bergen, A.A.B.; Vanakker, O.; Van Gils, M.; Knabbe, C.; et al. Genetic deletion of Abcc6 disturbs cholesterol homeostasis in mice. Sci. Rep. 2021, 11, 2137. [Google Scholar] [CrossRef]
- Pomozi, V.; Le Saux, O.; Brampton, C.; Apana, A.; Iliás, A.; Szeri, F.; Martin, L.; Monostory, K.; Paku, S.; Sarkadi, B.; et al. ABCC6 Is a Basolateral Plasma Membrane Protein. Circ. Res. 2013, 112, e148–e151. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nomenclature | Origin | Age | Sex | Lot | Surface Markers * |
|---|---|---|---|---|---|
| hMSCS-68m | Human bone marrow/femoral head | 68 | male | 467Z023.5 | CD73+, CD90+, CD105+, CD14−, CD34−, CD45−, CD19− |
| hMSCS-66f | Human bone marrow/femoral head | 66 | female | 451Z012.3 | CD73+, CD90+, CD105+, CD14−, CD34−, CD45−, CD19− |
| Gene | 5′-3′ Primer Sequences | TA (°C) | Efficiency | Product Size (bp) |
|---|---|---|---|---|
| hABCA1 | ATCCCCAGCACAGCCTAT TCTCCCCAAACCTTTCCA | 59 | 1.88 | 212 |
| hABCC6 | CCTGCTGATGTACGCCTT ACGCGAGCATTGTTCTGA | 60 | 1.92 | 267 |
| hATGL | CAGCGGTTTCATCCCCGTGT GCACATCTCTCGCAGCACCA | 61 | 2.00 | 283 |
| hELOVL3 | CAGCTTTGCAAGTCCGCGTTA GAGACGGAACAGAGCCGGGA | 66 | 1.75 | 136 |
| hHMBS | CTGCCAGAGAAGAGTGTG AGCTGTTGCCAGGATGAT | 63 | 1.92 | 165 |
| hLDLR | CGACTGCAAGGACAAATCTG AGTCATATTCCCGGTCACAC | 61 | 2.00 | 122 |
| hLEP | GGGAACCCTGTGCGGATTCTT GGAGGAGACTGACTGCGTG | 66 | 1.81 | 408 |
| hLPL | CCCTGCTCGTGCTGACTCTG CGCGGACACTGGGTAATGCT | 61 | 2.00 | 362 |
| hPLIN1 | CGGTCAGCCGGAGTGAGTG TACTGGAGGGCGGGGATCTT | 66 | 2.00 | 417 |
| hPPARg | ACCAAAGTGCAATCAAAGTGGA ATGAGGGAGTTGGAAGGCTCT | 66 | 1.96 | 100 |
| hRPL13 | CGGAAGGTGGTGGTCGTA CTCGGGAAGGGTTGGTGT | 63 | 1.87 | 115 |
| hSDHA | AACTCGCTCTTGGACCTG GAGTCGCAGTTCCGATGT | 63 | 1.93 | 177 |
| Gene | 5′-3′ Primer Sequences | TA (°C) | Product Size (bp) |
|---|---|---|---|
| ABCC6 | CTGTTCTCCGGGCATCAGAG GATGGACGGGGTGGTAGGAT | 59 | 554 |
| GRIN1 | GCGCCGCTAACCATAAACAA GAATCTCCTGCGGAGGGACG | 56 | 213 |
| HMGN5 | AGCAGATGCTTGTGCCAGTT CCCCACCCAAGGGGTTTAC | 56 | 218 |
| PIEZO2 | GCCCTGGAACTGGTGGTCTT AAAGGCTTCCCACTCTCAACT | 56 | 144 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plümers, R.; Osterhage, M.R.; Lindenkamp, C.; Knabbe, C.; Hendig, D. Targeting ABCC6 in Mesenchymal Stem Cells: Impairment of Mature Adipocyte Lipid Homeostasis. Int. J. Mol. Sci. 2022, 23, 9218. https://doi.org/10.3390/ijms23169218
Plümers R, Osterhage MR, Lindenkamp C, Knabbe C, Hendig D. Targeting ABCC6 in Mesenchymal Stem Cells: Impairment of Mature Adipocyte Lipid Homeostasis. International Journal of Molecular Sciences. 2022; 23(16):9218. https://doi.org/10.3390/ijms23169218
Chicago/Turabian StylePlümers, Ricarda, Michel R. Osterhage, Christopher Lindenkamp, Cornelius Knabbe, and Doris Hendig. 2022. "Targeting ABCC6 in Mesenchymal Stem Cells: Impairment of Mature Adipocyte Lipid Homeostasis" International Journal of Molecular Sciences 23, no. 16: 9218. https://doi.org/10.3390/ijms23169218
APA StylePlümers, R., Osterhage, M. R., Lindenkamp, C., Knabbe, C., & Hendig, D. (2022). Targeting ABCC6 in Mesenchymal Stem Cells: Impairment of Mature Adipocyte Lipid Homeostasis. International Journal of Molecular Sciences, 23(16), 9218. https://doi.org/10.3390/ijms23169218