
Metatranscriptomic Analysis Reveals Rich Mycoviral Diversity in Three Major Fungal Pathogens of Rice


Abstract
:1. Introduction
2. Results
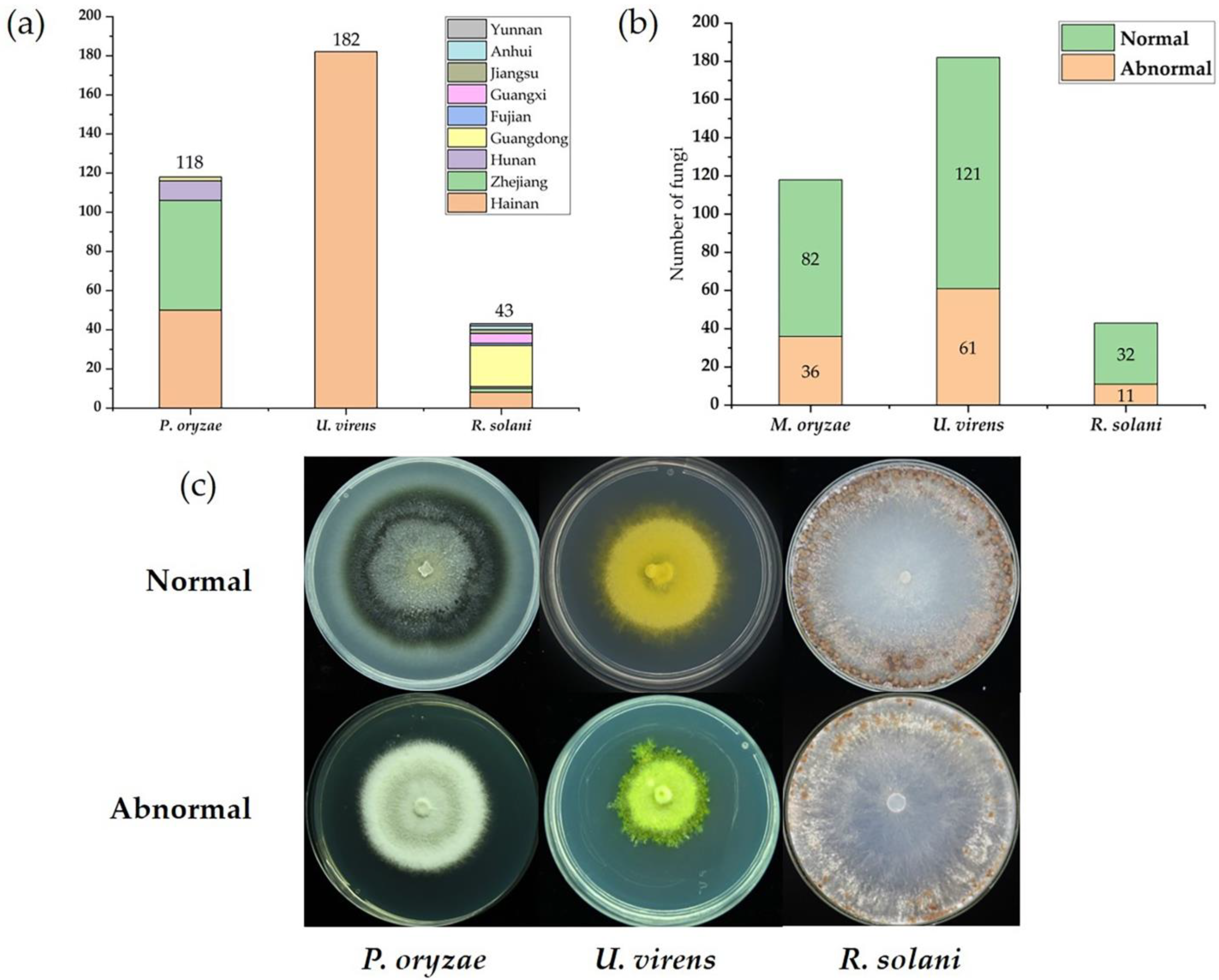
2.1. Collection and Analysis of Three Major Fungal Pathogens of Rice
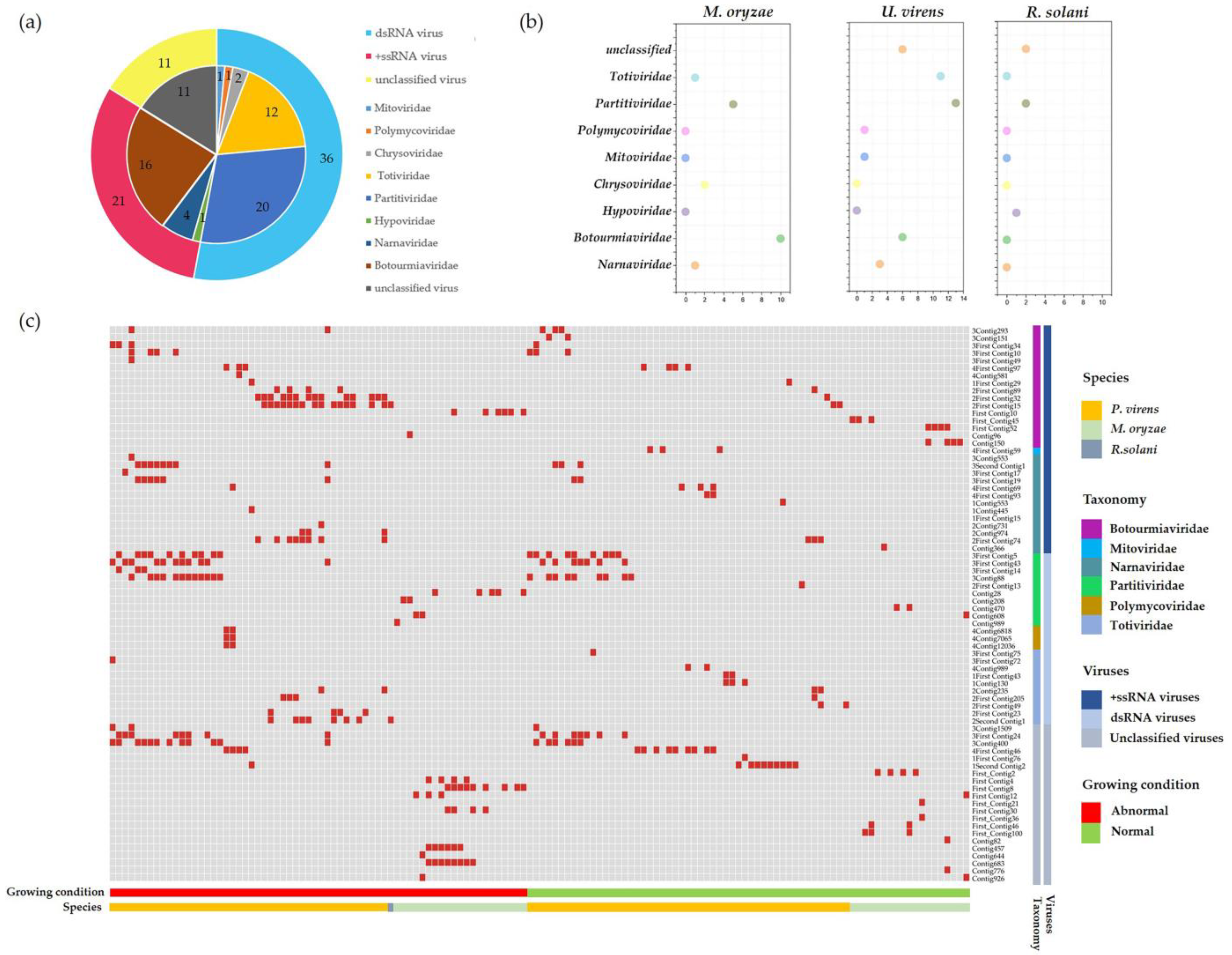
2.2. Metatranscriptomic Identification of Mycoviruses Infecting the Tested Strains
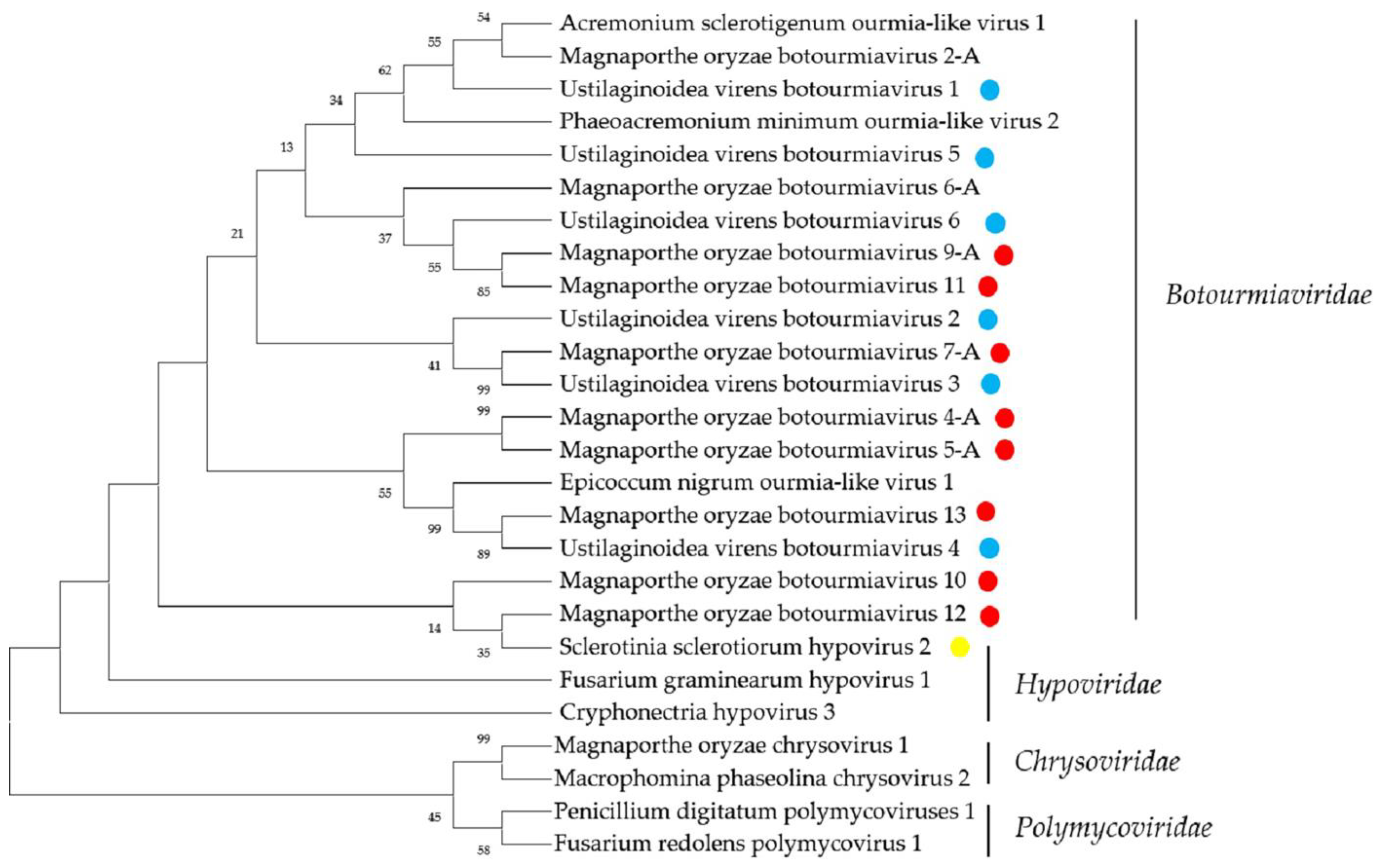
2.3. Sequences Related to Members of the Families Botourmiaviridae and Hypoviridae
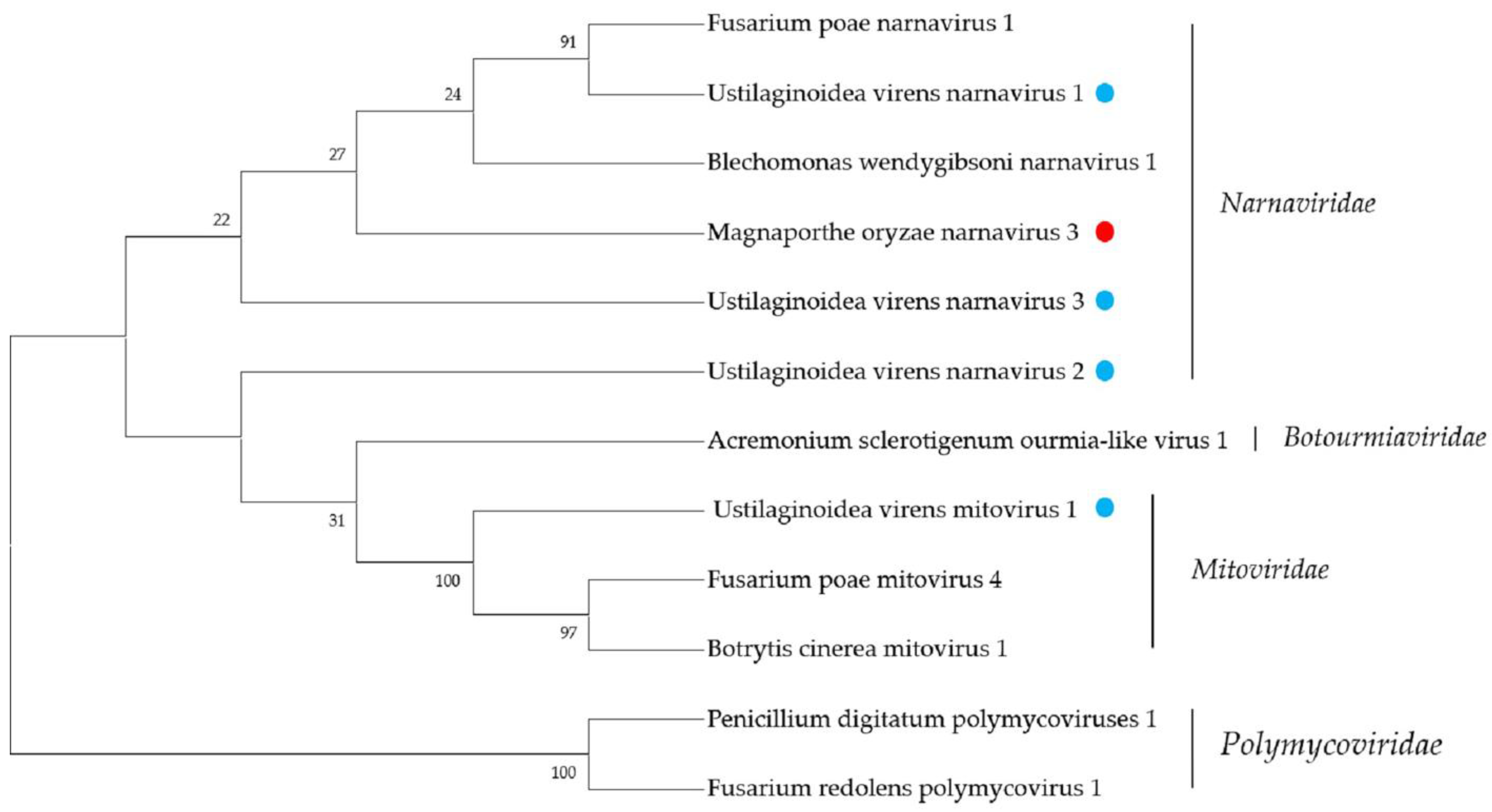
2.4. Narnaviridae- and Mitoviridae-Related Sequences
2.5. Sequences Related to Members of the Family Partitiviridae
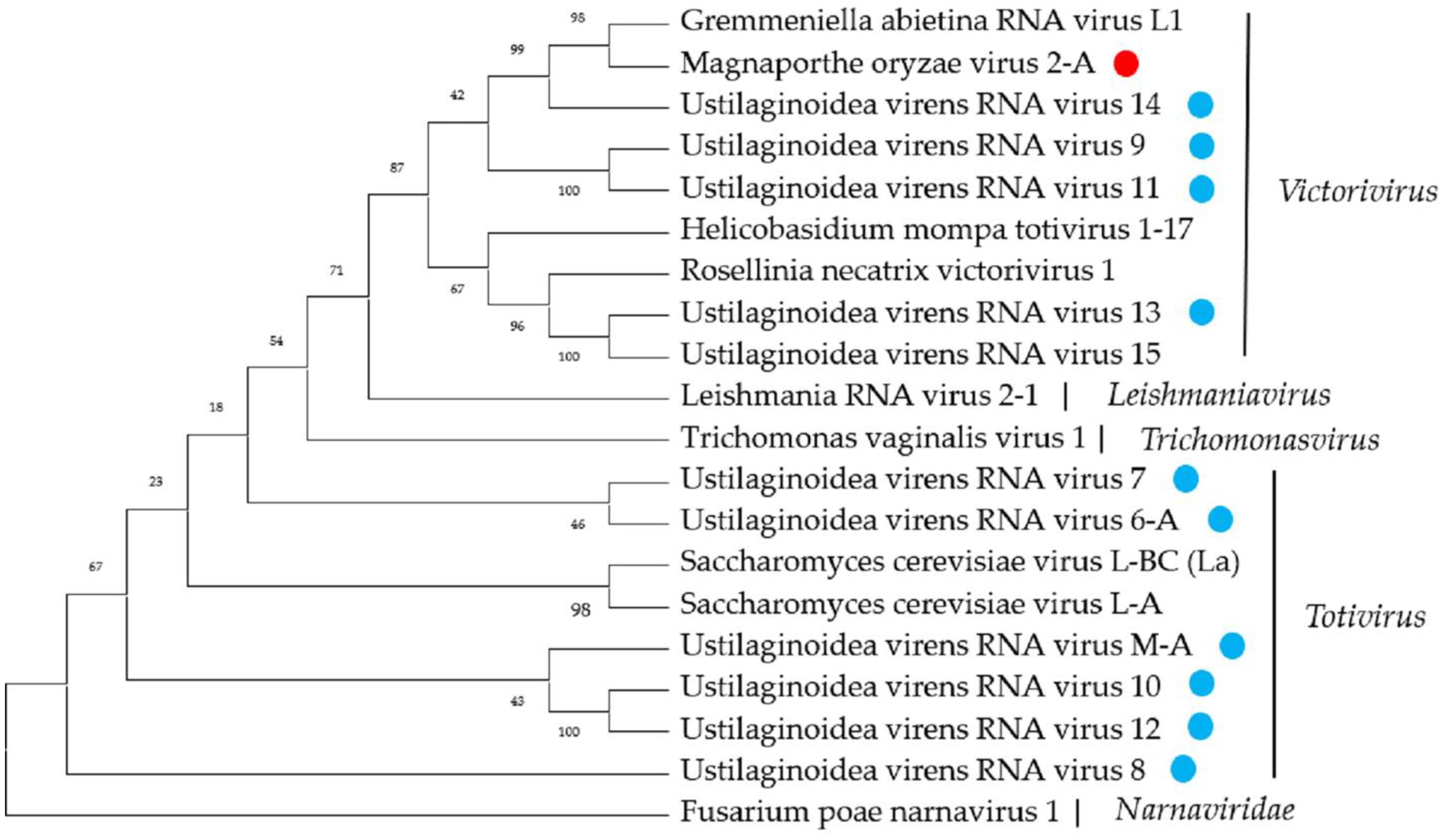
2.6. Twelve Predicted Novel Viruses in the Family Totiviridae
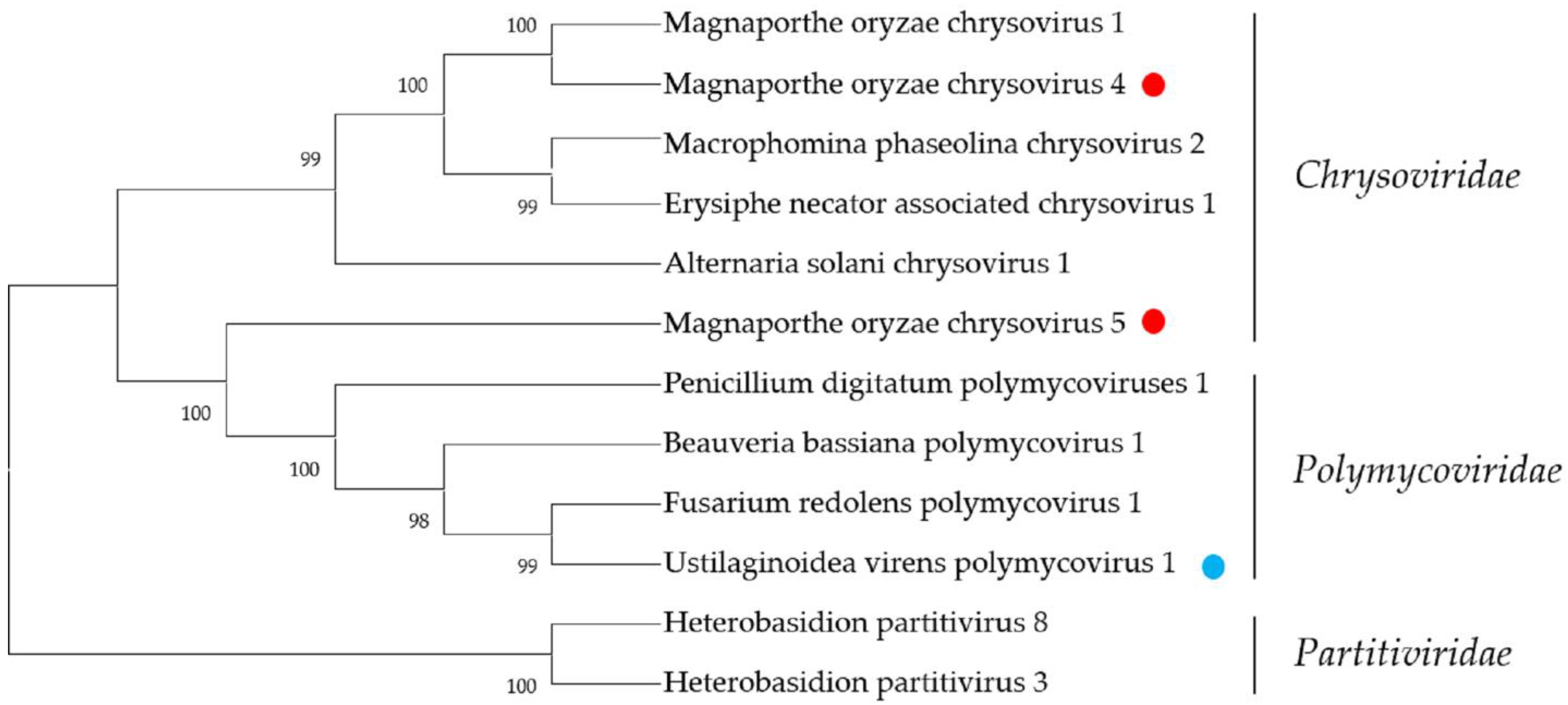
2.7. Chrysoviridae- and Polymycoviridae-Related Sequences
3. Discussion
4. Materials and Methods
4.1. Fungal Strains, Cultural Conditions, and RNA Extraction
4.2. Metatranscriptomic Sequencing and Bioinformatic Analysis
4.3. Putative Mycovirus Sequence Confirmation
4.4. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, X.; Ronald, P.C. Innate immunity in rice. Trends Plant Sci. 2011, 16, 451–459. [Google Scholar] [CrossRef]
- Khush, G.S. What it will take to feed 5.0 billion rice consumers in 2030. Plant Mol. Biol. 2005, 59, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, J.; Triplett, L.; Leach, J.E.; Wang, G.L. Novel insights into rice innate immunity against bacterial and fungal pathogens. Annu. Rev. Phytopathol. 2013, 52, 213–241. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H. Pathogen variability and host resistance in rice blast disease. Annu. Rev. Phytopathol. 1980, 18, 167–187. [Google Scholar] [CrossRef]
- Abdoulaye, A.H.; Foda, M.F.; Kotta-Loizou, I. Viruses infecting the plant pathogenic fungus Rhizoctonia solani. Viruses 2019, 11, 1113. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, T.; Luo, C.; Jiang, D.; Li, G.; Li, Q.; Tom, H.; Huang, J. Prevalence and diversity of mycoviruses infecting the plant pathogen Ustilaginoidea virens. Virus Res. 2015, 195, 47–56. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, L.; Zhang, X.; Zhang, Q.; Jia, Y.; Wang, G.; Li, S.; Tian, D.; Li, W.H.; Yang, S. Large-scale identification and functional analysis of NLR genes in blast resistance in the Tetep rice genome sequence. Proc. Natl. Acad. Sci. USA 2019, 116, 18479–18487. [Google Scholar] [CrossRef]
- Yin, J.; Zou, L.; Zhu, X.; Cao, Y.; He, M.; Chen, X. Fighting the enemy: How rice survives the blast pathogen’s attack. Crop J. 2021, 9, 543–552. [Google Scholar] [CrossRef]
- Ghabrial, S.A.; Castón, J.R.; Jiang, D.; Nibert, M.L.; Suzuki, N. 50-plus years of fungal viruses. Virology 2015, 479-480, 356–368. [Google Scholar] [CrossRef]
- Xie, J.; Jiang, D. New insights into mycoviruses and exploration for the biological control of crop fungal diseases. Annu. Rev. Phytopathol. 2014, 52, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Hollings, M. Viruses associated with a die-back disease of cultivated mushroom. Nature 1962, 196, 962–965. [Google Scholar] [CrossRef]
- Zhang, H.; Xie, J.; Fu, Y.; Cheng, J.; Qu, Z.; Zhao, Z.; Cheng, S.; Chen, T.; Li, B.; Wang, Q.; et al. A 2-kb mycovirus converts a pathogenic fungus into a beneficial endophyte for Brassica protection and yield enhancement. Mol. Plant 2020, 13, 1420–1433. [Google Scholar] [CrossRef]
- Jia, H.; Dong, K.; Zhou, L.; Wang, G.; Hong, N.; Jiang, D.; Xu, W. A dsRNA virus with filamentous viral particles. Nat. Commun. 2017, 8, 168. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, B.; Fu, Y.; Xie, J.; Cheng, J.; Ghabrial, S.A.; Li, G.; Yi, X.; Jiang, D. Extracellular transmission of a DNA mycovirus and its use as a natural fungicide. Proc. Natl. Acad. Sci. USA 2013, 110, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, S.; Zhang, L.; Qiu, D.; Guo, L. A tripartite ssDNA mycovirus from a plant pathogenic fungus is infectious as cloned DNA and purified virions. Sci. Adv. 2020, 6, eaay9634. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Yuan, Y.; Mao, J.; Zhu, Y.; Cao, Q.; Zhang, T.; Wang, S.; Liu, D. Characterization of two novel mycoviruses from Penicillium digitatum and the related fungicide resistance analysisand. Sci. Rep. 2018, 8, 5513. [Google Scholar] [CrossRef] [PubMed]
- Picarelli, M.A.S.C.; Forgia, M.; Rivas, E.B.; Nerva, L.; Chiapello, M.; Turina, M.; Colariccio, A. Extreme diversity of mycoviruses present in isolates of Rhizoctonia solani AG2-2 LP from Zoysia japonica from Brazil. Front. Cell. Infect. Microbiol. 2019, 9, 244. [Google Scholar] [CrossRef]
- Marzano, S.-Y.L.; Nelson, B.D.; Ajayi-Oyetunde, O.; Bradley, C.A.; Hughes, T.J.; Hartman, G.L.; Eastburn, D.M.; Domier, L.L. Identification of diverse mycoviruses through metatranscriptomics characterization of the viromes of five major fungal plant pathogens. J. Virol. 2016, 90, 6846–6863. [Google Scholar] [CrossRef]
- Kotta-Loizou, I.; Coutts, R.H. Studies on the virome of the entomopathogenic fungus Beauveria bassiana reveal novel dsRNA elements and mild hypervirulence. Plos Pathog. 2017, 13, e1006183. [Google Scholar] [CrossRef]
- He, W.; Hou, X.; Zhao, J.; Sun, J.; He, H.; Si, W.; Wang, J.; Jiang, Z.; Yan, Z.; Xing, G.; et al. Virome characterization of game animals in China reveals a spectrum of emerging pathogens. Cell 2022, 185, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Kavi Sidharthan, V.; Nagendran, K.; Baranwal, V.K. Exploration of plant transcriptomes reveals five putative novel poleroviruses and an enamovirus. Virus Genes 2022, 58, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Urayama, S.-I.; Sakoda, H.; Takai, R.; Katoh, Y.; Le, T.M.; Fukuhara, T.; Arie, T.; Teraoka, T.; Moriyama, H. A dsRNA mycovirus, Magnaporthe oryzae chrysovirus 1-B, suppresses vegetative growth and development of the rice blast fungus. Virology 2014, 448, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Higashiura, T.; Katoh, Y.; Urayama, S.I.; Hayashi, O.; Moriyama, H. Magnaporthe oryzae chrysovirus 1 strain D confers growth inhibition to the host fungus and exhibits multiform viral structural proteins. Virology 2019, 535, 241–254. [Google Scholar] [CrossRef]
- Urayama, S.; Kato, S.; Suzuki, Y.; Aoki, N.; Le, M.T.; Arie, T.; Teraoka, T.; Fukuhara, T.; Moriyama, H. Mycoviruses related to chrysovirus affect vegetative growth in the rice blast fungus Magnaporthe Oryzae. J. Gen. Virol. 2010, 91, 3085–3094. [Google Scholar] [CrossRef]
- Liu, C.; Zeng, M.; Zhang, M.; Shu, C.; Zhou, E. Complete nucleotide sequence of a partitivirus from Rhizoctonia solani AG-1 IA strain C24. Viruses 2018, 10, 703. [Google Scholar] [CrossRef]
- Zhang, M.; He, Z.; Huang, X.; Shu, C.; Zhou, E. Genome organizations and functional analyses of a novel Gammapartitivirus from Rhizoctonia solani AG-1 IA strain D122. Viruses 2021, 13, 2254. [Google Scholar] [CrossRef]
- Zheng, L.; Shu, C.; Zhang, M.; Yang, M.; Zhou, E. Molecular characterization of a novel Endornavirus conferring hypovirulence in rice sheath blight fungus Rhizoctonia solani AG-1 IA strain GD-2. Viruses 2019, 11, 178. [Google Scholar] [CrossRef]
- Zheng, L.; Zhang, M.; Chen, Q.; Zhu, M.; Zhou, E. A novel mycoviras closely related to viruses in the genus Alphapartitivirus confers hypovirulence in the phytopathogenic fungus. Rhizoctonia Solani. Virol. 2014, 456–457, 220–226. [Google Scholar] [CrossRef]
- Ayllón, M.A.; Turina, M.; Xie, J.; Nerva, L.; Marzano, S.Y.I.; Donaire, L.; Jiang, D. ICTV Report Consortium. ICTV virus taxonomy profile: Botourmiaviridae. J. Gen. Virol. 2020, 101, 454–455. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, L.; Esmael, A.; Duan, J.; Bian, X.; Jia, J.; Xie, J.; Cheng, J.; Fu, Y.; Jiang, D. Four novel botourmiaviruses co-infecting an isolate of the rice blast fungus Magnaporthe oryzae. Viruses 2020, 12, 1383. [Google Scholar] [CrossRef] [PubMed]
- Ohkita, S.; Lee, Y.; Nguyen, Q.; Ikeda, K.; Suzuki, N.; Nakayashiki, H. Three ourmia-like viruses and their associated RNAs in Pyricularia oryzae. Virology 2019, 534, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Ghabrial, S.A.; Kim, K.; Pearson, M.; Marzano, S.L.; Yaegashi, H.; Xie, J.; Guo, L.; Kondo, H.; Koloniuk, I.; et al. ICTV Report Consortium. ICTV virus taxonomy profile: Hypoviridae. J. Gen. Virol. 2018, 99, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Hillman, B.I.; Cai, G. The family Narnaviridae: Simplest of RNA viruses. Adv. Virus Res. 2013, 86, 149–176. [Google Scholar] [PubMed]
- Ghabrial, S.A.; Suzuki, N. Viruses of plant pathogenic fungi. Annu. Rev. Phytopathol. 2009, 47, 353–384. [Google Scholar] [CrossRef]
- Vainio, E.J.; Chiba, S.; Ghabrial, S.A.; Maiss, E.; Roossinck, M.; Sabanadzovic, S.; Suzuki, N.; Xie, J.; Nibert, M. ICTV Report Consortium. ICTV virus taxonomy profile: Partitiviridae. J. Gen. Virol. 2018, 99, 17–18. [Google Scholar] [CrossRef]
- Du, Y.; He, X.; Zhou, X.; Fang, S.; Deng, Q. Complete nucleotide sequence of Magnaporthe oryzae partitivirus 1. Arch. Virol. 2016, 161, 3295–3298. [Google Scholar] [CrossRef]
- Zhang, M.; Zheng, L.; Liu, C.; Shu, C.; Zhou, E. Characterization of a novel dsRNA mycovirus isolated from strain A105 of Rhizoctonia solani AG-1 IA. Arch. Virol. 2018, 163, 427–430. [Google Scholar] [CrossRef]
- Zhong, J.; Zhu, J.; Lei, X.; Chen, D.; Zhu, H.; Gao, B. Complete genome sequence and organization of a novel virus from the rice false smut fungus Ustilaginoidea virens. Virus Genes 2014, 48, 329–333. [Google Scholar] [CrossRef]
- Yokoi, T.; Yamashita, S.; Hibi, T. The nucleotide sequence and genome organization of Magnaporthe oryzae virus 1. Arch. Virol. 2007, 152, 2265–2269. [Google Scholar] [CrossRef]
- Maejima, K.; Himeno, M.; Komatsu, K.; Kakizawa, S.; Yamaji, Y.; Hamamoto, H.; Namba, S. Complete nucleotide sequence of a new double-stranded RNA virus from the rice blast fungus, Magnaporthe oryzae. Arch. Virol. 2008, 153, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Kotta-Loizou, I.; Castón, J.R.; Coutts, R.H.A.; Hillman, B.I.; Jiang, D.; Kim, D.-H.; Moriyama, H.; Suzuki, N. ICTV Report Consortium. ICTV Virus Taxonomy Profile: Chrysoviridae. J. Gen. Virol. 2020, 101, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Kanhayuwa, L.; Kotta-Loizou, I.; Özkan, S.; Coutts, R.H.A. A novel mycovirus from Aspergillus fumigatus contains four unique dsRNAs as its genome and is infectious as dsRNA. Proc. Natl. Acad. Sci. USA 2015, 111, 9100–9105. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Xiang, J.; Zhang, M.; Fu, M.; Yang, Z.; Hong, N.; Wang, G. Characterization of a novel double-stranded RNA mycovirus conferring hypovirulence from the phytopathogenic fungus Botryosphaeria dothidea. Virology 2016, 493, 75–85. [Google Scholar] [CrossRef]
- Mu, F.; Xie, J.; Cheng, S.; You, M.; Barbetti, M.J.; Jia, J.; Wang, Q.; Cheng, J.; Fu, Y.; Chen, T.; et al. Virome characterization of a collection of S. sclerotiorum from Australia. Front. Microbiol. 2017, 8, 2540. [Google Scholar] [CrossRef]
- Filippou, C.; Diss, R.M.; Daudu, J.O.; Coutts, R.H.A.; Kotta-Loizou, I. The polymycovirus-mediated growth enhancement of the entomopathogenic fungus Beauveria bassiana is dependent on carbon and nitrogen metabolism. Front. Microbiol. 2021, 12, 606366. [Google Scholar] [CrossRef]
- Nerva, L.; Vigani, G.; Silvestre, D.D.; Ciuffo, M.; Forgia, M.; Chitarra, W.; Turina, M. Biological and molecular characterization of Chenopodium quinoa mitovirus 1 reveals a distinct small RNA response compared to cytoplasmic RNA viruses. J. Virol. 2019, 93, e01998-18. [Google Scholar] [CrossRef]
- Vainio, E.J.; Müller, M.M.; Korhonen, K.; Piri, T.; Hantula, J. Viruses accumulate in aging infection centers of a fungal forest pathogen. ISME J. 2015, 9, 497–507. [Google Scholar] [CrossRef]
- Mu, F.; Li, B.; Cheng, S.; Jia, J.; Jiang, D.; Fu, y.; Cheng, J.; Lin, Y.; Chen, T.; Xie, J. Nine viruses from eight lineages exhibiting new evolutionary modes that co-infect a hypovirulent phytopathogenic fungus. Plos Pathog. 2021, 17, e1009823. [Google Scholar] [CrossRef]
- Herrero, N. A novel monopartite dsRNA virus isolated from the entomopathogenic and nematophagous fungus Purpureocillium lilacinum. Arch. Virol. 2016, 161, 3375–3384. [Google Scholar] [CrossRef]
- Herrero, N.; Duenas, E.; Quesada-Moraga, E.; Zabalgogeazcoa, I. Prevalence and diversity of viruses in the entomopathogenic fungus Beauveria bassiana. Appl. Environ. Microbiol. 2012, 78, 8523–8530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tan, X.; He, Y.; Xie, K.; Li, L.; Wang, R.; Hong, G.; Li, J.; Li, J.; Taliansky, M.; et al. Rice black–streaked dwarf virus P10 acts as either a synergistic or antagonistic determinant during superinfection with related or unrelated virus. Mol. Plant Pathol. 2019, 20, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Lee, K.M.; Cho, W.K.; Park, J.Y.; Kim, K.H. Differential contribution of RNA interference components in response to distinct Fusarium graminearum virus infections. J. Virol. 2018, 92, e01756-17. [Google Scholar] [CrossRef]
- Son, M.; Lee, K.M.; Yu, J.; Kang, M.; Park, J.M.; Kwon, S.J.; Kim, K.H. The HEX1 gene of Fusarium graminearum is required for fungal asexual reproduction and pathogenesis and for efficient viral RNA accumulation of Fusarium graminearum virus 1. J. Virol. 2013, 87, 10356–10367. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Putative Viruses | Contig Number | Accesson | Length | Best Match | aa Identity | Family |
|---|---|---|---|---|---|---|
| Magnaporthe oryzae narnavirus 3(MoNV3) | Contig366 | ON791620 | 2531 | Alternaria tenuissima narnavirus 1 | 72.864 | Narnaviridae |
| Magnaporthe oryzae botourmiavirus 6-A(MoBV6A) | Contig2016 | ON791624 | 1519 | Magnaporthe oryzae botourmiavirus 6 | 98.683 | Botourmiaviridae |
| Magnaporthe oryzae botourmiavirus 6-A(MoBV6A) | Contig2016 | ON791624 | 1519 | Magnaporthe oryzae botourmiavirus 6 | 98.683 | Botourmiaviridae |
| Magnaporthe oryzae botourmiavirus 4-A(MoBV4A) | First_Contig6 | ON791622 | 2092 | Magnaporthe oryzae ourmia-like virus 4 | 96.853 | Botourmiaviridae |
| Magnaporthe oryzae botourmiavirus 5-A(MoBV5A) | First_Contig10 | ON791623 | 3335 | Magnaporthe oryzae botourmiavirus 5 | 96.013 | Botourmiaviridae |
| Magnaporthe oryzae botourmiavirus 9-A(MoBV9A) | Contig527 | ON791626 | 2833 | Magnaporthe oryzae botourmiavirus 9 | 95.302 | Botourmiaviridae |
| Magnaporthe oryzae botourmiavirus 7-A(MoBV7A) | First_Contig140 | ON791625 | 2227 | Magnaporthe oryzae botourmiavirus 7 | 94.545 | Botourmiaviridae |
| Magnaporthe oryzae botourmiavirus 10(MoBV10) | Contig66 | ON791628 | 1180 | Epicoccum nigrum ourmia-like virus 1 | 85.294 | Botourmiaviridae |
| Magnaporthe oryzae botourmiavirus 11(MoBV11) | Contig150 | ON791627 | 2539 | Pyricularia oryzae ourmia-like virus 3 | 79.149 | Botourmiaviridae |
| Magnaporthe oryzae botourmiavirus 13(MoBV13) | Contig75 | ON791630 | 2209 | Sclerotinia sclerotiorum ourmia-like virus 2 | 47.3 | Botourmiaviridae |
| Magnaporthe oryzae botourmiavirus 12(MoBV12) | Contig102 | ON791629 | 645 | Botrytis ourmia-like virus | 31.3 | Botourmiaviridae |
| Magnaporthe oryzae chrysovirus 4(MoCV4) | Contig2839 | ON791617 | 3519 | Magnaporthe oryzae chrysovirus 1 | 91.408 | Chrysoviridae |
| Magnaporthe oryzae chrysovirus 5(MoCV5) | Contig5875 | ON791618 | 2793 | Magnaporthe oryzae chrysovirus 3 | 58.9 | Chrysoviridae |
| Magnaporthe oryzae partitivirus 2-A(MoPV2A) | Contig1078 | ON791632 | 1769 | Magnaporthe oryzae partitivirus 2 | 99.545 | Partitiviridae |
| Magnaporthe oryzae partitivirus 1-A(MoPV1A) | Contig673 | ON791631 | 1473 | Magnaporthe oryzae partitivirus 1 | 99.456 | Partitiviridae |
| Magnaporthe oryzae partitivirus 5(MoPV5) | Contig110 | ON791635 | 1563 | Penicillium aurantiogriseum partitivirus 1 | 91.358 | Partitiviridae |
| Magnaporthe oryzae partitivirus 3-A(MoPV3A) | Contig49 | ON791633 | 1800 | Magnaporthe oryzae partitivirus 3 | 86.697 | Partitiviridae |
| Magnaporthe oryzae partitivirus 4(MoPV4) | Contig208 | ON791634 | 1592 | Verticillium dahliae partitivirus 1 | 73.2 | Partitiviridae |
| Magnaporthe oryzae virus 2-A(MoV2A) | First_Contig297 | ON791619 | 5164 | Magnaporthe oryzae virus 2 | 99.167 | Totiviridae |
| Magnaporthe oryzae RNA virus 1(MoRV1) | First_Contig2 | ON791638 | 3165 | Magnaporthe oryzae RNA virus | 96.809 | unclassified |
| Magnaporthe oryzae mononegaambi virus 2 | First_Contig100 | ON791636 | 3681 | Magnaporthe oryzae mononegaambi virus 1 | 84.281 | unclassified |
| Magnaporthe oryzae mymonavirus 2(MoMV2) | First_Contig46 | ON791637 | 4224 | Magnaporthe oryzae mymonavirus 1 | 82.787 | unclassified |
| Rhizoctonia solani dsRNA virus 3(RsRV3) | Contig989 | NC032150 | 502 | Rhizoctonia solani dsRNA virus 3 | 100 | Partitiviridae |
| Rhizoctonia solani dsRNA virus 2(RsRV2) | Contig990 | MZ043917 | 712 | Rhizoctonia solani dsRNA virus 2 | 100 | Partitiviridae |
| Sclerotinia sclerotiorum hypovirus 2(SsHV2) | Contig995 | KF898354 | 628 | Sclerotinia sclerotiorum hypovirus 2 | 100 | Hypoviridae |
| Rhizoctonia fumigata mycovirus | Contig1079 | NC_026954 | 515 | Rhizoctonia fumigata mycovirus | 100 | unclassified |
| Rhizoctonia solani RNA virus HN008 | Contig1208 | NC_027529 | 4437 | Rhizoctonia solani RNA virus HN008 | 100 | unclassified |
| Ustilaginoidea virens botourmiavirus 6(UvBV6) | Contig25222 | ON791661 | 219 | Soybean leaf-associated ourmiavirus 2 | 58.3 | Botourmiaviridae |
| Ustilaginoidea virens botourmiavirus 4(UvBV4) | Contig293 | ON791659 | 2331 | Sclerotinia sclerotiorum ourmia-like virus 2 | 51.1 | Botourmiaviridae |
| Ustilaginoidea virens botourmiavirus 5(UvBV5) | First_Contig57 | ON791660 | 557 | Soybean leaf-associated ourmiavirus 1 | 50.8 | Botourmiaviridae |
| Ustilaginoidea virens botourmiavirus 2(UvBV2) | First_Contig29 | ON791657 | 2496 | Rhizoctonia solani ourmia-like virus 1 RNA 1 | 35.8 | Botourmiaviridae |
| Ustilaginoidea virens botourmiavirus 1(UvBV1) | Contig255 | ON791656 | 2314 | Magnaporthe oryzae ourmia-like virus | 35.6 | Botourmiaviridae |
| Ustilaginoidea virens botourmiavirus 3(UvBV3) | Contig1333 | ON791658 | 2268 | Sclerotinia sclerotiorum ourmia-like virus 1 RNA 1 | 28.6 | Botourmiaviridae |
| Ustilaginoidea virens mitovirus 1(UvMV1) | First_Contig59 | ON791662 | 2570 | Sclerotinia sclerotiorum mitovirus 6 | 47.2 | Mitoviridae |
| Ustilaginoidea virens narnavirus 3(UvNV3) | Contig731 | ON791665 | 2398 | Sanya narnavirus 7 | 72.3 | Narnaviridae |
| Ustilaginoidea virens narnavirus 2(UvNV2) | Contig553 | ON791664 | 2395 | Plasmopara viticola lesion associated narnavirus 43 | 67.05 | Narnaviridae |
| Ustilaginoidea virens narnavirus 1(UvNV1) | First_Contig93 | ON791663 | 1963 | Fusarium poae narnavirus 1 | 35.4 | Narnaviridae |
| Ustilaginoidea virens partitivirus 3A(UvPV3A) | First_Contig43 | ON791676 | 1134 | Ustilaginoidea virens partitivirus 3 | 98.9 | Partitiviridae |
| Ustilaginoidea virens partitivirus 2A(UvPV2A) | First_Contig80 | ON791675 | 2018 | Ustilaginoidea virens partitivirus 2 | 98.4 | Partitiviridae |
| Ustilaginoidea virens partitivirus 11(UvNV11) | First_Contig14 | ON791673 | 3270 | Ustilaginoidea virens nonsegmented virus 1 | 97.2 | Partitiviridae |
| Ustilaginoidea virens partitivirus 6(UvNV6) | Contig1995 | ON791668 | 354 | Botryotinia fuckeliana partitivirus 1 | 94.9 | Partitiviridae |
| Ustilaginoidea virens partitivirus 10(UvNV10) | Contig294 | ON791672 | 379 | Penicillium aurantiogriseum partitivirus 1 | 93.2 | Partitiviridae |
| Ustilaginoidea virens partitivirus 12(UvNV12) | Contig88 | ON791674 | 1823 | Ustilaginoidea virens partitivirus | 88.5 | Partitiviridae |
| Ustilaginoidea virens partitivirus 8(UvNV8) | First_Contig46 | ON791670 | 661 | Colletotrichum truncatum partitivirus 1 | 85.9 | Partitiviridae |
| Ustilaginoidea virens partitivirus 9(UvNV9) | Contig439 | ON791671 | 482 | Discula destructiva virus 1 | 83.3 | Partitiviridae |
| Ustilaginoidea virens partitivirus 13(UvPV13) | Contig6891 | ON791677 | 300 | Ustilaginoidea virens RNA virus | 80 | Partitiviridae |
| Ustilaginoidea virens partitivirus 7(UvNV7) | Contig337 | ON791669 | 588 | Colletotrichum partitivirus 1 | 76.6 | Partitiviridae |
| Ustilaginoidea virens partitivirus 5(UvNV5) | Contig711 | ON791667 | 1810 | Botryosphaeria dothidea partitivirus 1 | 74.3 | Partitiviridae |
| Ustilaginoidea virens partitivirus 14(UvPV14) | Contig321 | ON791678 | 1573 | Verticillium dahliae partitivirus 1 | 74.1 | Partitiviridae |
| Ustilaginoidea virens partitivirus 4(UvNV4) | Contig256 | ON791666 | 1625 | Botryosphaeria dothidea partitivirus 1 | 58.1 | Partitiviridae |
| Ustilaginoidea virens polymycovirus 1(UvPMV1) | Contig6818 | ON791679 | 1957 | Cladosporium cladosporioides virus 1 | 58.7 | Polymycoviridae |
| Ustilaginoidea virens RNA virus M-A(UvRVMA) | First_Contig38 | ON791647 | 3028 | Ustilaginoidea virens RNA virus M | 99.4 | Totiviridae |
| Ustilaginoidea virens RNA virus 6-A(UvRV6A) | First_Contig22 | ON791649 | 2157 | Ustilaginoidea virens RNA virus 6 | 96 | Totiviridae |
| Ustilaginoidea virens RNA virus 15(UvRV15) | Contig392 | ON791646 | 4627 | Ustilaginoidea virens RNA virus L | 81.9 | Totiviridae |
| Ustilaginoidea virens RNA virus 13(UvRV13) | Contig28 | ON791644 | 5227 | Ustilaginoidea virens RNA virus 1 | 76.7 | Totiviridae |
| Ustilaginoidea virens RNA virus 14(UvRV14) | First_Contig78 | ON791645 | 2507 | Ustilaginoidea virens RNA virus 3 | 75.2 | Totiviridae |
| Ustilaginoidea virens RNA virus 11(UvRV11) | Contig21259 | ON791642 | 250 | Phomopsis vexans RNA virus | 50 | Totiviridae |
| Ustilaginoidea virens RNA virus 7(UvRV7) | First_Contig27 | ON791648 | 4661 | Ustilaginoidea virens RNA virus 5 | 49.1 | Totiviridae |
| Ustilaginoidea virens RNA virus 10(UvRV10) | Contig94 | ON791641 | 910 | Penicillium aurantiogriseum totivirus 1 | 47.1 | Totiviridae |
| Ustilaginoidea virens RNA virus 8(UvRV8) | Contig3788 | ON791639 | 710 | Beauveria bassiana victorivirus 1 | 45 | Totiviridae |
| Ustilaginoidea virens RNA virus 12(UvRV12) | First_Contig136 | ON791643 | 939 | Rosellinia necatrix victorivirus 1 | 37.9 | Totiviridae |
| Ustilaginoidea virens RNA virus 9(UvRV9) | First_Contig40 | ON791640 | 4956 | Gremmeniella abietina RNA virus L1 | 35.9 | Totiviridae |
| Ustilaginoidea virens unassigned RNA virus HNND-1-A | Contig508 | ON791651 | 2912 | Ustilaginoidea virens unassigned RNA virus HNND-1 | 97.1 | unclassified |
| Ustilaginoidea virens mycovirus 1 | First_Contig11 | ON791650 | 1149 | Ustilaginoidea virens mycovirus | 94.9 | unclassified |
| Ustilaginoidea virens virus 1(UvV1) | Contig21350 | ON791655 | 533 | Alternaria tenuissima virus | 68.9 | unclassified |
| Ustilaginoidea virens narna-like virus 3(UvNLV3) | Contig1170 | ON791654 | 3128 | Beihai narna-like virus 23 | 37 | unclassified |
| Ustilaginoidea virens narna-like virus 1(UvNLV1) | Second_Contig2 | ON791652 | 3099 | Beihai narna-like virus 21 | 34.3 | unclassified |
| Ustilaginoidea virens narna-like virus 2(UvNLV2) | First_Contig75 | ON791653 | 689 | Beihai narna-like virus 22 | 27.1 | unclassified |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Z.; Huang, X.; Fan, Y.; Yang, M.; Zhou, E. Metatranscriptomic Analysis Reveals Rich Mycoviral Diversity in Three Major Fungal Pathogens of Rice. Int. J. Mol. Sci. 2022, 23, 9192. https://doi.org/10.3390/ijms23169192
He Z, Huang X, Fan Y, Yang M, Zhou E. Metatranscriptomic Analysis Reveals Rich Mycoviral Diversity in Three Major Fungal Pathogens of Rice. International Journal of Molecular Sciences. 2022; 23(16):9192. https://doi.org/10.3390/ijms23169192
Chicago/Turabian StyleHe, Zhenrui, Xiaotong Huang, Yu Fan, Mei Yang, and Erxun Zhou. 2022. "Metatranscriptomic Analysis Reveals Rich Mycoviral Diversity in Three Major Fungal Pathogens of Rice" International Journal of Molecular Sciences 23, no. 16: 9192. https://doi.org/10.3390/ijms23169192
APA StyleHe, Z., Huang, X., Fan, Y., Yang, M., & Zhou, E. (2022). Metatranscriptomic Analysis Reveals Rich Mycoviral Diversity in Three Major Fungal Pathogens of Rice. International Journal of Molecular Sciences, 23(16), 9192. https://doi.org/10.3390/ijms23169192