
Development of Hydroxamic Acid Compounds for Inhibition of Metallo-β-Lactamase from Bacillus anthracis

 , ,
, , _Kim.png)
Abstract
:1. Introduction
2. Results
2.1. Synthesis of Compound 4 and 6
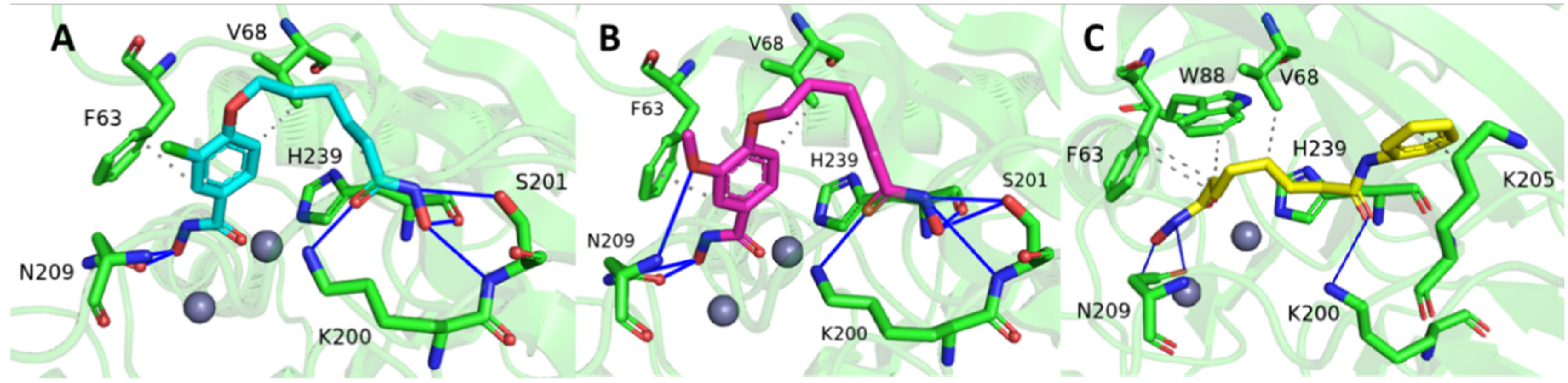
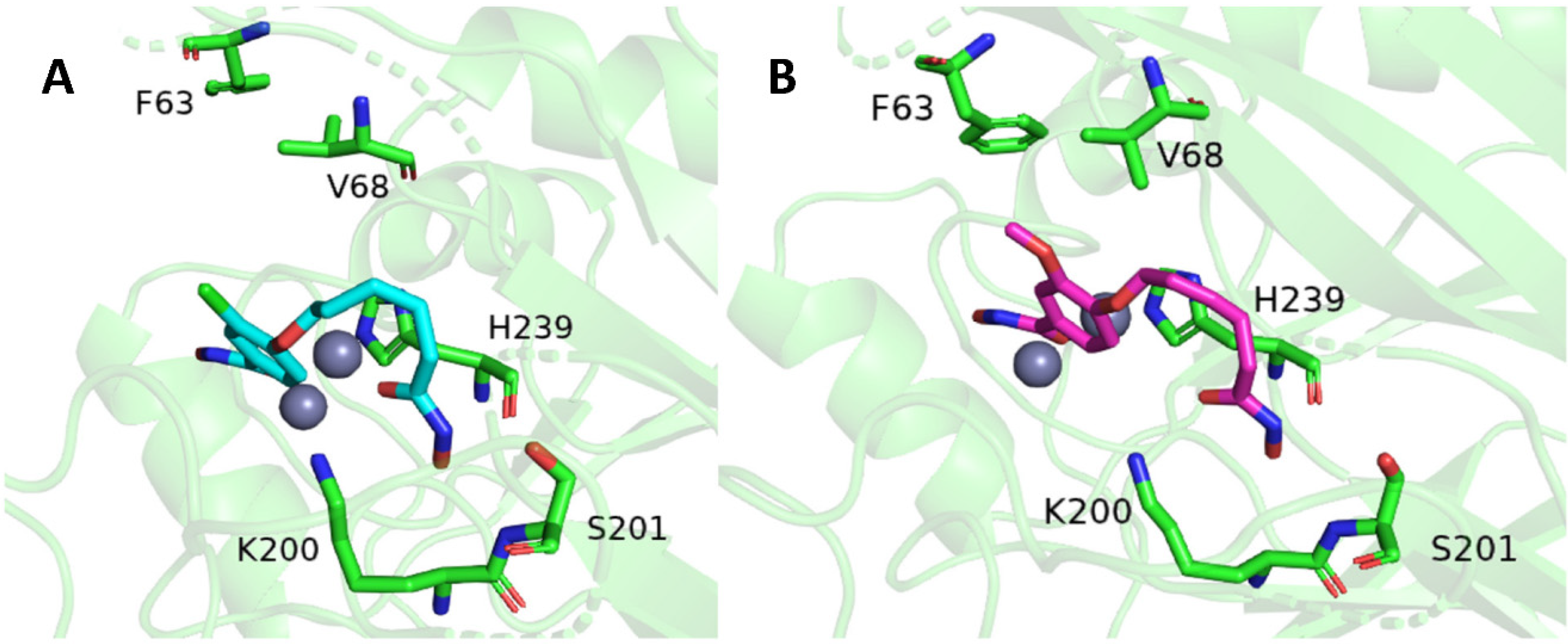
2.2. Molecular Docking
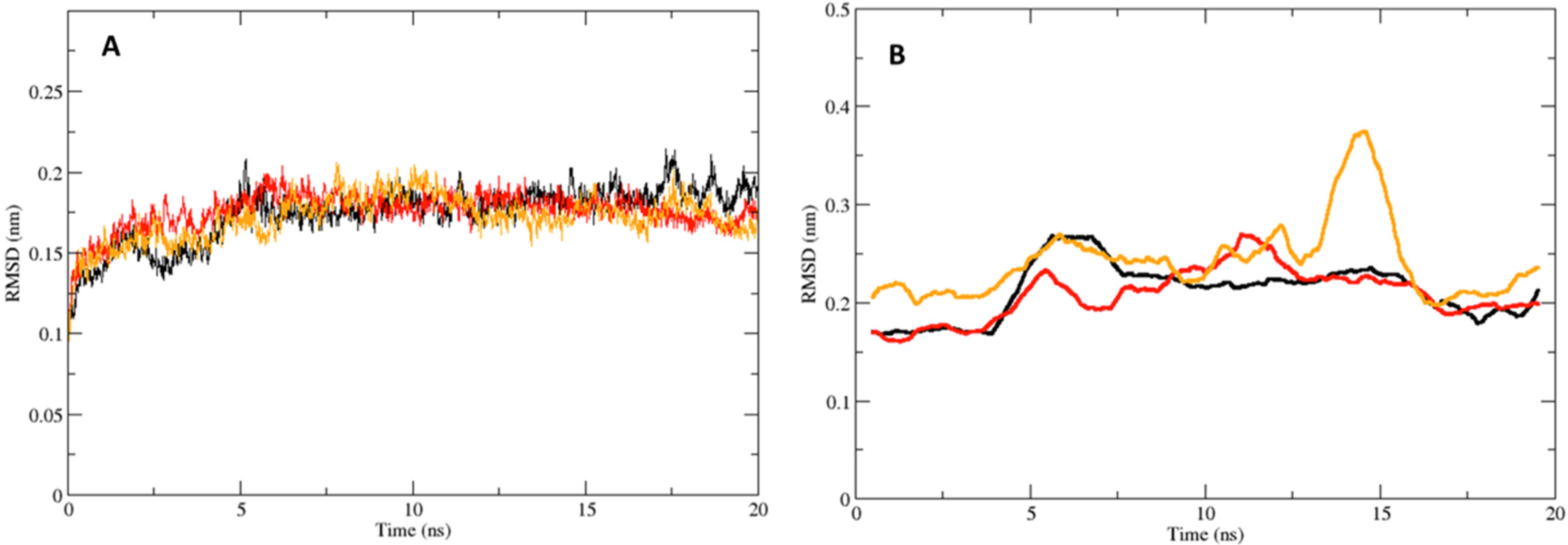
2.3. Molecular Dynamics
2.4. Inhibition Tests
3. Discussion
4. Materials and Methods
4.1. General Procedures
4.2. Molecular Docking Simulations
4.3. Molecular Dynamics Simulations
4.4. Free Energy Calculations
4.5. Inhibition Tests
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Collier, R.J.; Young, J.A.T. Anthrax Toxin. Annu. Rev. Cell Dev. Biol. 2003, 19, 45–70. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.K. Anthrax: A Disease of Biowarfare and Public Health Importance. World J. Clin. Cases 2015, 3, 20–33. [Google Scholar] [CrossRef]
- Hawk, M.J.; Breece, R.M.; Hajdin, C.E.; Bender, K.M.; Hu, Z.; Costello, A.L.; Bennett, B.; Tierney, D.L.; Crowder, M.W. Differential Binding of Co(II) and Zn(II) to Metallo-β-Lactamase Bla2 from Bacillus Anthracis. J. Am. Chem. Soc. 2009, 131, 10753–10762. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, S.R.; Kim, S.G.; Lee, J.S.; Kim, S.K. Purification Development and Characterization of the Zinc-Dependent Metallo-β-Lactamase from Bacillus anthracis. Biotechnol. Lett. 2011, 33, 1417–1422. [Google Scholar] [CrossRef]
- Kim, S.-K.; Demuth, M.; Schlesinger, S.R.; Kim, S.J.; Urbanczyk, J.; Shaw, R.W.; Shin, H. Inhibition of Bacillus anthracis Metallo-β-Lactamase by Compounds with Hydroxamic Acid Functionality. J. Enzym. Inhib. Med. Chem. 2016, 31, 132–137. [Google Scholar] [CrossRef]
- Materon, I.C.; Queenan, A.M.; Koehler, T.M.; Bush, K.; Palzkill, T. Biochemical Characterization of Beta-Lactamases Bla1 and Bla2 from Bacillus anthracis. Antimicrob. Agents Chemother. 2003, 47, 2040–2042. [Google Scholar] [CrossRef]
- Crowder, M.W.; Spencer, J.; Vila, A.J. Metallo-β-Lactamases: Novel Weaponry for Antibiotic Resistance in Bacteria. Acc. Chem. Res. 2006, 39, 721–728. [Google Scholar] [CrossRef]
- Puerta, D.T.; Cohen, S.M. Elucidating Drug-Metalloprotein Interactions with Tris(Pyrazolyl)Borate Model Complexes. Inorg. Chem. 2002, 41, 5075–5082. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.W.; Valladares, M.H.; Adlington, R.M.; Amicosante, G.; Baldwin, J.E.; Frère, J.-M.; Galleni, M.; Rossolini, G.M.; Schofield, C.J. Hydroxamate Inhibitors OfAeromonas HydrophilaAE036 Metallo-B-Lactamase. Bioorg. Chem. 1999, 27, 35–40. [Google Scholar] [CrossRef]
- Liénard, B.M.R.; Horsfall, L.E.; Galleni, M.; Frère, J.-M.; Schofield, C.J. Inhibitors of the FEZ-1 Metallo-β-Lactamase. Bioorg. Med. Chem. Lett. 2007, 17, 964–968. [Google Scholar] [CrossRef]
- Payne, D.J.; Bateson, J.H.; Gasson, B.C.; Proctor, D.; Khushi, T.; Farmer, T.H.; Tolson, D.A.; Bell, D.; Skett, P.W.; Marshall, A.C.; et al. Inhibition of Metallo-Beta-Lactamases by a Series of Mercaptoacetic Acid Thiol Ester Derivatives. Antimicrob. Agents Chemother. 1997, 41, 135–140. [Google Scholar] [CrossRef]
- Toney, J.H.; Hammond, G.G.; Fitzgerald, P.M.; Sharma, N.; Balkovec, J.M.; Rouen, G.P.; Olson, S.H.; Hammond, M.L.; Greenlee, M.L.; Gao, Y.D. Succinic Acids as Potent Inhibitors of Plasmid-Borne IMP-1 Metallo-Beta-Lactamase. J. Biol. Chem. 2001, 276, 31913–31918. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.; Walsh, T.R. A New Approach to the Inhibition of Metallo-β-Lactamases. Angew. Chem. Int. Ed. 2006, 45, 1022–1026. [Google Scholar] [CrossRef]
- Beharry, Z.; Chen, H.; Gadhachanda, V.R.; Buynak, J.D.; Palzkill, T. Evaluation of Penicillin-based Inhibitors of the Class A and B beta-lactamase from Bacillus anthracis. Biochem. Biophys. Res. Commun. 2004, 313, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Brem, J.; van Berkel, S.S.; Zollman, D.; Lee, S.Y.; Gileadi, O.; McHugh, P.J.; Walsh, T.R.; McDonough, M.A.; Schofield, C.J. Structural Basis of Metallo-β-Lactamase Inhibition by Captopril Stereoisomers. Antimicrob. Agents Chemother. 2016, 60, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the Scope of the Protein–Ligand Interaction Profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]
- Macchiagodena, M.; Pagliai, M.; Andreini, C.; Rosato, A.; Procacci, P. Upgrading and Validation of the AMBER Force Field for Histidine and Cysteine Zinc(II)-Binding Residues in Sites with Four Protein Ligands. J. Chem. Inf. Model. 2019, 59, 3803–3816. [Google Scholar] [CrossRef] [PubMed]
- Macchiagodena, M.; Pagliai, M.; Andreini, C.; Rosato, A.; Procacci, P. Upgraded AMBER Force Field for Zinc-Binding Residues and Ligands for Predicting Structural Properties and Binding Affinities in Zinc-Proteins. ACS Omega 2020, 5, 15301–15310. [Google Scholar] [CrossRef] [PubMed]
- Sousa Da Silva, A.W.; Vranken, W.F. ACPYPE -AnteChamber PYthon Parser InterfacE. Da Silva Vranken BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. Gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Fast, W.; Wang, Z.; Benkovic, S.J. Familial Mutations and Zinc Stoichiometry Determine the Rate-Limiting Step of Nitrocefin Hydrolysis by Metallo-Beta-Lactamase from Bacteroides Fragilis. Biochemistry 2001, 40, 1640–1650. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Autodock Binding Affinity (kcal/mol) | Average End-State Free Energy (kcal/mol) | |
|---|---|---|
| Compound 4 | −7.5 | −27.18 ± 0.65 |
| Compound 6 | −7.7 | −30.08 ± 1.12 |
| SAHA | −6.7 | −27.82 ± 0.51 |
| IC50 (μM) | Ki (μM) | |
|---|---|---|
| Compound 4 | 20.0 ± 5.0 | 6.4 ± 1.7 |
| Compound 6 | 14.9 ± 9.8 | 4.7 ± 1.4 |
| SAHA | >100 | N.D. * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huckleby, A.E.; Saul, J.G.; Shin, H.; Desmarais, S.; Bokka, A.; Jeon, J.; Kim, S.-K. Development of Hydroxamic Acid Compounds for Inhibition of Metallo-β-Lactamase from Bacillus anthracis. Int. J. Mol. Sci. 2022, 23, 9163. https://doi.org/10.3390/ijms23169163
Huckleby AE, Saul JG, Shin H, Desmarais S, Bokka A, Jeon J, Kim S-K. Development of Hydroxamic Acid Compounds for Inhibition of Metallo-β-Lactamase from Bacillus anthracis. International Journal of Molecular Sciences. 2022; 23(16):9163. https://doi.org/10.3390/ijms23169163
Chicago/Turabian StyleHuckleby, Andrew E., Jhawn G. Saul, Hyunshun Shin, Staci Desmarais, Apparao Bokka, Junha Jeon, and Sung-Kun Kim. 2022. "Development of Hydroxamic Acid Compounds for Inhibition of Metallo-β-Lactamase from Bacillus anthracis" International Journal of Molecular Sciences 23, no. 16: 9163. https://doi.org/10.3390/ijms23169163
APA StyleHuckleby, A. E., Saul, J. G., Shin, H., Desmarais, S., Bokka, A., Jeon, J., & Kim, S.-K. (2022). Development of Hydroxamic Acid Compounds for Inhibition of Metallo-β-Lactamase from Bacillus anthracis. International Journal of Molecular Sciences, 23(16), 9163. https://doi.org/10.3390/ijms23169163