
Smoking Cessation in Mice Does Not Switch off Persistent Lung Inflammation and Does Not Restore the Expression of HDAC2 and SIRT1

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Effects of Cigarette Smoke on Lung Parenchyma
2.2. Emphysema Lesions Do Progress after Smoking Cessation
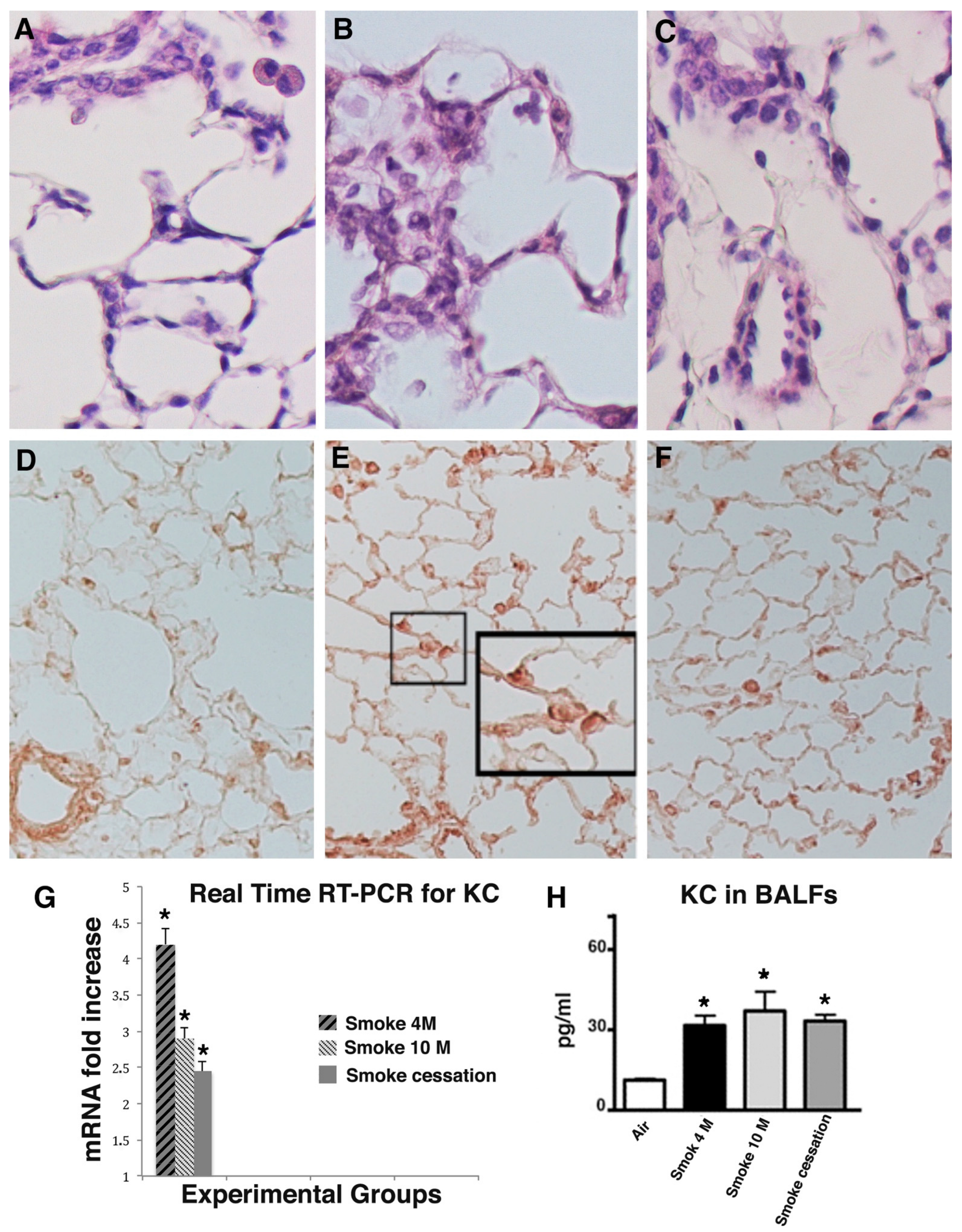
2.3. Lung Inflammation Does Persist for a Long Time after Smoking Cessation
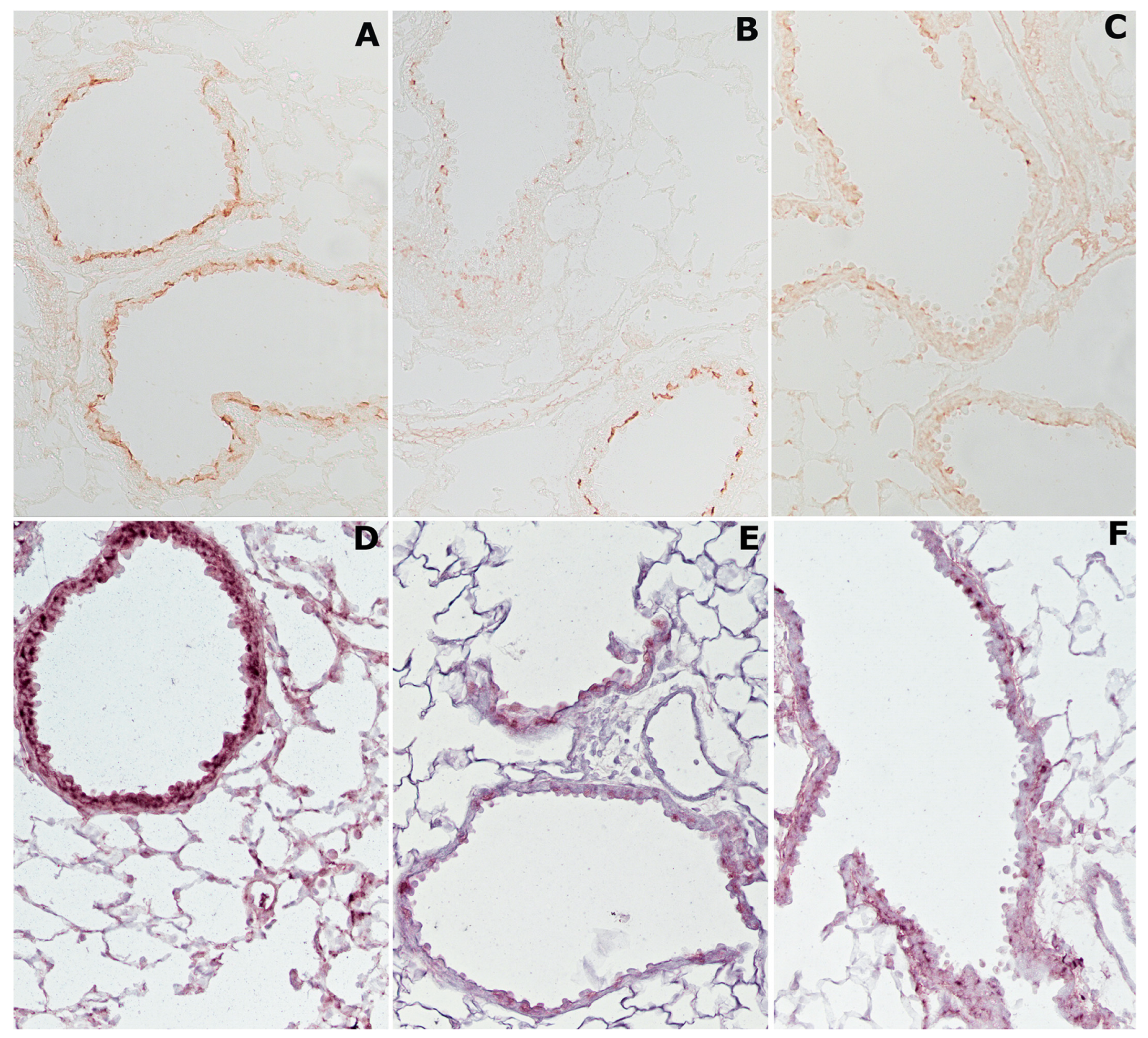
2.4. Cigarette Smoke Causes Changes in Histone Deacetylases HDAC2 and Sirtuin-1 (SIRT1)
2.5. Cigarette Smoke Induces p38 MAPK and pSer10 Phosphorylation
2.6. Neutrophilic Inflammation Is Accompanied by Intense Staining for Neutrophil Elastase, Metalloprotease 9 (MMP-9), and 8-Oxo-7,8-Dihydro-2′-Deoxyguanosine (8-OHdG)
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Exposure to Cigarette Smoke
4.3. Morphology and Morphometry
4.4. Inflammatory Cells Profile in BALF
4.5. Immunohistochemistry
4.6. Determination of Desmosine
4.7. RNA Isolation and cDNA Synthesis
4.8. Real-Time RT-PCR
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mannino, D.M.; Wylam, M.E.; Ten, R.; Prakash, U.B.S.; Nadrous, H.F.; Clawson, M.L.; Anderson, P.M. The natural history of chronic obstructive pulmonary disease. Eur. Respir. J. 2006, 27, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Petrache, I. Pathogenesis of chronic obstructive pulmonary disease. J. Clin. Investig. 2012, 122, 2749–2755. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Inflammatory endotypes in COPD. Allergy 2019, 74, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Cigarette smoking and health. American Thoracic Society. Am. J. Respir. Crit. Care Med. 1996, 153, 861–865. [CrossRef]
- Fletcher, C.; Peto, R. The natural history of chronic airflow obstruction. BMJ 1977, 1, 1645–1648. [Google Scholar] [CrossRef]
- Kirkham, P.A.; Barnes, P.J. Oxidative Stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef]
- Hikichi, M.; Mizumura, K.; Maruoka, S.; Gon, Y. Pathogenesis of chronic obstructive pulmonary disease (COPD) induced by cigarette smoke. J. Thorac Dis. 2019, 11 (Suppl. S17), S2129–S2140. [Google Scholar] [CrossRef]
- Culpitt, S.V.; Maziak, W.; Loukidis, S.; Nightingale, J.A.; Matthews, J.L.; Barnes, P.J. Effect of High Dose Inhaled Steroid on Cells, Cytokines, and Proteases in Induced Sputum in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 1999, 160, 1635–1639. [Google Scholar] [CrossRef]
- Barnes, P.J. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2013, 131, 636–645. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P. Roflumilast: A Review in COPD. Drugs 2015, 75, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, P.D.; Connett, J.E.; Waller, L.A.; Altose, M.D.; Bailey, W.C.; Buist, A.S.; Tashkin, D.P. Lung Health Study Research Group Smoking Cessation and Lung Function in Mild-to-Moderate Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2000, 161, 381–390. [Google Scholar] [CrossRef]
- Simmons, M.S.; Connett, J.E.; Nides, M.A.; Lindgren, P.G.; Kleerup, E.C.; Murray, R.P.; Bjornson, W.M.; Tashkin, D.P. Smoking reduction and the rate of decline in FEV1: Results from the Lung Health Study. Eur. Respir. J. 2005, 25, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Willemse, B.W.M.; Hacken, N.H.T.T.; Rutgers, B.; Lesman-Leegte, I.G.A.T.; Postma, D.S.; Timens, W. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur. Respir. J. 2005, 26, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Braber, S.; Henricks, P.A.J.; Nijkamp, F.P.; Kraneveld, A.D.; Folkerts, G. Inflammatory changes in the airways of mice caused by cigarette smoke exposure are only partially reversed after smoking cessation. Respir. Res. 2010, 11, 99. [Google Scholar] [CrossRef]
- Dhariwal, J.; Tennant, R.C.; Hansell, D.M.; Westwick, J.; Walker, C.; Ward, S.P.; Pride, N.; Barnes, P.J.; Kon, O.M.; Hansel, T.T. Smoking Cessation in COPD Causes a Transient Improvement in Spirometry and Decreases Micronodules on High-Resolution CT Imaging. Chest 2014, 145, 1006–1015. [Google Scholar] [CrossRef]
- Rahman, I.; De Cunto, G.; Sundar, I.K.; Lungarella, G. Vulnerability and Genetic Susceptibility to Cigarette Smoke–Induced Emphysema in Mice. Am. J. Respir. Cell Mol. Biol. 2017, 57, 270–271. [Google Scholar] [CrossRef]
- De Cunto, G.; Brancaleone, V.; Riemma, M.A.; Cerqua, I.; Vellecco, V.; Spaziano, G.; Cavarra, E.; Bartalesi, B.; D’Agostino, B.; Lungarella, G.; et al. Functional contribution of sphingosine-1-phosphate to airway pathology in cigarette smoke-exposed mice. J. Cereb. Blood Flow Metab. 2019, 177, 267–281. [Google Scholar] [CrossRef]
- De Cunto, G.; Cavarra, E.; Bartalesi, B.; Lungarella, G.; Lucattelli, M. Alveolar Macrophage Phenotype and Compartmentalization Drive Different Pulmonary Changes in Mouse Strains Exposed to Cigarette Smoke. COPD J. Chronic Obstr. Pulm. Dis. 2020, 17, 429–443. [Google Scholar] [CrossRef]
- De Cunto, G.; Cavarra, E.; Bartalesi, B.; Lucattelli, M.; Lungarella, G. Innate Immunity and Cell Surface Receptors in the Pathogenesis of COPD: Insights from Mouse Smoking Models. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 1143–1154. [Google Scholar] [CrossRef]
- De Cunto, G.; Bartalesi, B.; Cavarra, E.; Balzano, E.; Lungarella, G.; Lucattelli, M. Ongoing Lung Inflammation and Disease Progression in Mice after Smoking Cessation. Am. J. Pathol. 2018, 188, 2195–2206. [Google Scholar] [CrossRef] [PubMed]
- Gamble, E.; Grootendorst, D.C.; Hattotuwa, K.; O’Shaughnessy, T.; Ram, F.S.F.; Qiu, Y.; Zhu, J.; Vignola, A.M.; Kroegel, C.; Morell, F.; et al. Airway mucosal inflammation in COPD is similar in smokers and ex-smokers: A pooled analysis. Eur. Respir. J. 2007, 30, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Lapperre, T.S.; Postma, D.S.; Gosman, M.M.; Snoeck-Stroband, J.B.; Hacken, N.H.T.T.; Hiemstra, P.S.; Timens, W.; Sterk, P.J.; Mauad, T. Relation between duration of smoking cessation and bronchial inflammation in COPD. Thorax 2006, 61, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Pantano, S.; Natoli, G. p38-dependent marking of inflammatory genes for increased NF-κB recruitment. Nat. Immunol. 2001, 3, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Marwick, J.A.; Kirkham, P.A.; Stevenson, C.S.; Danahay, H.; Giddings, J.; Butler, K.; Donaldson, K.; MacNee, W.; Rahman, I. Cigarette Smoke Alters Chromatin Remodeling and Induces Proinflammatory Genes in Rat Lungs. Am. J. Respir. Cell Mol. Biol. 2004, 31, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I. Oxidative Stress and Gene Transcription in Asthma and Chronic Obstructive Pulmonary Disease: Antioxidant Therapeutic Targets. Curr. Drug Target-Inflamm. Allergy 2002, 1, 291–315. [Google Scholar] [CrossRef] [PubMed]
- Montuschi, P.; Collins, J.V.; Ciabattoni, G.; Lazzeri, N.; Corradi, M.; Kharitonov, S.A.; Barnes, P.J. Exhaled 8-Isoprostane as an In Vivo Biomarker of Lung Oxidative Stress in Patients with COPD and Healthy Smokers. Am. J. Respir. Crit. Care Med. 2000, 162, 1175–1177. [Google Scholar] [CrossRef]
- Lagente, V.; Planquois, J.-M.; Leclerc, O.; Schmidlin, F.; Bertrand, C.P. Oxidative stress is an important component of airway inflammation in mice exposed to cigarette smoke or lipopolysaccharide. Clin. Exp. Pharmacol. Physiol. 2008, 35, 601–605. [Google Scholar] [CrossRef]
- Victoni, T.; Barreto, E.; Lagente, V.; Carvalho, V.F. Oxidative Imbalance as a Crucial Factor in Inflammatory Lung Diseases: Could Antioxidant Treatment Constitute a New Therapeutic Strategy? Oxidative Med. Cell. Longev. 2021, 2021, 6646923. [Google Scholar] [CrossRef]
- Rahman, I.; van Schadewijk, A.A.M.; Crowther, A.J.L.; Hiemstra, P.S.; Stolk, J.; MacNee, W.; De Boer, W.I. 4-Hydroxy-2-Nonenal, a Specific Lipid Peroxidation Product, Is Elevated in Lungs of Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2002, 166, 490–495. [Google Scholar] [CrossRef]
- Petrache, I.; Medler, T.R.; Richter, A.T.; Kamocki, K.; Chukwueke, U.; Zhen, L.; Gu, Y.; Adamowicz, J.; Schweitzer, K.S.; Hubbard, W.C.; et al. Superoxide dismutase protects against apoptosis and alveolar enlargement induced by ceramide. Am. J. Physiol. Cell. Mol. Physiol. 2008, 295, L44–L53. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, T.; Cho, C.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke–induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Rangasamy, T.; Thimmulappa, R.K.; Lee, H.; Osburn, W.O.; Brigelius-Flohé, R.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Glutathione Peroxidase 2, the Major Cigarette Smoke–Inducible Isoform of GPX in Lungs, Is Regulated by Nrf2. Am. J. Respir. Cell Mol. Biol. 2006, 35, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef]
- To, Y.; Ito, K.; Kizawa, Y.; Failla, M.; Ito, M.; Kusama, T.; Elliott, W.M.; Hogg, J.C.; Adcock, I.M.; Barnes, P.J. Targeting Phosphoinositide-3-Kinase-δ with Theophylline Reverses Corticosteroid Insensitivity in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2010, 182, 897–904. [Google Scholar] [CrossRef]
- Osoata, G.O.; Yamamura, S.; Ito, M.; Vuppusetty, C.; Adcock, I.M.; Barnes, P.J.; Ito, K. Nitration of distinct tyrosine residues causes inactivation of histone deacetylase 2. Biochem. Biophys. Res. Commun. 2009, 384, 366–371. [Google Scholar] [CrossRef]
- Baker, J.R.; Vuppusetty, C.; Colley, T.; Papaioannou, A.I.; Fenwick, P.; Donnelly, L.; Ito, K.; Barnes, P.J. Oxidative stress dependent microRNA-34a activation via PI3Kα reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci. Rep. 2016, 6, 35871. [Google Scholar] [CrossRef]
- Pouwels, S.D.; Hesse, L.; Faiz, A.; Lubbers, J.; Bodha, P.K.; Hacken, N.H.T.T.; Van Oosterhout, A.J.M.; Nawijn, M.; Heijink, I.H. Susceptibility for cigarette smoke-induced DAMP release and DAMP-induced inflammation in COPD. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L881–L892. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Zhang, H.; Davies, K.J.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef]
- Cavarra, E.; Bartalesi, B.; Lucattelli, M.; Fineschi, S.; Lunghi, B.; Gambelli, F.; Ortiz, L.A.; Martorana, P.A.; Lungarella, G. Effects of Cigarette Smoke in Mice with Different Levels of α1-Proteinase Inhibitor and Sensitivity to Oxidants. Am. J. Respir. Crit. Care Med. 2001, 164, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Percival, K.; Radi, Z.A. A modified Verhoeff-van Gieson elastin histochemical stain to enable pulmonary arterial hypertension model characterization. Eur. J. Histochem. 2016, 60, 2588. [Google Scholar] [CrossRef] [PubMed]
- Thurlbeck, W.M. Measurement of pulmonary emphysema. Am. Rev. Respir. Dis. 1967, 95, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Cagle, P.T.; Berend, N.; Thurlbeck, W.M. The “Destructive Index” in Nonemphysematous and Emphysematous Lungs: Morphologic Observations and Correlation with Function. Am. Rev. Respir. Dis. 1989, 139, 308–312. [Google Scholar] [CrossRef]
- Campbell, H.; Tomkeieff, S.I. Calculation of the internal surface of a lung. Nature 1952, 170, 116–117. [Google Scholar] [CrossRef]
- Cocci, F.; Miniati, M.; Monti, S.; Cavarra, E.; Gambelli, F.; Battolla, L.; Lucattelli, M.; Lungarella, G. Urinary desmosine excretion is inversely correlated with the extent of emphysema in patients with chronic obstructive pulmonary disease. Int. J. Biochem. Cell Biol. 2002, 34, 594–604. [Google Scholar] [CrossRef]
- Winer, J.; Jung, C.K.S.; Shackel, I.; Williams, P. Development and Validation of Real-Time Quantitative Reverse Transcriptase–Polymerase Chain Reaction for Monitoring Gene Expression in Cardiac Myocytesin Vitro. Anal. Biochem. 1999, 270, 41–49. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Groups | 4 Months | 10 Months | ||
|---|---|---|---|---|
| Lm (μm) | ISA (cm2) | Lm (μm) | ISA (cm2) | |
| Air | 39.90 ± 1.1 | 1112 ± 34 | 40.12 ± 1.1 | 1020 ± 35 |
| CS | 42.70 ± 1.1 * | 1020 ± 45 * | 45.90 ± 2.0 *† | 928 ± 48 *† |
| CS + SC | 44.98 ± 2.1 *† | 947 ± 33 *† | ||
| Air-Exposed | Smoke-Exposed | Smoke Cessation | |
|---|---|---|---|
| Total cell count, ×105 | 1.34 ± 0.3 | 1.87 ± 0.42 * | 1.55 ± 0.16 |
| Differential cell count, ×105 | |||
| Macrophages | 1.12 ± 0.2 | 1.29 ± 0.39 | 1.13 ± 0.11 |
| Neutrophils | 0.15 ± 0.06 | 0.47 ± 0.12 * | 0.33 ± 0.04 *# |
| Lymphocytes | 0.07 ± 0.03 | 0.11 ± 0.03 | 0.09 ± 0.01 # |
| Primer Sequence | Probe | Amplicon Length (nt) | |
|---|---|---|---|
| KC (CXCL1) | Fw: 5’-AGACTCCAGCCACACTCCAA-3’ Rev: TGACAGCGCAGCTCATTG-3’ | #1 | 86 |
| rRNA 18S | FW: 5’-AAATCAGTTATGGTTCCTTTGGTC-3’ Rev: 5’-GCTCTAGAATTACCACAGTTATCCAA-3’ | #55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Cunto, G.; De Meo, S.; Bartalesi, B.; Cavarra, E.; Lungarella, G.; Lucattelli, M. Smoking Cessation in Mice Does Not Switch off Persistent Lung Inflammation and Does Not Restore the Expression of HDAC2 and SIRT1. Int. J. Mol. Sci. 2022, 23, 9104. https://doi.org/10.3390/ijms23169104
De Cunto G, De Meo S, Bartalesi B, Cavarra E, Lungarella G, Lucattelli M. Smoking Cessation in Mice Does Not Switch off Persistent Lung Inflammation and Does Not Restore the Expression of HDAC2 and SIRT1. International Journal of Molecular Sciences. 2022; 23(16):9104. https://doi.org/10.3390/ijms23169104
Chicago/Turabian StyleDe Cunto, Giovanna, Simone De Meo, Barbara Bartalesi, Eleonora Cavarra, Giuseppe Lungarella, and Monica Lucattelli. 2022. "Smoking Cessation in Mice Does Not Switch off Persistent Lung Inflammation and Does Not Restore the Expression of HDAC2 and SIRT1" International Journal of Molecular Sciences 23, no. 16: 9104. https://doi.org/10.3390/ijms23169104
APA StyleDe Cunto, G., De Meo, S., Bartalesi, B., Cavarra, E., Lungarella, G., & Lucattelli, M. (2022). Smoking Cessation in Mice Does Not Switch off Persistent Lung Inflammation and Does Not Restore the Expression of HDAC2 and SIRT1. International Journal of Molecular Sciences, 23(16), 9104. https://doi.org/10.3390/ijms23169104