
Restoring TRAILR2/DR5-Mediated Activation of Apoptosis upon Endoplasmic Reticulum Stress as a Therapeutic Strategy in Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. UPR Signaling Branches
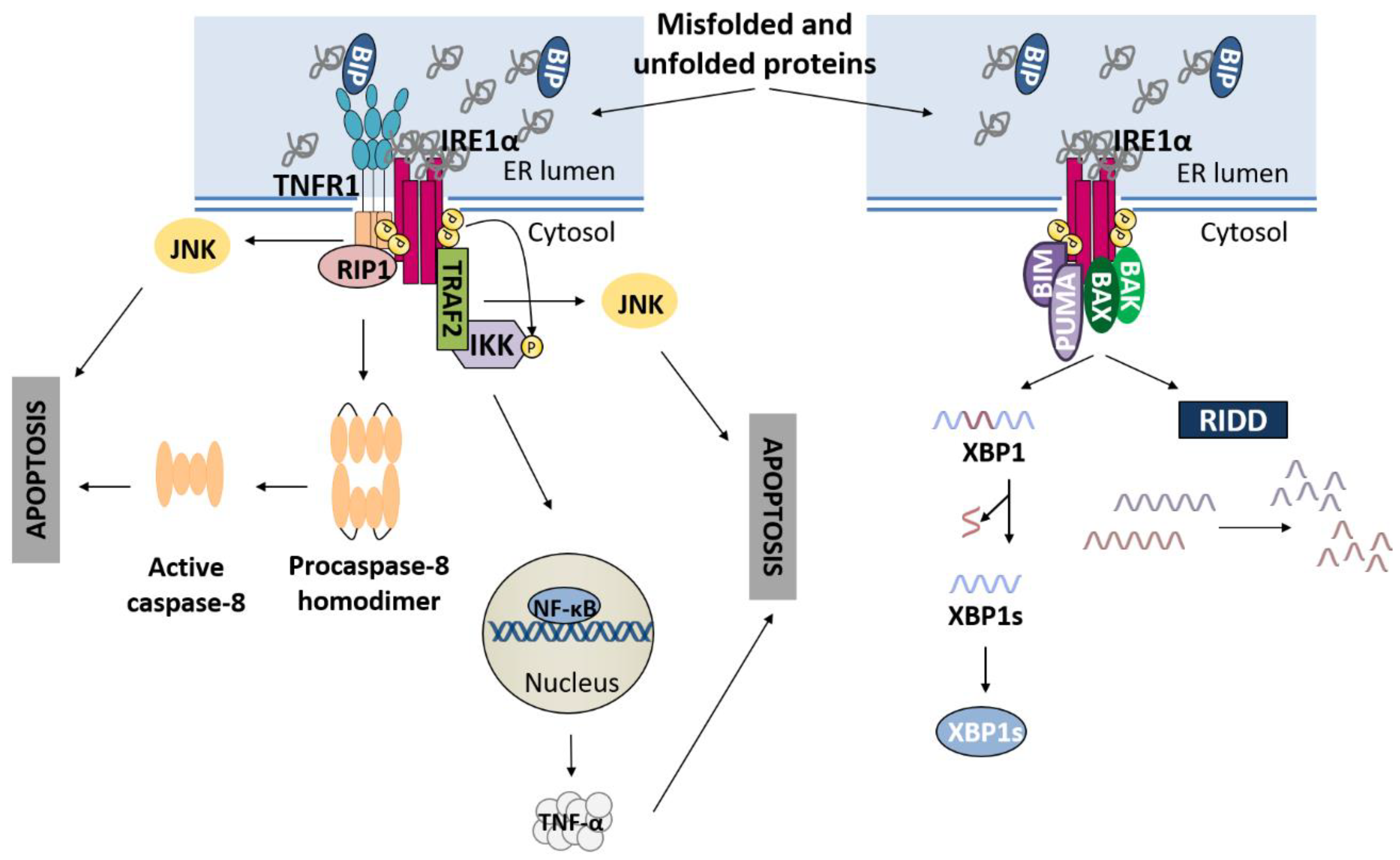
2.1. IRE1α Pathway
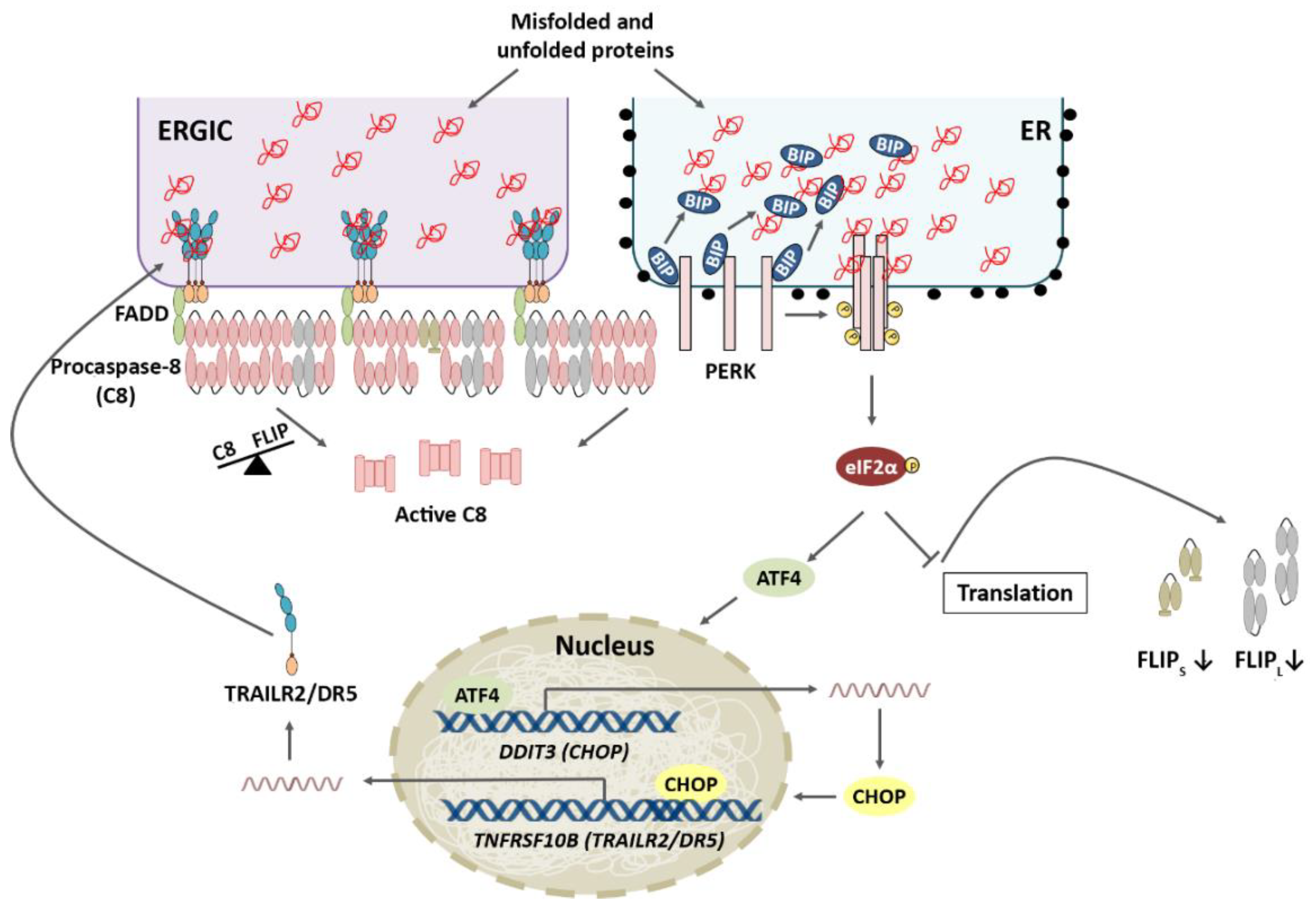
2.2. PERK Pathway
2.3. ATF6 Pathway
3. UPR Activation: Restore ER Homeostasis or Die in the Attempt
3.1. IRE1α Pathway and Apoptosis
3.2. PERK Pathway and Apoptosis
3.3. ATF6 Pathway and Apoptosis
4. Role of the TRAIL-R2-Activated Extrinsic Apoptotic Pathway in the Control of Tumor Progression
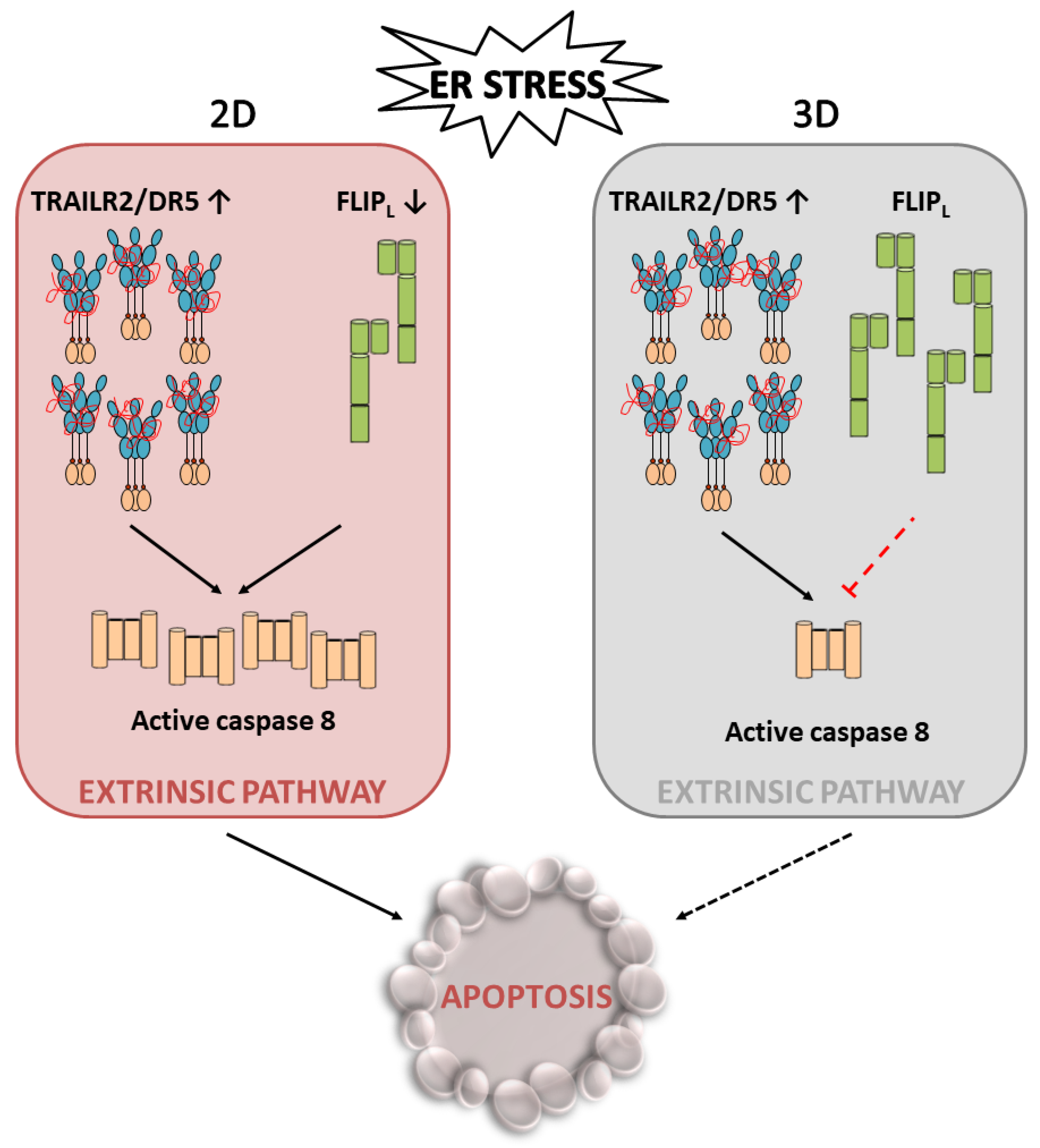
4.1. TRAIL-R2 Upregulation in Cells Undergoing ER Stress
4.2. Role of Cellular FLICE-like Inhibitory Protein (FLIP) in Apoptosis Regulation upon ER Stress
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic Reticulum and the Unfolded Protein Response. Dynamics and Metabolic Integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. Protein Misfolding in the Endoplasmic Reticulum as a Conduit to Human Disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Needham, P.G.; Guerriero, C.J.; Brodsky, J.L. Chaperoning Endoplasmic Reticulum–Associated Degradation (ERAD) and Protein Conformational Diseases. Cold Spring Harb. Perspect. Biol. 2019, 11, a033928. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Rapoport, T.A. Mechanistic Insights into ER-Associated Protein Degradation. Curr. Opin. Cell Biol. 2018, 53, 22–28. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.Y.; Kaufman, R.J. ATF6α Optimizes Long-Term Endoplasmic Reticulum Function to Protect Cells from Chronic Stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A Link between the Unfolded Protein Response, Lipid Biosynthesis, and Biogenesis of the Endoplasmic Reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 Controls Diverse Cell Type- and Condition-Specific Transcriptional Regulatory Networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef] [PubMed]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The EIF2α/ATF4 Pathway is Essential for Stress-Induced Autophagy Gene Expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic Interaction of BiP and ER Stress Transducers in the Unfolded-Protein Response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Karagöz, G.E.; Acosta-Alvear, D.; Nguyen, H.T.; Lee, C.P.; Chu, F.; Walter, P. An Unfolded Protein-Induced Conformational Switch Activates Mammalian IRE1. eLife 2017, 6, e30700. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, J.; Tao, J.; Sha, B. The Luminal Domain of the ER Stress Sensor Protein PERK Binds Misfolded Proteins and Thereby Triggers PERK Oligomerization. J. Biol. Chem. 2018, 293, 4110–4121. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional Induction of Genes Encoding Endoplasmic Reticulum Resident Proteins Requires a Transmembrane Protein Kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The Impact of the Endoplasmic Reticulum Protein-Folding Environment on Cancer Development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Sriburi, R.; Bommiasamy, H.; Buldak, G.L.; Robbins, G.R.; Frank, M.; Jackowski, S.; Brewer, J.W. Coordinate Regulation of Phospholipid Biosynthesis and Secretory Pathway Gene Expression in XBP-1(S)-Induced Endoplasmic Reticulum Biogenesis. J. Biol. Chem. 2007, 282, 7024–7034. [Google Scholar] [CrossRef]
- Wang, J.M.; Qiu, Y.; Yang, Z.Q.; Li, L.; Zhang, K. Inositol-Requiring Enzyme 1 Facilitates Diabetic Wound Healing through Modulating MicroRNAs. Diabetes 2017, 66, 177–192. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1a Cleaves Select MicroRNAs During ER Stress to Derepress Translation of Proapoptotic Caspase-2. Science 2012, 338, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.B.; Koong, A.C.; Niwa, M. Ire1 Has Distinct Catalytic Mechanisms for XBP1/HAC1 Splicing and RIDD. Cell Rep. 2014, 9, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein Translation and Folding Are Coupled by an Endoplasmic-Reticulum-Resident Kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation Reinitiation at Alternative Open Reading Frames Regulates Gene Expression in an Integrated Stress Response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Vattem, K.M.; Wek, R.C. Reinitiation Involving Upstream ORFs Regulates ATF4 MRNA Translation in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2004, 10, 11269–11274. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Fawcett, T.W.; Martindale, J.L.; Guyton, K.Z.; Hai, T.; Holbrook, N.J. Complexes Containing Activating Transcription Factor (ATF)/CAMP-Responsive-Element-Binding Protein (CREB) Interact with the CCAAT/Enhancer-Binding Protein (C/EBP)-ATF Composite Site to Regulate Gadd153 Expression during the Stress Response. Biochem. J. 1999, 339, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-Stress-Induced Transcriptional Regulation Increases Protein Synthesis Leading to Cell Death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP Induces Death by Promoting Protein Synthesis and Oxidation in the Stressed Endoplasmic Reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hendershot, L.M. Delineation of a Negative Feedback Regulatory Loop That Controls Protein Translation during Endoplasmic Reticulum Stress. J. Biol. Chem. 2003, 278, 34864–34873. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Wang, H.G. CHOP is Involved in Endoplasmic Reticulum Stress-Induced Apoptosis by Enhancing DR5 Expression in Human Carcinoma Cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal Apoptosis Induced by Endoplasmic Reticulum Stress is Regulated by ATF4-CHOP-Mediated Induction of the Bcl-2 Homology 3-Only Member PUMA. J. Neurosci. 2010, 30, 16938–16948. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a Novel ER Stress-Inducible Gene, is Induced via ATF4-CHOP Pathway and is Involved in Cell Death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.-O.; Aw, T.-Y.; Holbrook, N.J. Gadd153 Sensitizes Cells to Endoplasmic Reticulum Stress by Down-Regulating Bcl2 and Perturbing the Cellular Redox State. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor ATF6 is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Schindler, A.J.; Schekman, R. In Vitro Reconstitution of ER-Stress Induced ATF6 Transport in COPII Vesicles. Proc. Natl. Acad. Sci. USA 2009, 106, 17775–17780. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER Stress Induces Cleavage of Membrane-Bound ATF6 by the Same Proteases That Process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic Reticulum Stress Signals in the Tumour and Its Microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Woehlbier, U.; Hetz, C. Modulating Stress Responses by the UPRosome: A Matter of Life and Death. Trends Biochem. Sci. 2011, 36, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Cano-González, A.; Mauro-Lizcano, M.; Iglesias-Serret, D.; Gil, J.; López-Rivas, A. Involvement of Both Caspase-8 and Noxa-Activated Pathways in Endoplasmic Reticulum Stress-Induced Apoptosis in Triple-Negative Breast Tumor Cells Article. Cell Death Dis. 2018, 9, 134. [Google Scholar] [CrossRef] [PubMed]
- Martín-Pérez, R.; Palacios, C.; Yerbes, R.; Cano-González, A.; Iglesias-Serret, D.; Gil, J.; Reginato, M.J.; López-Rivas, A. Activated ERBB2/HER2 Licenses Sensitivity to Apoptosis upon Endoplasmic Reticulum Stress through a PERK-Dependent Pathway. Cancer Res. 2014, 74, 1766–1777. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.; Rojas-Rivera, D.; Hetz, C. Integrating Stress Signals at the Endoplasmic Reticulum: The BCL-2 Protein Family Rheostat. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 564–574. [Google Scholar] [CrossRef]
- Zong, W.X.; Li, C.; Hatzivassiliou, G.; Lindsten, T.; Yu, Q.C.; Yuan, J.; Thompson, C.B. Bax and Bak Can Localize to the Endoplasmic Reticulum to Initiate Apoptosis. J. Cell Biol. 2003, 162, 59–69. [Google Scholar] [CrossRef]
- Urra, H.; Pihán, P.; Hetz, C. The UPRosome—Decoding Novel Biological Outputs of IRE1α Function. J. Cell Sci. 2020, 133, jcs218107. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.Z.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Yang, Q.; Kim, Y.S.; Lin, Y.; Lewis, J.; Neckers, L.; Liu, Z.G. Tumour Necrosis Factor Receptor 1 Mediates Endoplasmic Reticulum Stress-Induced Activation of the MAP Kinase JNK. EMBO Rep. 2006, 7, 622–627. [Google Scholar] [CrossRef]
- Hu, P.; Han, Z.; Couvillon, A.D.; Kaufman, R.J.; Exton, J.H. Autocrine Tumor Necrosis Factor Alpha Links Endoplasmic Reticulum Stress to the Membrane Death Receptor Pathway through IRE1α-Mediated NF-ΚB Activation and Down-Regulation of TRAF2 Expression. Mol. Cell. Biol. 2006, 26, 3071–3084. [Google Scholar] [CrossRef]
- Estornes, Y.; Aguileta, M.A.; Dubuisson, C.; de Keyser, J.; Goossens, V.; Kersse, K.; Samali, A.; Vandenabeele, P.; Bertrand, M.J.M. RIPK1 Promotes Death Receptor-Independent Caspase-8-Mediated Apoptosis under Unresolved ER Stress Conditions. Cell Death Dis. 2014, 5, e555. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Bernasconi, P.; Fisher, J.; Lee, A.H.; Bassik, M.C.; Antonsson, B.; Brandt, G.S.; Iwakoshi, N.N.; Schrinzel, A.; Glimcher, L.H.; et al. Proapoptotic BAX and BAK Modulate the Unfolded Protein Response by a Direct Interaction with IRE1α. Science 2006, 312, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.A.; Zamorano, S.; Lisbona, F.; Rojas-Rivera, D.; Urra, H.; Cubillos-Ruiz, J.R.; Armisen, R.; Henriquez, D.R.; Cheng, E.H.; Letek, M.; et al. BH3-Only Proteins Are Part of a Regulatory Network That Control the Sustained Signalling of the Unfolded Protein Response Sensor IRE1α. EMBO J. 2012, 31, 2322–2335. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.P.; Austgen, K.; Nishino, M.; Coakley, K.M.; Hagen, A.; Han, D.; Papa, F.R.; Oakes, S.A. Caspase-2 Cleavage of BID is a Critical Apoptotic Signal Downstream of Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2008, 28, 3943–3951. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.K.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death under Irremediable ER Stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Lisbona, F.; Rojas-Rivera, D.; Hetz, C. When ER Stress Reaches a Dead End. Biochim. Biophys. Acta-Mol. Cell Res. 2013, 1833, 3507–3517. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.K.; Lawrence, D.A.; Lu, M.; Tan, J.; Harnoss, J.M.; Marsters, S.A.; Liu, P.; Sandoval, W.; Martin, S.E.; Ashkenazi, A. Coordination between Two Branches of the Unfolded Protein Response Determines Apoptotic Cell Fate. Mol. Cell 2018, 71, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 Activated by Proteolysis Binds in the Presence of NF-Y (CBF) Directly to the cis-Acting Element Responsible for the Mammalian Unfolded Protein Response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef]
- Morishima, N.; Nakanishi, K.; Nakano, A. Activating Transcription Factor-6 (ATF6) Mediates Apoptosis with Reduction of Myeloid Cell Leukemia Sequence 1 (Mcl-1) Protein via Induction of WW Domain Binding Protein. J. Biol. Chem. 2011, 286, 35227–35235. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.; Marsters, S.; Ashkenazi, A.; Walter, P. Misfolded Proteins Bind and Activate Death Receptor 5 to Trigger Apoptosis during Unresolved Endoplasmic Reticulum Stress. eLife 2020, 9, e52291. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Püschel, F.; León-Annicchiarico, C.L.; O’Connor, H.; Martin, S.J.; Palou-Gramón, D.; Lucendo, E.; Muñoz-Pinedo, C. Glucose Deprivation Induces ATF4-Mediated Apoptosis through TRAIL Death Receptors. Mol. Cell. Biol. 2017, 37, e00479-16. [Google Scholar] [CrossRef] [PubMed]
- Martín-Pérez, R.; Yerbes, R.; Mora-Molina, R.; Cano-González, A.; Arribas, J.; Mazzone, M.; López-Rivas, A.; Palacios, C. Oncogenic P95HER2/611CTF Primes Human Breast Epithelial Cells for Metabolic Stress-Induced down-Regulation of FLIP and Activation of TRAIL-R/Caspase-8-Dependent Apoptosis. Oncotarget 2017, 8, 93688–93703. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Day, T.W.; Sinn, A.L.; Huang, S.; Pollok, K.E.; Sandusky, G.E.; Safa, A.R. C-FLIP Gene Silencing Eliminates Tumor Cells in Breast Cancer Xenografts without Affecting Stromal Cells. Anticancer Res. 2009, 29, 3883–3886. [Google Scholar] [PubMed]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.R.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-Operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, L.M.; Fox, J.P.; Higgins, C.A.; Majkut, J.; Sessler, T.; McLaughlin, K.; McCann, C.; Roberts, J.Z.; Crawford, N.T.; McDade, S.S.; et al. A Revised Model of TRAIL -R2 DISC Assembly Explains How FLIP (L) Can Inhibit or Promote Apoptosis. EMBO Rep. 2020, 21, e49254. [Google Scholar] [CrossRef]
- Palacios, C.; Yerbes, R.; López-Rivas, A. Flavopiridol Induces Cellular FLICE-Inhibitory Protein Degradation by the Proteasome and Promotes TRAIL-Induced Early Signaling and Apoptosis in Breast Tumor Cells. Cancer Res. 2006, 66, 8858–8869. [Google Scholar] [CrossRef]
- Wilson, T.R.; McLaughlin, K.M.; McEwan, M.; Sakai, H.; Rogers, K.M.A.; Redmond, K.M.; Johnston, P.G.; Longley, D.B. C-FLIP: A Key Regulator of Colorectal Cancer Cell Death. Cancer Res. 2007, 67, 5754–5762. [Google Scholar] [CrossRef] [PubMed]
- Marini, E.S.; Giampietri, C.; Petrungaro, S.; Conti, S.; Filippini, A.; Scorrano, L.; Ziparo, E. The Endogenous Caspase-8 Inhibitor c-FLIPL Regulates ER Morphology and Crosstalk with Mitochondria. Cell Death Differ. 2015, 22, 1131–1143. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mora-Molina, R.; Stöhr, D.; Rehm, M.; López-Rivas, A. CFLIP Downregulation is an Early Event Required for Endoplasmic Reticulum Stress-Induced Apoptosis in Tumor Cells. Cell Death Dis. 2022, 13, 111. [Google Scholar] [CrossRef]
- Millard, M.; Yakavets, I.; Zorin, V.; Kulmukhamedova, A.; Marchal, S.; Bezdetnaya, L. Drug Delivery to Solid Tumors: The Predictive Value of the Multicellular Tumor Spheroid Model for Nanomedicine Screening. Int. J. Nanomed. 2017, 12, 7993–8007. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Devi, G.R. Three-Dimensional Culture Systems in Cancer Research: Focus on Tumor Spheroid Model. Pharmacol. Ther. 2016, 163, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. FLIP the Switch: Regulation of Apoptosis and Necroptosis by CFLIP. Int. J. Mol. Sci. 2015, 16, 30321–30341. [Google Scholar] [CrossRef] [PubMed]
- Riedl, A.; Schlederer, M.; Pudelko, K.; Stadler, M.; Walter, S.; Unterleuthner, D.; Unger, C.; Kramer, N.; Hengstschläger, M.; Kenner, L.; et al. Comparison of Cancer Cells in 2D vs 3D Culture Reveals Differences in AKT-MTOR-S6K Signaling and Drug Responses. J. Cell Sci. 2017, 130, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Lo, A.T.; Park, C.C.; Gray, J.W.; Bissell, M.J. HER2 Signaling Pathway Activation and Response of Breast Cancer Cells to HER2-Targeting Agents is Dependent Strongly on the 3D Microenvironment. Breast Cancer Res. Treat. 2010, 122, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Algeciras-Schimnich, A.; Griffith, T.S.; Lynch, D.H.; Paya, C.V. Cell Cycle-Dependent Regulation of FLIP Levels and Susceptibility to Fas-Mediated Apoptosis. J. Immunol. 1999, 162, 5205–5211. [Google Scholar]
- Fukazawa, T.; Fujiwara, T.; Uno, F.; Teraishi, F.; Kadowaki, Y.; Itoshima, T.; Takata, Y.; Kagawa, S.; Roth, J.A.; Tschopp, J.; et al. Accelerated Degradation of Cellular FLIP Protein through the Ubiquitin-Proteasome Pathway in P53-Mediated Apoptosis of Human Cancer Cells. Oncogene 2001, 20, 5225–5231. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Tran, T.; Sobkoviak, R.; Pope, R.M. Activation-Induced Degradation of FLIPL is Mediated via the Phosphatidylinositol 3-Kinase/Akt Signaling Pathway in Macrophages. J. Biol. Chem. 2009, 284, 14513–14523. [Google Scholar] [CrossRef] [PubMed]
- Wilkie-Grantham, R.P.; Matsuzawa, S.I.; Reed, J.C. Novel Phosphorylation and Ubiquitination Sites Regulate Reactive Oxygen Species-Dependent Degradation of Anti-Apoptotic c-FLIP Protein. J. Biol. Chem. 2013, 288, 12777–12790. [Google Scholar] [CrossRef]
- Yang, B.F.; Xiao, C.; Roa, W.H.; Krammer, P.H.; Hao, C. Calcium/Calmodulin-Dependent Protein Kinase II Regulation of c-FLIP Expression and Phosphorylation in Modulation of Fas-Mediated Signaling in Malignant Glioma Cells. J. Biol. Chem. 2003, 278, 7043–7050. [Google Scholar] [CrossRef] [PubMed]
- Chanvorachote, P.; Nimmannit, U.; Wang, L.; Stehlik, C.; Lu, B.; Azad, N.; Rojanasakul, Y. Nitric Oxide Negatively Regulates Fas CD95-Induced Apoptosis through Inhibition of Ubiquitin-Proteasome-Mediated Degradation of FLICE Inhibitory Protein. J. Biol. Chem. 2005, 280, 42044–42050. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Kamata, H.; Solinas, G.; Luo, J.L.; Maeda, S.; Venuprasad, K.; Liu, Y.C.; Karin, M. The E3 Ubiquitin Ligase Itch Couples JNK Activation to TNFα-Induced Cell Death by Inducing c-FLIPL Turnover. Cell 2006, 124, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, J.; Zhang, Y.; Qu, J.; Xu, L.; Zheng, H.; Liu, Y.; Qu, X. Cbl-b-Dependent Degradation of FLIPL is Involved in ATO-Induced Autophagy in Leukemic K562 and Gastric Cancer Cells. FEBS Lett. 2012, 586, 3104–3110. [Google Scholar] [CrossRef] [PubMed]
- Abedini, M.R.; Muller, E.J.; Brun, J.; Bergeron, R.; Gray, D.A.; Tsang, B.K. Cisplatin Induces P53-Dependent FLICE-like Inhibitory Protein Ubiquitination in Ovarian Cancer Cells. Cancer Res. 2008, 68, 4511–4517. [Google Scholar] [CrossRef] [PubMed]
- Haimerl, F.; Erhardt, A.; Sass, G.; Tiegs, G. Down-Regulation of the de-Ubiquitinating Enzyme Ubiquitin-Specific Protease 2 Contributes to Tumor Necrosis Factor-α-Induced Hepatocyte Survival. J. Biol. Chem. 2009, 284, 495–504. [Google Scholar] [CrossRef]
- Santini, S.; Stagni, V.; Giambruno, R.; Fianco, G.; di Benedetto, A.; Mottolese, M.; Pellegrini, M.; Barilà, D. ATM Kinase Activity Modulates ITCH E3-Ubiquitin Ligase Activity. Oncogene 2014, 33, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Gonulcu, S.C.; Unal, B.; Bassorgun, I.C.; Ozcan, M.; Coskun, H.S.; Elpek, G.O. Expression of Notch Pathway Components (Numb, Itch, and Siah-1) in Colorectal Tumors: A Clinicopathological Study. World J. Gastroenterol. 2020, 26, 3814–3833. [Google Scholar] [CrossRef]
- Hsu, T.S.; Mo, S.T.; Hsu, P.N.; Lai, M.Z. C-FLIP is a Target of the E3 Ligase Deltex1 in Gastric Cancer. Cell Death Dis. 2018, 9, 135. [Google Scholar] [CrossRef]
- Scudiero, I.; Zotti, T.; Ferravante, A.; Vessichelli, M.; Reale, C.; Masone, M.C.; Leonardi, A.; Vito, P.; Stilo, R. Tumor Necrosis Factor (TNF) Receptor-Associated Factor 7 is Required for TNFα-Induced Jun NH2-Terminal Kinase Activation and Promotes Cell Death by Regulating Polyubiquitination and Lysosomal Degradation of c-FLIP Protein. J. Biol. Chem. 2012, 287, 6053–6061. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Joo, D.; Liu, G.; Tu, H.; You, J.; Jin, J.; Zhao, X.; Hung, M.C.; Lin, X. Linear Ubiquitination of CFLIP Induced by LUBAC Contributes to TNF-Induced Apoptosis. J. Biol. Chem. 2018, 293, 20062–20072. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Sun, W.; Hao, X.; Li, T.; Su, L.; Liu, X. Down-Regulation of Cellular FLICE-Inhibitory Protein (Long Form) Contributes to Apoptosis Induced by Hsp90 Inhibition in Human Lung Cancer Cells. Cancer Cell Int. 2012, 12, 54. [Google Scholar] [CrossRef]
- Roberts, J.Z.; Holohan, C.; Sessler, T.; Fox, J.; Crawford, N.; Riley, J.S.; Khawaja, H.; Majkut, J.; Evergren, E.; Humphreys, L.M.; et al. The SCFSkp2 Ubiquitin Ligase Complex Modulates TRAIL-R2-Induced Apoptosis by Regulating FLIP(L). Cell Death Differ. 2020, 27, 2726–2741. [Google Scholar] [CrossRef] [PubMed]
- Kerr, E.; Holohan, C.; McLaughlin, K.M.; Majkut, J.; Dolan, S.; Redmond, K.; Riley, J.; McLaughlin, K.; Stasik, I.; Crudden, M.; et al. Identification of an Acetylation-Dependant Ku70/FLIP Complex That Regulates FLIP Expression and HDAC Inhibitor-Induced Apoptosis. Cell Death Differ. 2012, 19, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- McLornan, D.P.; Barrett, H.L.; Cummins, R.; McDermott, U.; McDowell, C.; Conlon, S.J.; Coyle, V.M.; van Schaeybroeck, S.; Wilson, R.; Kay, E.W.; et al. Prognostic Significance of TRAIL Signaling Molecules in Stage II and III Colorectal Cancer. Clin. Cancer Res. 2010, 16, 3442–3451. [Google Scholar] [CrossRef] [PubMed]
- Ryu, B.K.; Lee, M.G.; Chi, S.G.; Kim, Y.W.; Park, J.H. Increased Expression of CFLIPL in Colonic Adenocarcinoma. J. Pathol. 2001, 194, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R.; Pollok, K.E. Targeting the Anti-Apoptotic Protein c-FLIP for Cancer Therapy. Cancers 2011, 3, 1639–1671. [Google Scholar] [CrossRef] [PubMed]
- Ullenhag, G.J.; Mukherjee, A.; Watson, N.F.S.; Al-Attar, A.H.; Scholefield, J.H.; Durrant, L.G. Overexpression of FLIPL is an Independent Marker of Poor Prognosis in Colorectal Cancer Patients. Clin. Cancer Res. 2007, 13, 5070–5075. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mora-Molina, R.; López-Rivas, A. Restoring TRAILR2/DR5-Mediated Activation of Apoptosis upon Endoplasmic Reticulum Stress as a Therapeutic Strategy in Cancer. Int. J. Mol. Sci. 2022, 23, 8987. https://doi.org/10.3390/ijms23168987
Mora-Molina R, López-Rivas A. Restoring TRAILR2/DR5-Mediated Activation of Apoptosis upon Endoplasmic Reticulum Stress as a Therapeutic Strategy in Cancer. International Journal of Molecular Sciences. 2022; 23(16):8987. https://doi.org/10.3390/ijms23168987
Chicago/Turabian StyleMora-Molina, Rocío, and Abelardo López-Rivas. 2022. "Restoring TRAILR2/DR5-Mediated Activation of Apoptosis upon Endoplasmic Reticulum Stress as a Therapeutic Strategy in Cancer" International Journal of Molecular Sciences 23, no. 16: 8987. https://doi.org/10.3390/ijms23168987
APA StyleMora-Molina, R., & López-Rivas, A. (2022). Restoring TRAILR2/DR5-Mediated Activation of Apoptosis upon Endoplasmic Reticulum Stress as a Therapeutic Strategy in Cancer. International Journal of Molecular Sciences, 23(16), 8987. https://doi.org/10.3390/ijms23168987