
Efficient Base-Catalyzed Kemp Elimination in an Engineered Ancestral Enzyme



Abstract
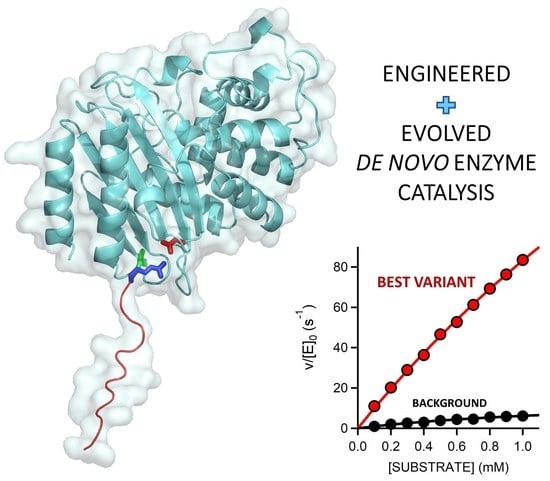
:
1. Introduction
2. Results
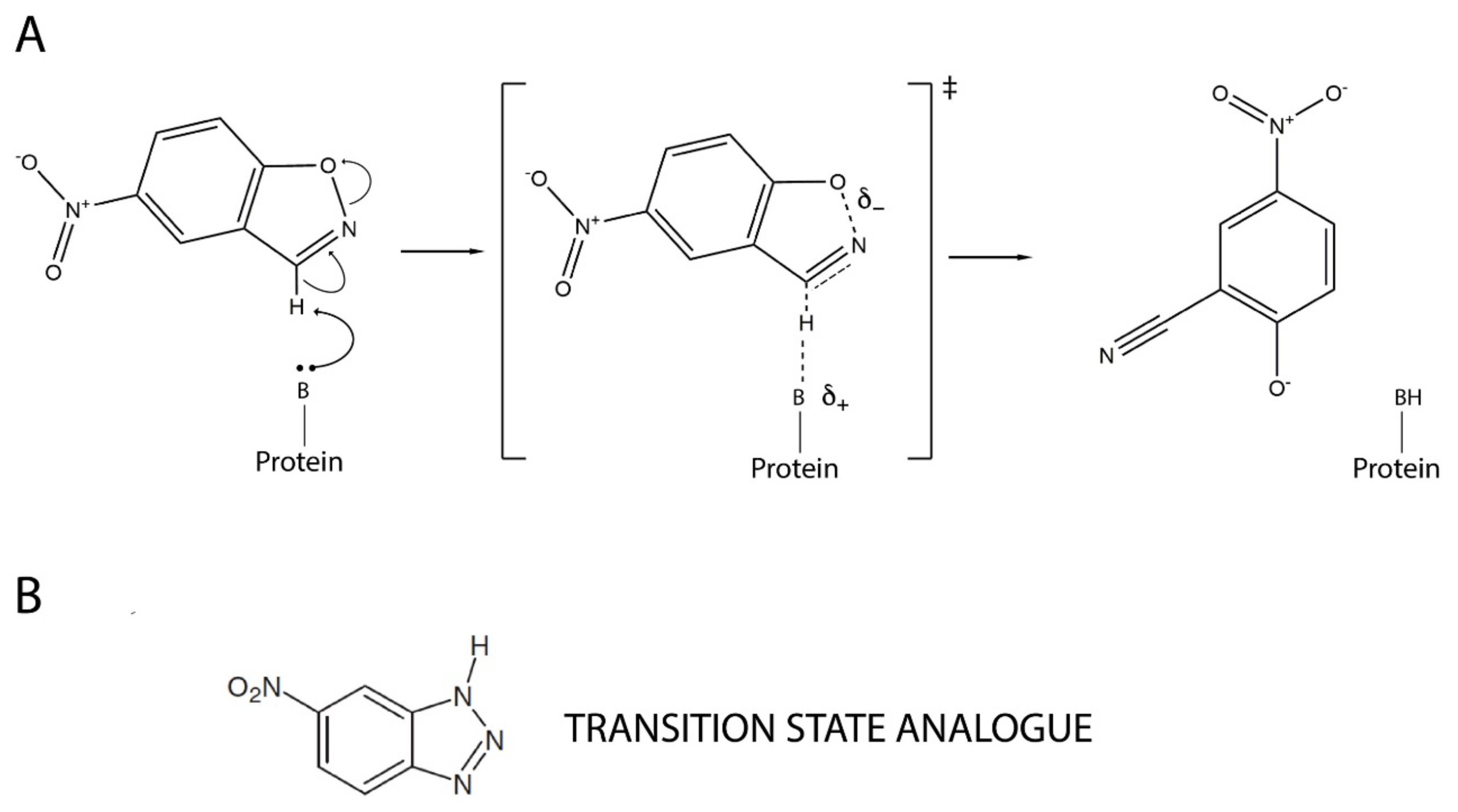
2.1. Kemp Eliminase Variants Used as Starting Point for This Work
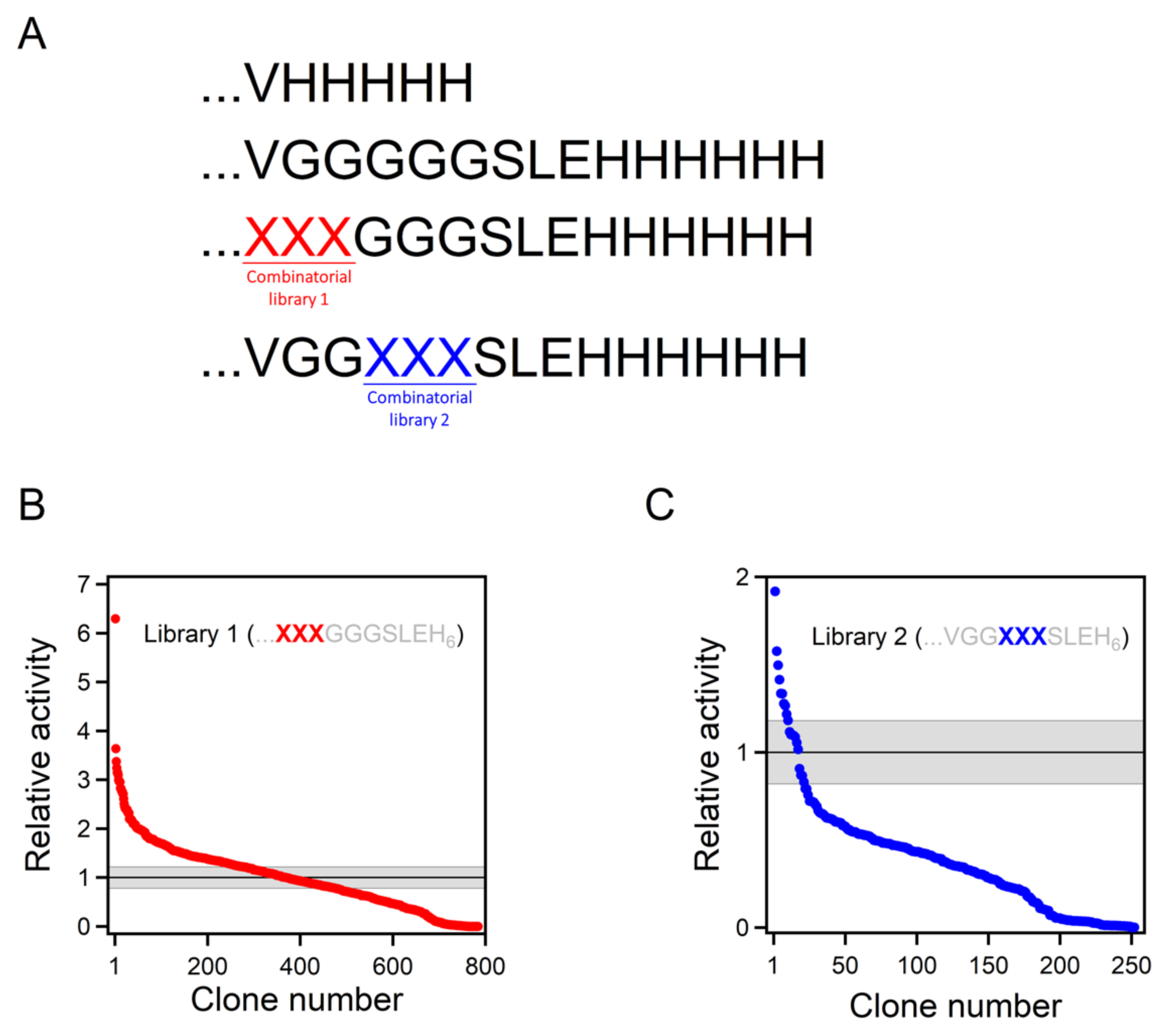
2.2. Combinatorial Library Design and Screening
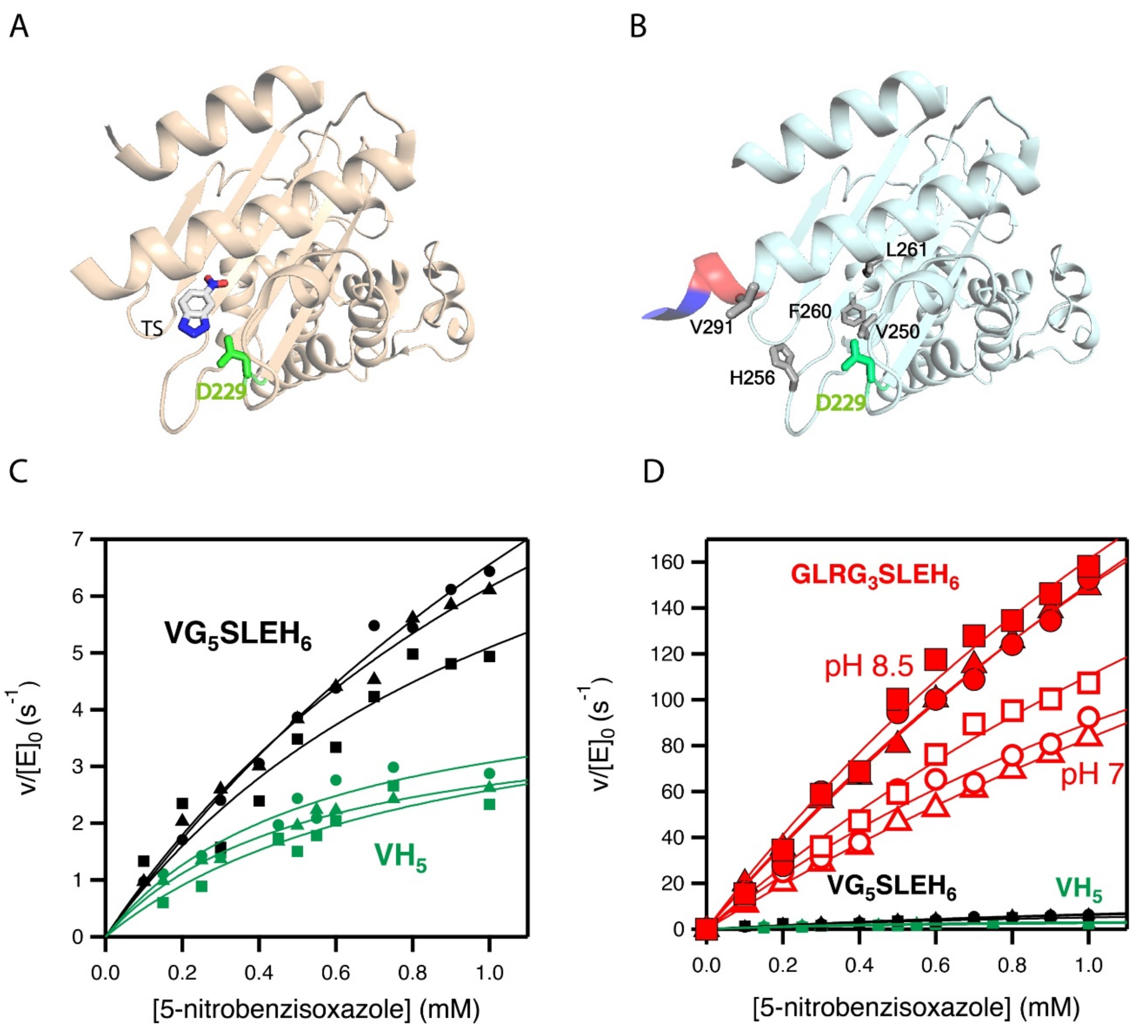
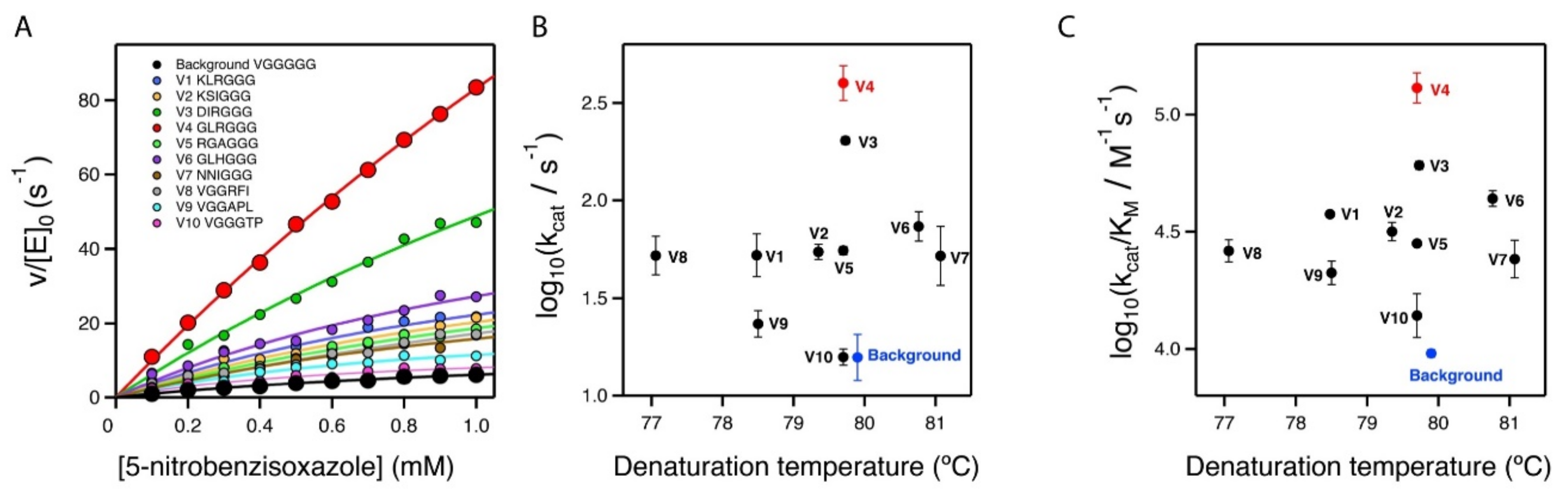
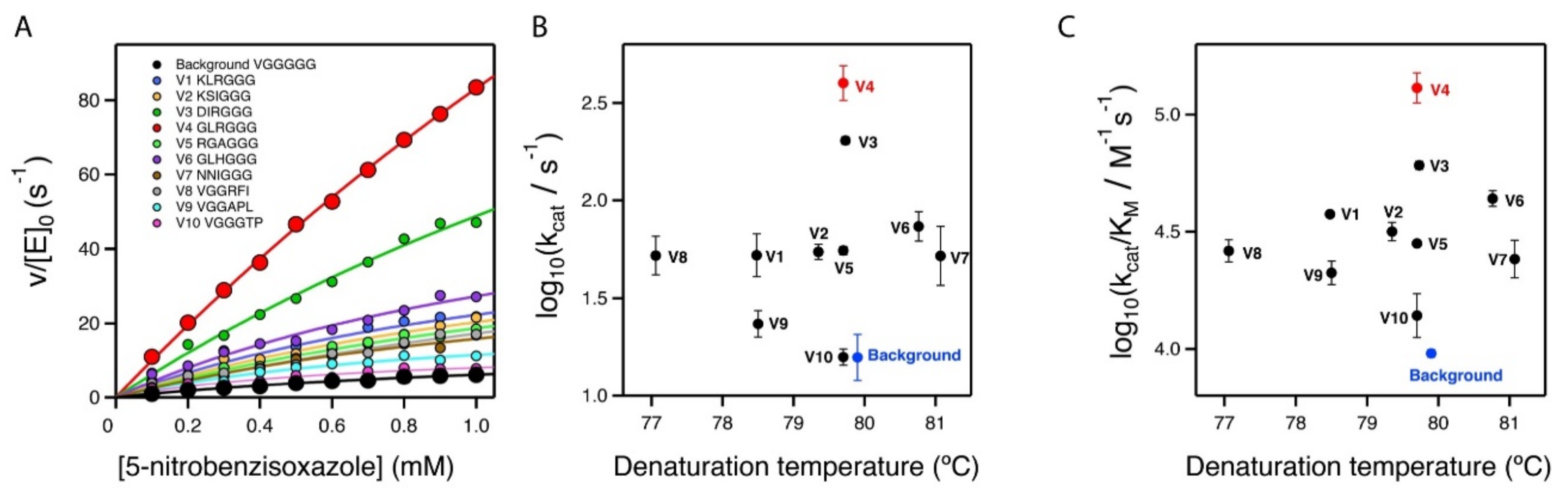
2.3. Stability and Catalytic Parameters for the Improved Kemp Eliminases
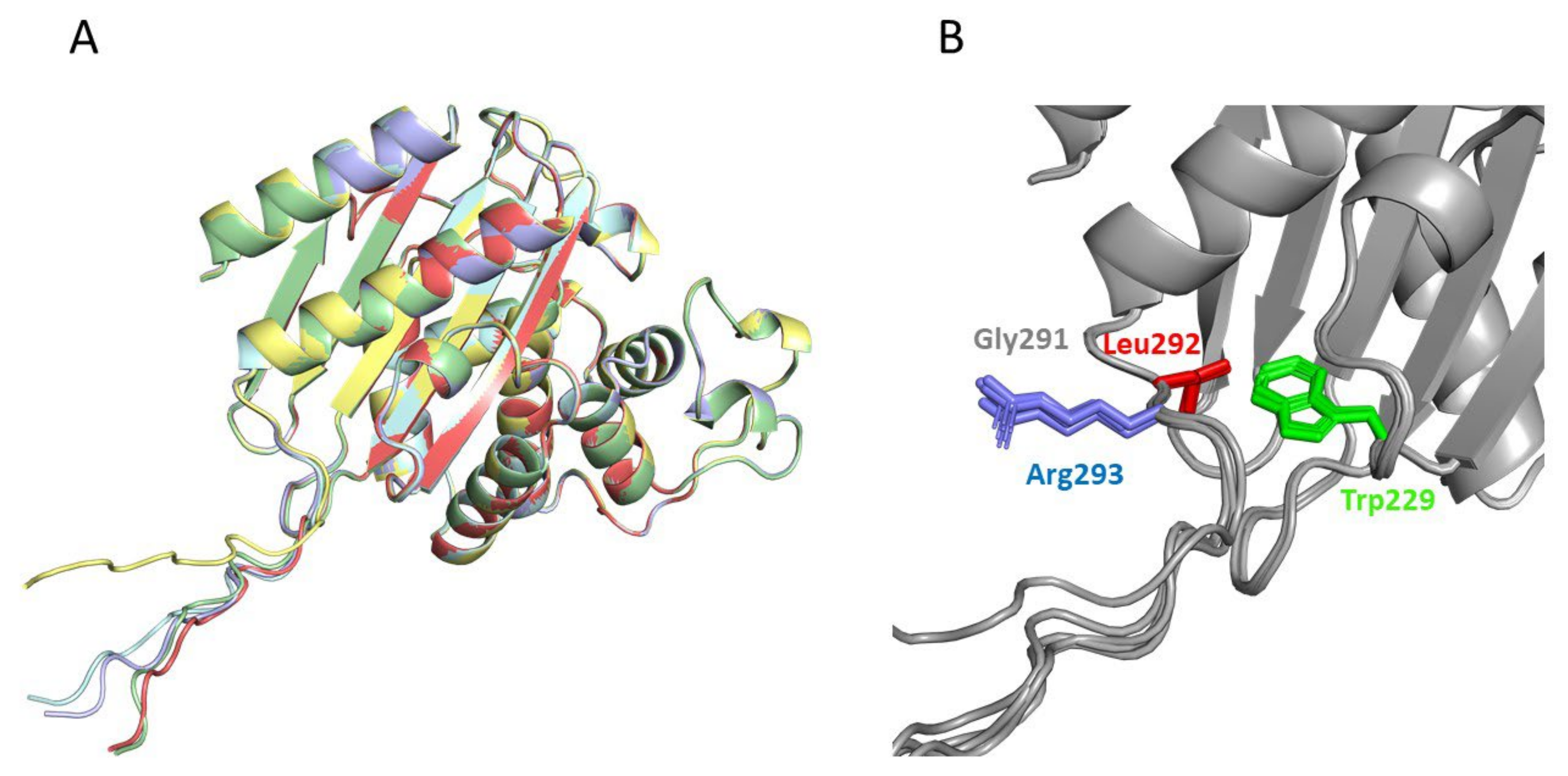
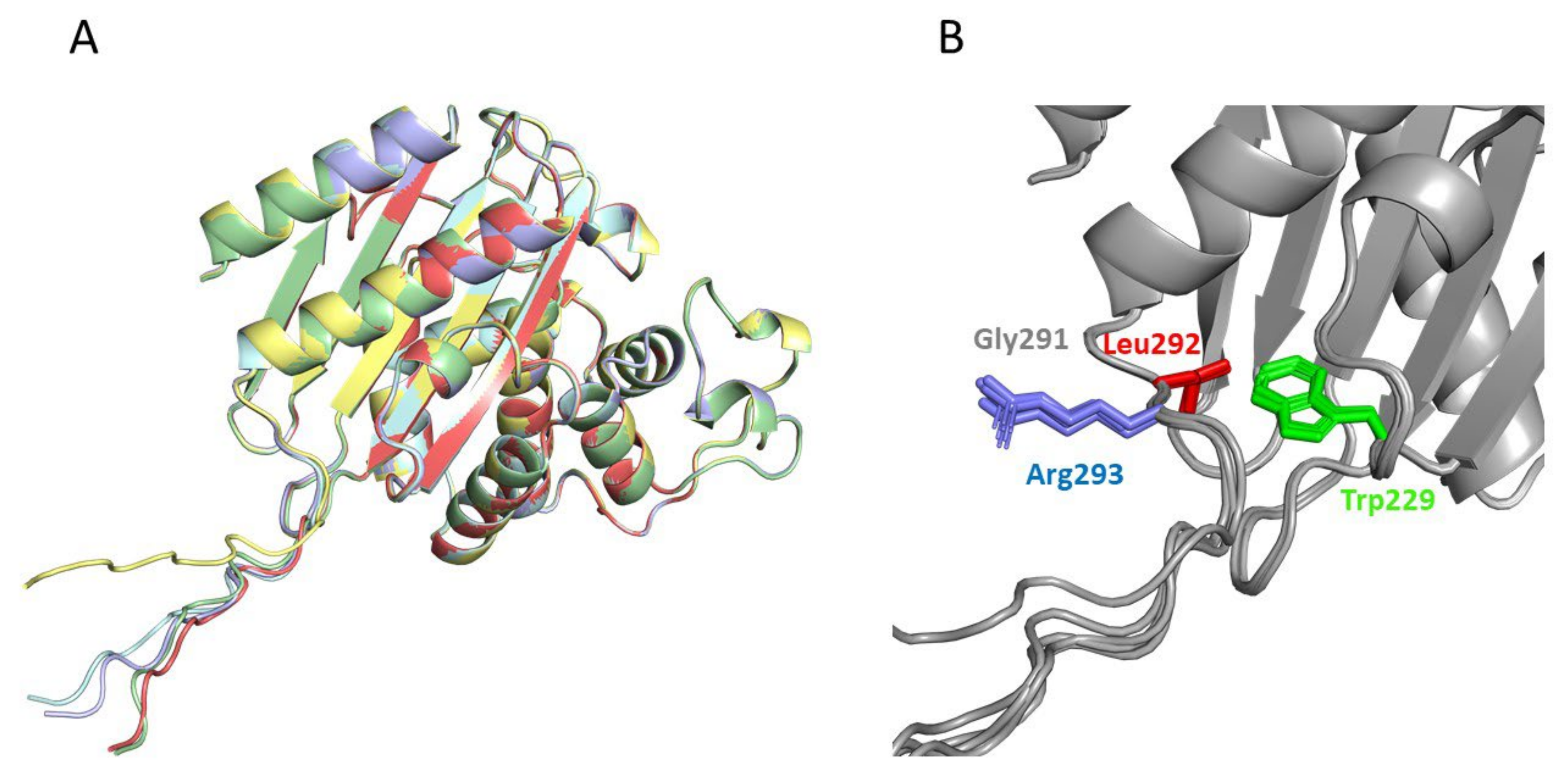
2.4. Structural Analysis of the Catalysis Enhancement
2.5. On the Possibility of Further Enhancements of Kemp Eliminase Activity
3. Discussion
4. Methods and Materials
4.1. Site-Saturation Mutagenic Libraries
4.2. Library Screening
4.3. Kemp Eliminase Assay for Library Screening
4.4. Protein Expression and Purification
4.5. Determination of Profiles of Rate versus Substrate Concentration for Kemp Eliminases
4.6. Protein Stability Determinations
4.7. Protein Structure Prediction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zuckerkandl, E.; Pauling, L. Molecular disease, evolution, and genic heterogeneity. In Horizons in Biochemistry; Kasha, A., Pullman, B., Eds.; Academic Press: New York, NY, USA, 1962; pp. 189–225. [Google Scholar]
- Pauling, L.; Zuckerkandl, E. Chemical paleogenetics. Molecular “restoration studies” of extinct forms of life. Acta Chem. Scan. 1963, 17, S9–S16. [Google Scholar]
- Benner, S.A.; Sassi, S.O.; Gaucher, E.A. Molecular Paleoscience: Systems biology from the past. In Advances in Enzymology and Related Areas of Molecular Biology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; Volume 75, pp. 1–132. [Google Scholar]
- Hochberg, G.K.A.; Thornton, J.W. Reconstructing ancient proteins to understand the causes of structure and function. Annu. Rev. Biochem. 2017, 46, 247–269. [Google Scholar] [CrossRef]
- Gumulya, Y.; Gillam, E.M. Exploring the past and the future of protein evolution with ancestral sequence reconstruction: The ′retro′ approach to protein engineering. Biochem. J. 2017, 474, 1–19. [Google Scholar] [CrossRef]
- Gaucher, E.A.; Govindarajan, S.; Ganesh, O.K. Palaeotemperature trend for Precambrian life inferred from resurrected proteins. Nature 2008, 451, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Risso, V.A.; Gavira, J.A.; Mejia-Carmona, D.F.; Gaucher, E.A.; Sanchez-Ruiz, J.M. Hypersability and substrate promiscuity in laboratory resurrections of Precambrian β-lactamases. J. Am. Chem. Soc. 2013, 135, 2899–2902. [Google Scholar] [CrossRef]
- Akanuma, S.; Nakajima, Y.; Kimura, M.; Nemoto, N.; Mase, T.; Miyazono, K.; Takonura, M.; Yamagishi, A. Experimental evidence for the thermophilicity of ancestral life. Proc. Natl. Acad. Sci. USA 2013, 110, 11067–11072. [Google Scholar] [CrossRef] [Green Version]
- Devamani, T.; Rauwerdink, A.M.; Lunzer, M.; Jones, B.J.; Mooney, J.L.; Tan, M.A.O.; Zhang, Z.-J.; Xu, J.-H.; Dean, A.M.; Kazalauslas, R.J. Catalytic promiscuity of ancestral esterases and hydroxynitrile lyases. J. Am. Chem. Soc. 2016, 138, 1046–1056. [Google Scholar] [CrossRef] [Green Version]
- Siddiq, M.A.; Hochberg, G.K.; Thornton, J.W. Evolution of protein specificity: Insights from ancestral protein reconstruction. Curr. Opin. Struct. Biol. 2017, 47, 113–122. [Google Scholar] [CrossRef]
- Nguyen, V.; Wilson, C.; Hoemberger, M.; Stiller, J.B.; Agafonov, R.V.; Kutter, S.; English, J.; Theobald, D.L.; Kern, D. Evolutionary drivers of thermoadpatation in enzyme catalysis. Science 2017, 355, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Risso, V.A.; Sanchez-Ruiz, J.M.; Ozkan, S.B. Biotechnological and protein engineering implications of ancestral protein resurrection. Curr. Opin. Struct. Biol. 2018, 51, 106–115. [Google Scholar] [CrossRef]
- Trudeau, D.L.; Tawfik, D.S. Protein engineers turned evolutionists—The quest for the optimal starting point. Curr. Opin. Struct. Biol. 2019, 60, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Spence, M.A.; Kaczmarski, J.A.; Saunders, J.W.; Jackson, C.J. Ancestral sequence resonctruction for protein engineers. Curr. Opin. Struct. Biol. 2021, 69, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Gamiz-Arco, G.; Gutierrez-Rus, L.; Risso, V.A.; Ibarra-Molero, B.; Hoshino, Y.; Petrovic, D.; Justicia, J.; Cuerva, J.M.; Romero-Rivera, A.; Seelig, B.; et al. Heme-binding enables allosteric modulation in an ancient TIM-barrel glycosidase. Nat. Commun. 2021, 12, 380. [Google Scholar] [CrossRef]
- Gamiz-Arco, G.; Risso, V.A.; Gaucher, E.A.; Gavira, J.A.; Naganathan, A.N.; Ibarra-Molero, B.; Sanchez-Ruiz, J.M. Combining ancestral reconstruction with folding-landscape simulations to engineer heterologous protein expression. J. Mol. Biol. 2021, 433, 167321. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.A. Enzyme recruitment in evolution of new function. Annu. Rev. Microbiol. 1976, 30, 409–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, V.E.; Gaucher, E.A. Engineering proteins by reconstructing evolutionary adaptive paths. Methods Mol. Biol. 2014, 1179, 353–363. [Google Scholar]
- Zakas, P.M.; Brown, H.C.; Knight, K.; Meeks, S.L.; Spencer, H.T.; Gaucher, E.A.; Doering, C.B. Enhancing the pharmaceutical properties of protein drugs by ancestral sequence reconstruction. Nat. Biotechnol. 2017, 35, 35–37. [Google Scholar] [CrossRef] [Green Version]
- Alcalde, M. When directed evolution met ancestral enzyme resurrection. Microb. Biotechnol. 2017, 10, 22–24. [Google Scholar] [CrossRef]
- Delgado, A.; Arco, R.; Ibarra-Molero, B.; Sanchez-Ruiz, J.M. Using resurrected ancestral protein to engineer virus resistance. Cell Rep. 2017, 19, 1247–1256. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Fernandez, B.J.; Risso, V.A.; Rueda, A.; Sanchez-Ruiz, J.M.; Alcalde, M. Ancestral resurrection and directed evolution of fungal mesozoic laccases. Appl. Environ. Microbiol. 2020, 86, e00778-20. [Google Scholar] [CrossRef]
- Khersonsky, O.; Tawfik, D.S. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010, 79, 471–505. [Google Scholar] [PubMed]
- James, L.C.; Tawfik, D.S. Conformational diversity and protein evolution—A 60-year old hypothesis revisited. Trends Biochem. Sci. 2003, 28, 361–368. [Google Scholar] [CrossRef]
- Copley, S.D. An evolutionary biochemist’s perspective on promiscuity. Trends Biochem. Sci. 2015, 40, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Pabis, A.; Risso, V.A.; Sanchez-Ruiz, J.M.; Kamerlin, S.C.L. Cooperativity and flexibility in enzyme evolution. Curr. Opin. Struct. Biol. 2018, 48, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, D.; Risso, V.A.; Kamerlin, S.C.L.; Sanchez-Ruiz, J.M. Conformational dynamics and enzyme evolution. J. R. Soc. Interface 2018, 15, 20180330. [Google Scholar] [CrossRef]
- Bloom, J.D.; Labthavikul, S.T.; Otey, C.R.; Arnold, F.H. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. USA 2006, 103, 5869–5874. [Google Scholar] [CrossRef] [Green Version]
- Jaenicke, R. Do ultrastable proteins from hyperthermophiles have high or low conformational rigidity. Proc. Natl. Acad. Sci. USA 2000, 97, 2962–2964. [Google Scholar] [CrossRef] [Green Version]
- Zou, T.; Risso, V.A.; Gavira, J.A.; Sanchez-Ruiz, J.M.; Ozkan, B. Evolution of conformational dynamics determines the conversion of a promiscuous generalist into a specialist enzyme. Mol. Biol. Evol. 2015, 119, 1323–1333. [Google Scholar] [CrossRef] [Green Version]
- Modi, T.; Risso, V.A.; Martinez-Rodriguez, S.; Gavira, J.A.; Mebrat, M.D.; Van Horn, W.D.; Sanchez-Ruiz, J.M.; Ozkan, B. Hinge-shift mechanism as a protein design principle for the evolution of β-lactamases from substrate promiscuity to specificity. Nat. Commun. 2021, 12, 1852. [Google Scholar] [CrossRef]
- Lovelock, S.L.; Crawshaw, R.; Basler, S.; Levy, C.; Baker, D.; Hilvert, D.; Green, A.P. The road to fully programmable protein catalysis. Nature 2022, 606, 49–58. [Google Scholar] [CrossRef]
- Risso, V.A.; Martinez-Rodriguez, S.; Candel, A.M.; Krüger, D.M.; Pantoja-Uceda, D.; Ortega-Muñoz, M.; Santoyo-Gonzalez, F.; Gaucher, E.A.; Kamerlin, S.C.L.; Bruix, M.; et al. De novo active sites for resurrected Precambrian enzymes. Nat. Commun. 2017, 8, 16113. [Google Scholar] [CrossRef] [PubMed]
- Risso, V.A.; Romero-Rivera, A.; Gutierrez-Rus, L.; Ortega-Muñoz, M.; Santoyo-Gonzalez, F.; Gavira, J.A.; Sanchez-Ruiz, J.M.; Kamerlin, S.C.L. Enhancing a de novo enzyme activity by computationally-focused ultra-low-throughput screening. Chem. Sci. 2020, 11, 6134–6148. [Google Scholar] [CrossRef] [PubMed]
- Bar-Even, A.; Noor, E.; Savir, Y.; Liebermeister, W.; Davidi, D.; Tawfik, D.S.; Milo, R. The moderately efficient enzyme: Evolutionary trends and physicochemical trends shaping enzyme parameters. Biochemsitry 2011, 50, 4402–4410. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, R.; Kries, H.; Pinkas, D.M.; Mittl, P.R.E.; Grütter, M.G.; Provett, H.K.; Mayo, S.L.; Hilvert, D. Precision is essential for efficient catalysis in an evolved enzyme. Nature 2013, 503, 418–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, J.M.; Biler, M.; Risso, V.A.; Sanchez-Ruiz, J.M.; Kamerlin, S.C.L. Manipulating conformational dynamics to repurpose ancient proteins for modern catalytic functions. ACS Cat. 2020, 10, 4863–4870. [Google Scholar] [CrossRef]
- Chen, X.; Zaro, J.; Shen, W.-C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Ouzonis, C.A.; Kunin, V.; Darzentas, N.; Goldovsky, L. A minimal estimate for the gene content of the last universal ancestor—Exobiology from a terrestrial perspective. Res. Microbiol. 2006, 157, 57–68. [Google Scholar] [CrossRef]
- Weiss, M.C.; Sousa, F.L.; Mmjavac, N.; Neukirken, S.; Roettger, M.; Nelson-Sathi, S.; Martin, W.F. The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 2016, 1, 16116. [Google Scholar] [CrossRef]
- Holm, R.H.; Kennepohl, P.; Solomon, E.I. Structural and Functional Aspects of Metal Sites in Biology. Chem. Rev. 1996, 96, 2239–2314. [Google Scholar] [CrossRef] [PubMed]
- Waldron, K.J.; Robinson, N.J. How do bacterial cells ensure that metalloproteins get the correct metal? Nat. Rev. Microbiol. 2009, 7, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Preiswerk, N.; Beck, T.; Schulz, J.D. Impact of scaffold rigidity on the design and evolution of an artificial Diels-Alderase. Proc. Natl. Acad. Sci. USA 2014, 111, 8013–8018. [Google Scholar] [CrossRef] [Green Version]
- Obexer, R.; Godina, A.; Garrabou, X.; Mittl, P.R.E.; Baker, D.; Griffiths, A.D.; Hilvert, D. Emergence of a catalytic tetrad during evolution of a highly active artificial aldolase. Nat. Chem. 2017, 9, 50–56. [Google Scholar] [CrossRef]
- Crawshaw, R.; Crossley, A.E.; Johannissen, L.; Burke, A.J.; Hay, S.; Levy, C.; Baker, D.; Lovelock, S.L.; Green, A.P. Engineering an efficient and enantioselective enzyme for the Morita-Baylis-Hillman reaction. Nat. Chem. 2022, 14, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Röthlisberger, D.; Khersonsky, O.; Wollacott, A.M.; Jiang, L.; DeChancie, J.; Betker, J.; Gallaher, J.L.; Althoff, E.A.; Xangehllini, A.; Dym, O.; et al. Kemp elimination catalysis by computational enzyme design. Nature 2008, 453, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Wang, B.; Ilie, A.; Dubey, K.D.; Bange, G.; Korendovych, I.V.; Shaik, S.; Reetz, M.T. A redox-mediated Kemp eliminase. Nat. Commun. 2017, 8, 14876. [Google Scholar] [CrossRef] [Green Version]
- Korendovych, I.; Bhattacharya, S.; Margheritis, E.; Takahashi, K.; Kulesha, A.; D’Souza, A.; Kim, I.; Tame, J.; Yoon, J.; Volkov, A.; et al. NMR-guided directed evolution. 2021; preprint from research square. [Google Scholar] [CrossRef]
- Privett, H.K.; Kiss, G.; Lee, T.M.; Blomberg, R.; Chica, R.A.; Thomas, L.M.; Hilvert, D.; Houk, K.N.; Mayo, S.L. Iterative approach to computational enzyme design. Proc. Natl. Acad. Sci. USA 2012, 109, 3790–3795. [Google Scholar] [CrossRef] [Green Version]
- Khersonsky, O.; Lipsh, R.; Avizemer, Z.; Ashani, Y.; Goldsmith, M.; Leader, H.; Dym, O.; Rogotner, S.; Trudeaum, D.L.; Prikusky, J.; et al. Automated design of efficient and functionally diverse enzyme repertoires. Mol. Cell 2018, 72, 178–186.e5. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Lutz, S. Circular permutation: A different way to engineer enzyme structure and function. Trends Botechnol. 2011, 29, 18–25. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Sequence a | kcat (s−1) b | KM (mM) b | kcat/KM (s−1 M−1) b | Tm (Cº) c |
|---|---|---|---|---|---|
| V4 at pH 8.5 | …GLRGGG… | 635.0 ± 59.1 | 3.14 ± 0.49 | (2.0 ± 0.1) × 105 | - |
| V4 | …GLRGGG… | 407.5 ± 76.7 | 3.35 ± 0.91 | (1.3 ± 0.2) × 105 | 79.7 |
| V3 | …DIRGGG… | 202.9 ± 8.9 | 3.35 ± 0.21 | (6.1 ± 0.3) × 104 | 79.7 |
| V6 | …GLHGGG… | 74.7 ± 13.5 | 1.74 ± 0.46 | (4.4 ± 0.3) × 104 | 80.8 |
| V1 | …KLRGGG… | 54.1 ± 13.2 | 1.45 ± 0.37 | (3.6 ± 0.1) × 104 | 78.5 |
| V2 | …KSIGGG… | 54.7 ± 4.8 | 1.75 ± 0.32 | (3.2 ± 0.3) × 104 | 79.4 |
| V5 | …RGAGGG… | 55.4 ± 2.7 | 1.97 ± 0.1 | (2.82 ± 0.01) × 104 | 79.7 |
| V8 | …VGGRFI… | 53.5 ± 11.0 | 2.10 ± 0.61 | (2.6 ± 0.3) × 104 | 77.0 |
| V7 | …NNIGGG… | 55.4 ± 20.8 | 2.50 ± 1.43 | (2.5 ± 0.4) × 104 | 81.1 |
| V9 | …VGGAPL… | 23.7 ± 3.8 | 1.12 ± 0.16 | (2.1 ± 0.2) × 104 | 78.5 |
| V10 | …VGGGTP… | 15.9 ± 1.5 | 1.19 ± 0.32 | (1.4 ± 0.3) × 104 | 79.7 |
| BACKGROUND | …VGGGGG… | 16.3 ± 4.4 | 1.71 ± 0.48 | (9.6 ± 0.4) × 103 | 79.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutierrez-Rus, L.I.; Alcalde, M.; Risso, V.A.; Sanchez-Ruiz, J.M. Efficient Base-Catalyzed Kemp Elimination in an Engineered Ancestral Enzyme. Int. J. Mol. Sci. 2022, 23, 8934. https://doi.org/10.3390/ijms23168934
Gutierrez-Rus LI, Alcalde M, Risso VA, Sanchez-Ruiz JM. Efficient Base-Catalyzed Kemp Elimination in an Engineered Ancestral Enzyme. International Journal of Molecular Sciences. 2022; 23(16):8934. https://doi.org/10.3390/ijms23168934
Chicago/Turabian StyleGutierrez-Rus, Luis I., Miguel Alcalde, Valeria A. Risso, and Jose M. Sanchez-Ruiz. 2022. "Efficient Base-Catalyzed Kemp Elimination in an Engineered Ancestral Enzyme" International Journal of Molecular Sciences 23, no. 16: 8934. https://doi.org/10.3390/ijms23168934
APA StyleGutierrez-Rus, L. I., Alcalde, M., Risso, V. A., & Sanchez-Ruiz, J. M. (2022). Efficient Base-Catalyzed Kemp Elimination in an Engineered Ancestral Enzyme. International Journal of Molecular Sciences, 23(16), 8934. https://doi.org/10.3390/ijms23168934