
A Novel Homozygous Variant in DYSF Gene Is Associated with Autosomal Recessive Limb Girdle Muscular Dystrophy R2/2B

 , ,
, ,
Abstract
:1. Introduction
2. Results
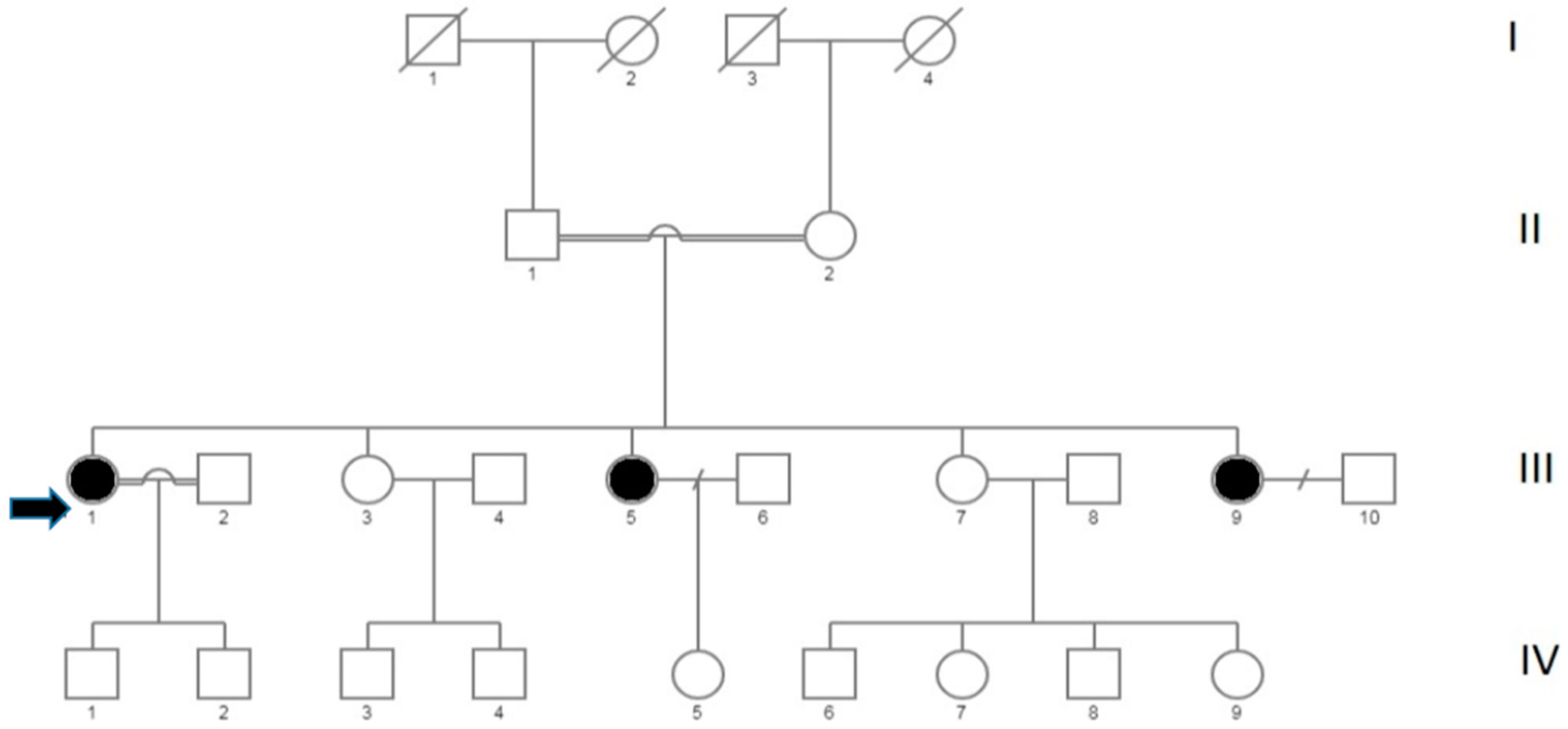
2.1. Targeted Next Generation Sequencing
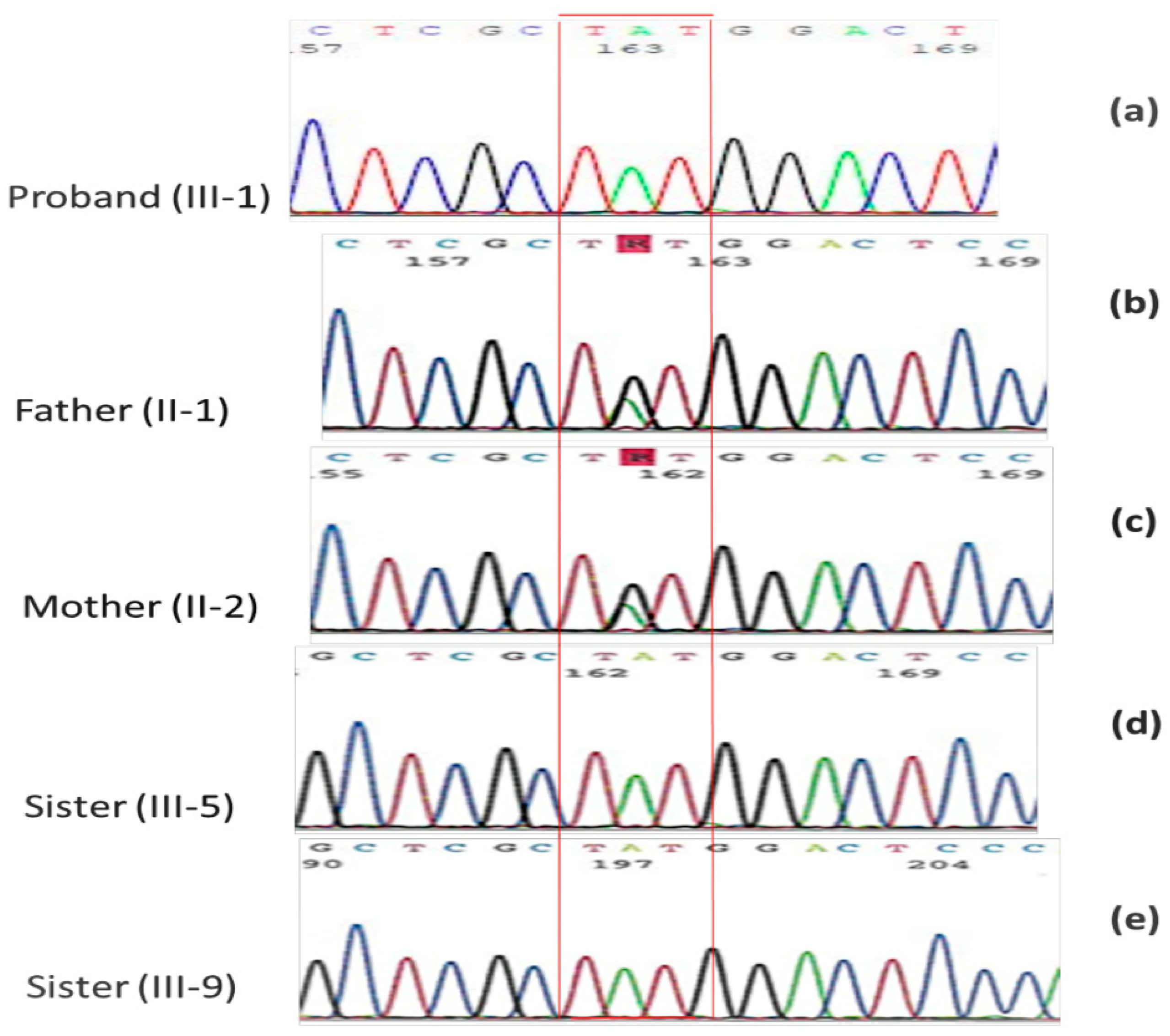
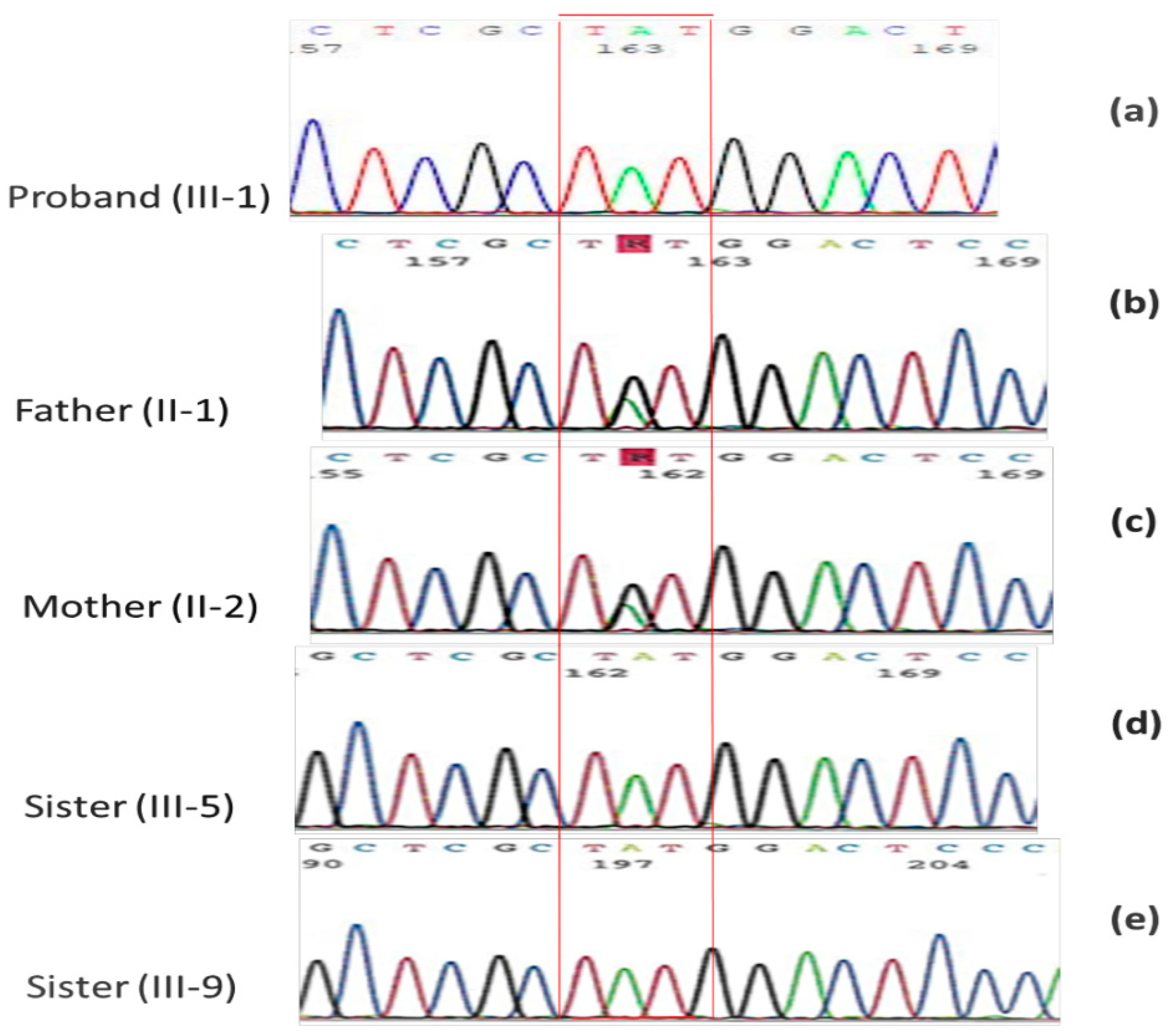
2.2. Family Mutational Screening by Sanger Sequencing
2.3. Functional Prediction of the Variant c.5033G>A
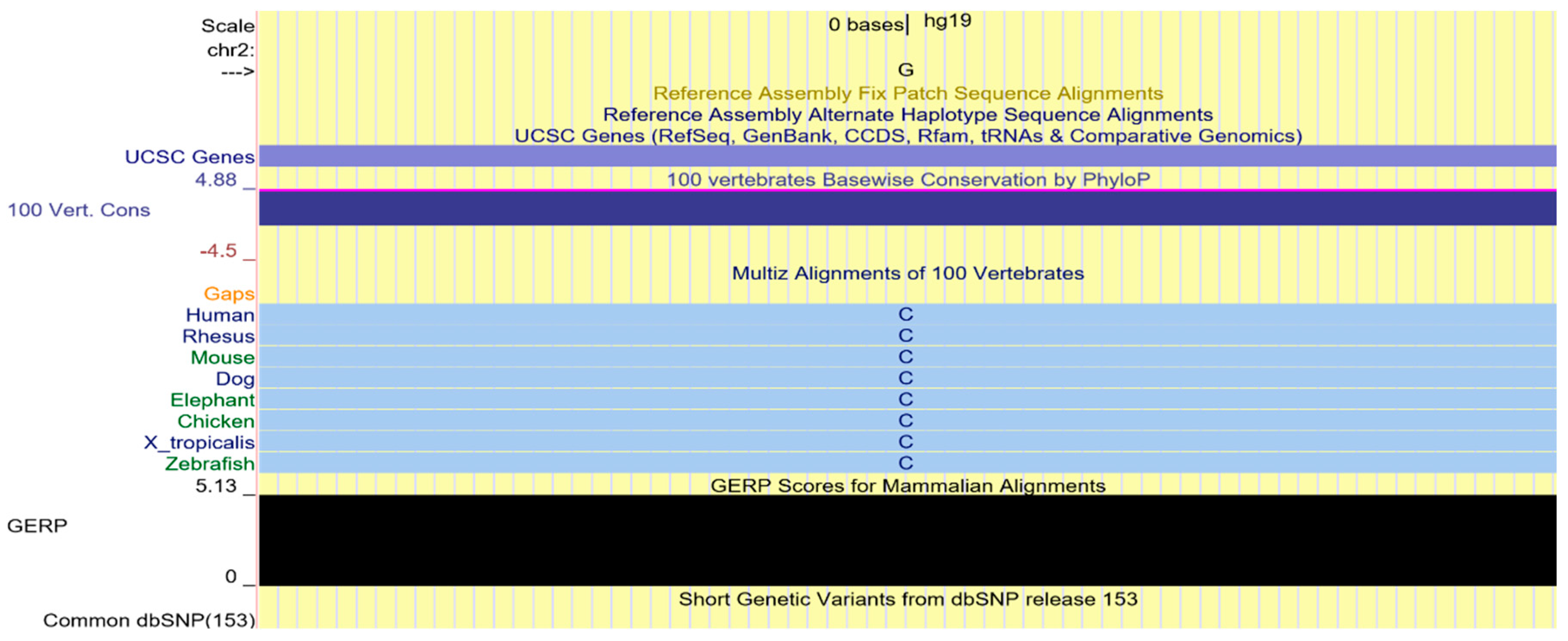
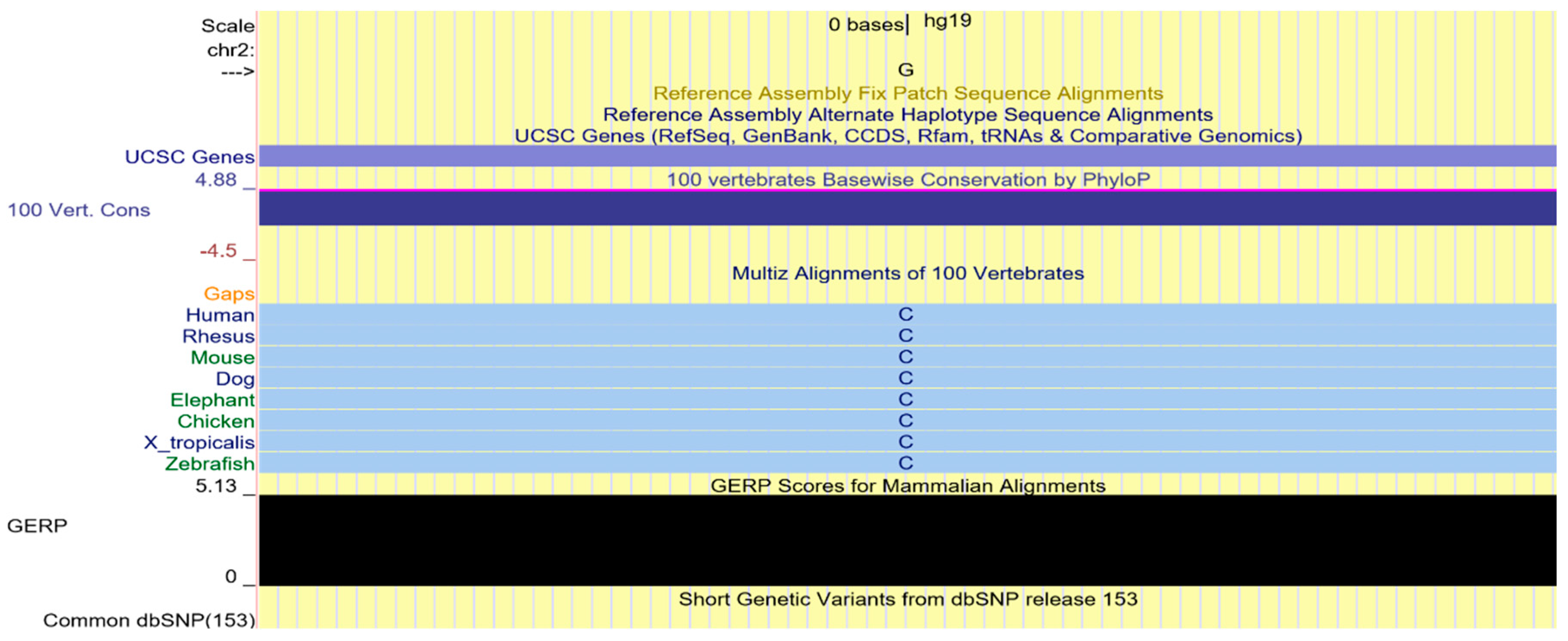
2.4. Multi-Species Alignment
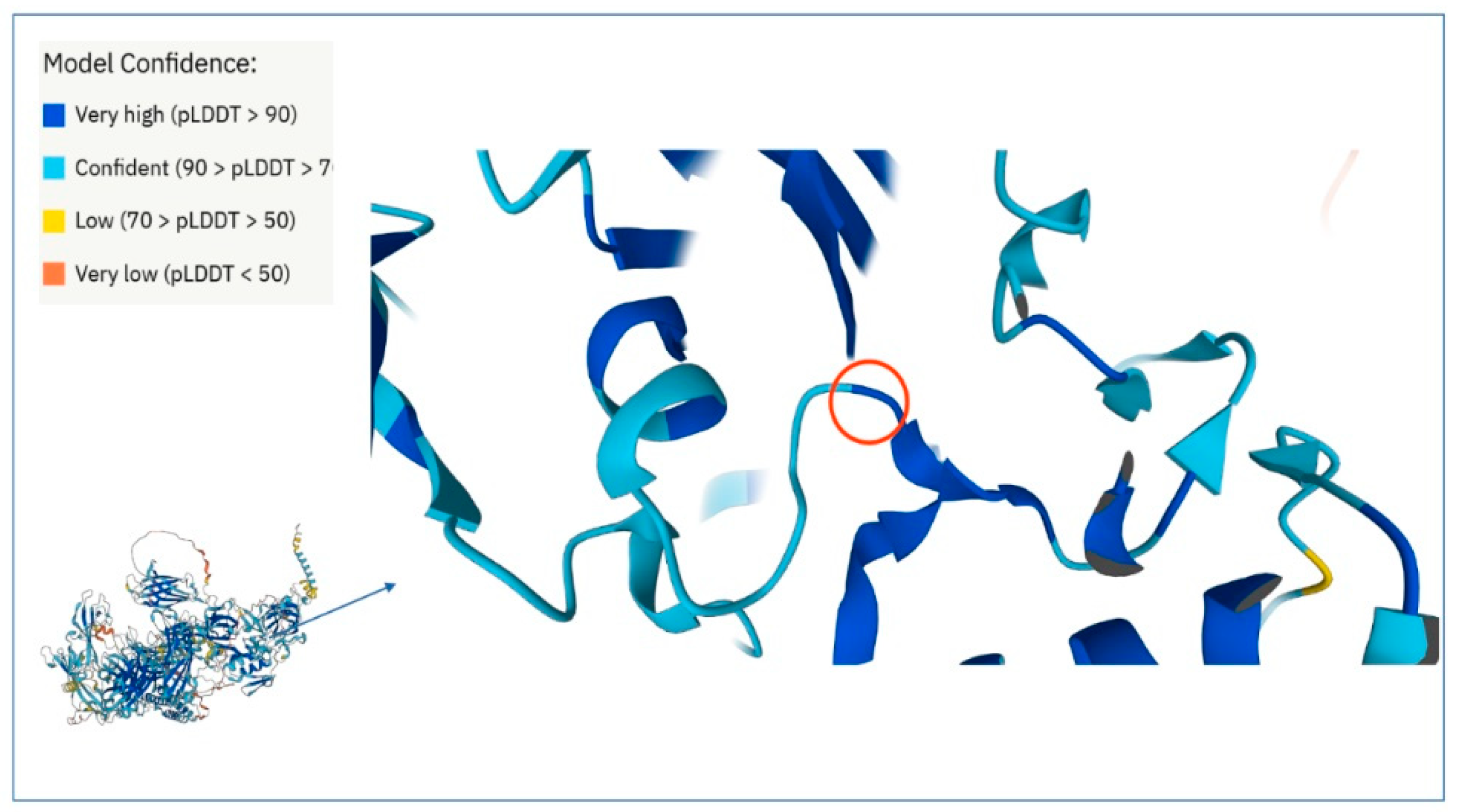
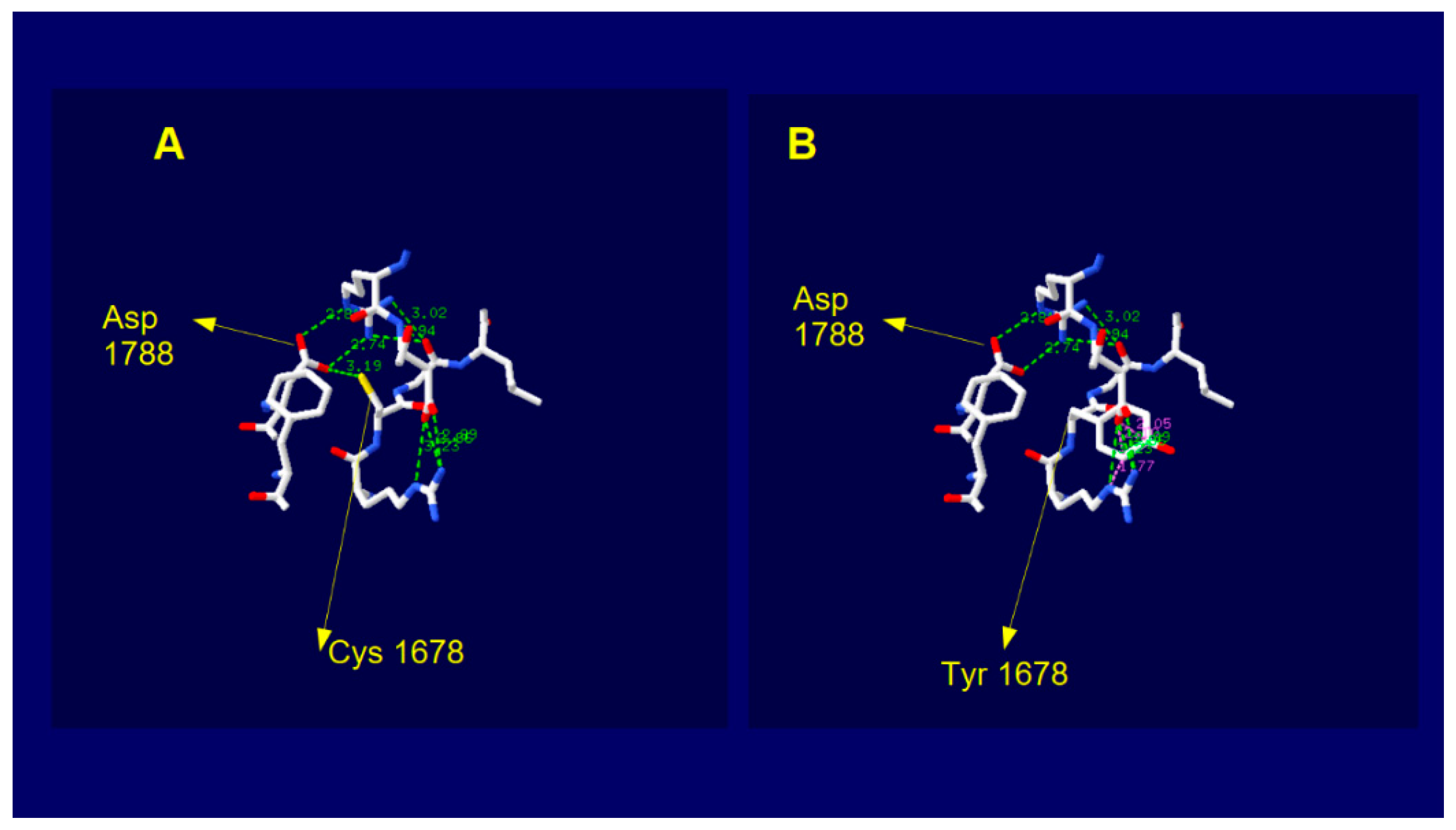
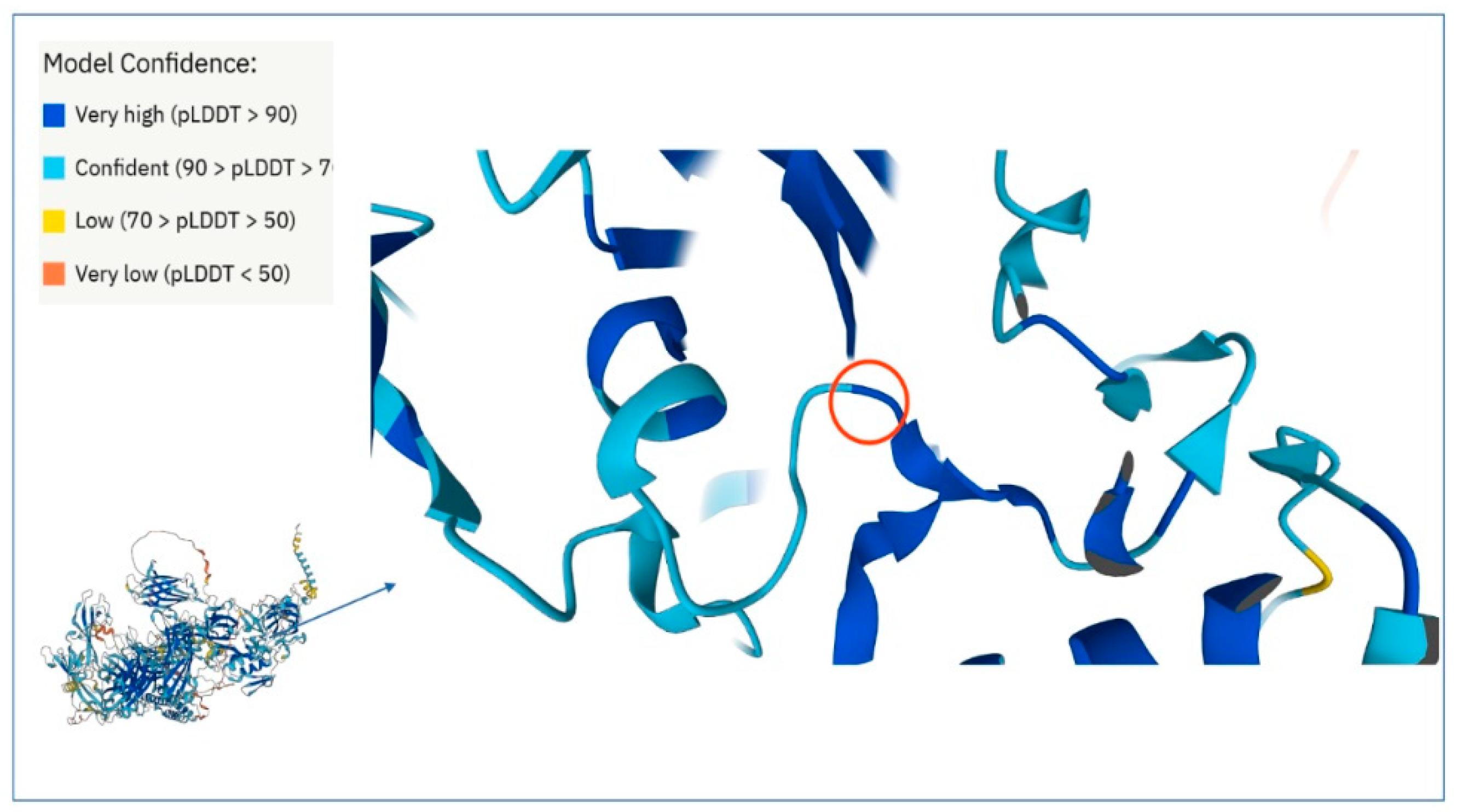
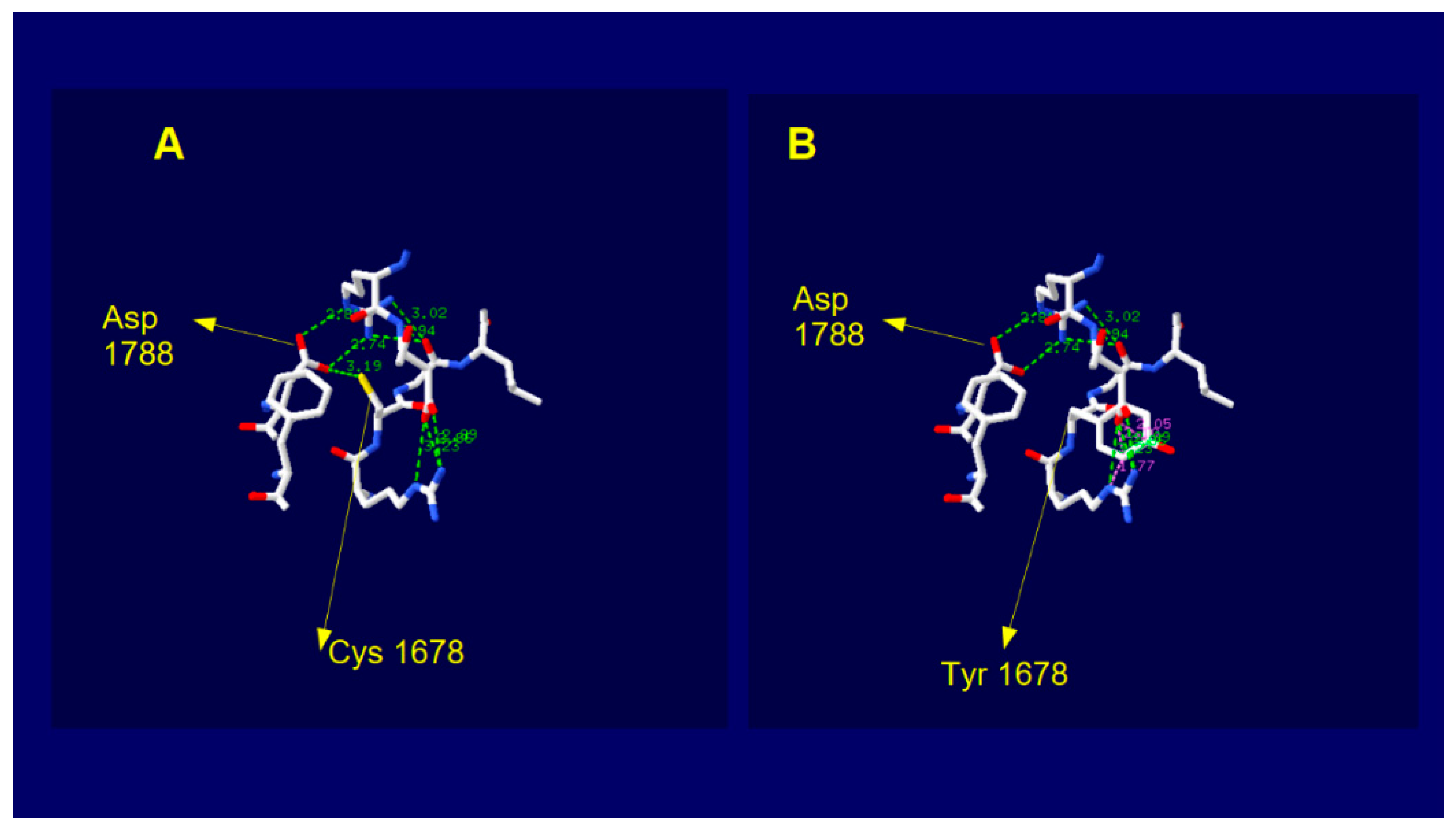
2.5. Dysferlin C2E Model
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Muscle Biopsy
4.3. NGS Data Analysis
4.4. PCR and Sanger Sequencing
4.5. Bioinformatic Software
4.6. In Silico 3D Structure Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cárdenas, A.M.; González-Jamett, A.M.; Cea, L.A.; Bevilacqua, J.A.; Caviedes, P. Dysferlin function in skeletal muscle: Possible pathological mechanisms and therapeutical targets in dysferlinopathies. Exp. Neurol. 2016, 283 Pt A, 246–254. [Google Scholar] [CrossRef]
- Okubo, M.; Iida, A.; Hayashi, S.; Mori-Yoshimura, M.; Oya, Y.; Watanabe, A.; Arahata, H.; El Sherif, R.; Noguchi, S.; Nishino, I. Three novel recessive DYSF mutations identified in three patients with muscular dystrophy, limb-girdle, type 2B. J. Neurol. Sci. 2018, 395, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Fanin, M.; Nascimbeni, A.C.; Aurino, S.; Tasca, E.; Pegoraro, E.; Nigro, V.; Angelini, C. Frequency of LGMD gene mutations in Italian patients with distinct clinical phenotypes. Neurology 2009, 72, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Magri, F.; Nigro, V.; Angelini, C.; Mongini, T.; Mora, M.; Moroni, I.; Toscano, A.; D’angelo, M.G.; Tomelleri, G.; Siciliano, G.; et al. The Italian Limb Girdle Muscular Dystrophy Registry: Relative frequency, clinical features, and differential diagnosis. Muscle Nerve 2017, 55, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Murphy, A.; Udd, B. LGMD workshop study group 229th ENMC international workshop: Limb girdle muscular dystrophies—nomenclature and reformed classification Naarden, The Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushby, K.M.; Beckmann, J.S. The limb-girdle muscular dystrophies-proposal for a new nomenclature. Neuromuscul. Disord. 1995, 5, 337–343. [Google Scholar] [CrossRef]
- Takahashi, T.; Aoki, M.; Suzuki, N.; Tateyama, M.; Yaginuma, C.; Sato, H.; Hayasaka, M.; Sugawara, H.; Ito, M.; Abe-Kondo, E.; et al. Clinical features and a mutation with late onset of limb girdle muscular dystrophy 2B. J. Neurol. Neurosurg. Psychiatry 2013, 84, 433–440. [Google Scholar] [CrossRef]
- Díaz, J.; Woudt, L.; Suazo, L.; Garrido, C.; Caviedes, P.; Cárdenas, A.M.; Castiglioni, C.; Bevilacqua, J.A. Broadening the imaging phenotype of dysferlinopathy at different disease stages. Muscle Nerve 2016, 54, 203–210. [Google Scholar] [CrossRef]
- Ueyama, H.; Kumamoto, T.; Horinouchi, H.; Fujimoto, S.; Aono, H.; Tsuda, T. Clinical heterogeneity in dysferlinopathy. Intern. Med. 2002, 41, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Guglieri, M.; Straub, V.; Bushby, K.; Lochmüller, H. Limb-girdle muscular dystrophies. Curr. Opin. Neurol. 2008, 21, 576–584. [Google Scholar] [CrossRef]
- Rosales, X.Q.; Gastier-Foster, J.M.; Lewis, S.; Vinod, M.; Thrush, D.L.; Astbury, C.; Pyatt, R.; Reshmi, S.; Sahenk, Z.; Mendell, J.R. Novel diagnostic features of dysferlinopathies. Muscle Nerve 2010, 42, 14–21. [Google Scholar] [CrossRef]
- Woudt, L.; Di Capua, G.A.; Krahn, M.; Castiglioni, C.; Hughes, R.; Campero, M.; Trangulao, A.; González-Hormazábal, P.; Godoy-Herrera, R.; Lévy, N.; et al. Toward an objective measure of functional disability in dysferlinopathy. Muscle Nerve 2016, 53, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Bashir, R.; Britton, S.; Strachan, T.; Keers, S.; Vafiadaki, E.; Lako, M.; Richard, I.; Marchand, S.; Bourg, N.; Argov, Z.; et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat. Genet. 1998, 20, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Aoki, M.; Illa, I.; Wu, C.; Fardeau, M.; Angelini, C.; Serrano, C.; Urtizberea, J.A.; Hentati, F.; Hamida, M.B.; et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998, 20, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M.; Liu, J.; Richard, I.; Bashir, R.; Britton, S.; Keers, S.M.; Oeltjen, J.; Brown, H.E.; Marchand, S.; Bourg, N.; et al. Genomic organization of the dysferlin gene and novel mutations in Miyoshi myopathy. Neurology 2001, 57, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Algahtani, H.; Shirah, B.; Alassiri, A.H.; Habib, B.A.; Almuhanna, R.; Ahamed, M.F. Limb-girdle muscular dystrophy type 2B: An unusual cause of proximal muscular weakness in Saudi Arabia. J. Back Musculoskelet. Rehabil. 2018, 31, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.V.; Davison, K.; Moss, J.A.; Young, C.; Cullen, M.J.; Walsh, J.; Johnson, M.A.; Bashir, R.; Britton, S.; Keers, S.; et al. Dysferlin is a plasma membrane protein and is expressed early in human development. Hum. Mol. Genet. 1999, 8, 855–861, Erratum in Hum. Mol. Genet. 1999, 8, 1141. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Weisleder, N.; Ko, J.K.; Komazaki, S.; Sunada, Y.; Nishi, M.; Takeshima, H.; Ma, J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 2009, 284, 15894–15902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeil, P.L. Repairing a torn cell surface: Make way, lysosomes to the rescue. J. Cell. Sci. 2002, 115 Pt 5, 873–879. [Google Scholar] [CrossRef]
- Bulankina, A.V.; Thoms, S. Functions of Vertebrate Ferlins. Cells 2020, 9, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britton, S.; Freeman, T.; Vafiadaki, E.; Keers, S.; Harrison, R.; Bushby, K.; Bashir, R. The third human FER-1-like protein is highly similar to dysferlin. Genomics 2000, 68, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009, 11, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Pan, Z.; Nishi, M.; Komazaki, S.; Takeshima, H.; Ma, J. MG53 regulates membrane budding and exocytosis in muscle cells. J. Biol. Chem. 2009, 284, 3314–3322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; Tang, R.H.; Weisleder, N.; Xiao, B.; Yuan, Z.; Cai, C.; Zhu, H.; Lin, P.; Qiao, C.; Li, J.; et al. Enhancing muscle membrane repair by gene delivery of MG53 ameliorates muscular dystrophy and heart failure in δ-Sarcoglycan-deficient hamsters. Mol. Ther. 2012, 20, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Blazek, A.D.; Paleo, B.J.; Weisleder, N. Plasma Membrane Repair: A Central Process for Maintaining Cellular Homeostasis. Physiology 2015, 30, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Lin, P.; De, G.; Choi, K.H.; Takeshima, H.; Weisleder, N.; Ma, J. Polymerase transcriptase release factor (PTRF) anchors MG53 protein to cell injury site for initiation of membrane repair. J. Biol. Chem. 2011, 286, 12820–12824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.; Zhu, H.; Cai, C.; Wang, X.; Cao, C.; Xiao, R.; Pan, Z.; Weisleder, N.; Takeshima, H.; Ma, J. Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J. 2012, 26, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H., Jr. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittel, D.C.; Chandra, G.; Tirunagri, L.M.S.; Deora, A.B.; Medikayala, S.; Scheffer, L.; Defour, A.; Jaiswal, J.K. Annexin A2 Mediates Dysferlin Accumulation and Muscle Cell Membrane Repair. Cells 2020, 9, 1919. [Google Scholar] [CrossRef] [PubMed]
- Boye, T.L.; Maeda, K.; Pezeshkian, W.; Sønder, S.L.; Haeger, S.C.; Gerke, V.; Simonsen, A.C.; Nylandsted, J. Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nat. Commun. 2017, 8, 1623. [Google Scholar] [CrossRef] [Green Version]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Blebbing confers resistance against cell lysis. Cell Death Differ. 2011, 18, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potez, S.; Luginbühl, M.; Monastyrskaya, K.; Hostettler, A.; Draeger, A.; Babiychuk, E.B. Tailored protection against plasmalemmal injury by annexins with different Ca2+ sensitivities. J. Biol. Chem. 2011, 286, 17982–17991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, V.; Savarese, M. Genetic basis of limb-girdle muscular dystrophies: The 2014 update. Acta Myol. 2014, 33, 1–12. [Google Scholar] [PubMed]
- Savarese, M.; Di Fruscio, G.; Torella, A.; Fiorillo, C.; Magri, F.; Fanin, M.; Ruggiero, L.; Ricci, G.; Astrea, G.; Passamano, L.; et al. The genetic basis of undiagnosed muscular dystrophies and myopathies: Results from 504 patients. Neurology 2016, 87, 71–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georganopoulou, D.G.; Moisiadis, V.G.; Malik, F.A.; Mohajer, A.; Dashevsky, T.M.; Wuu, S.T.; Hu, C.K. A Journey with LGMD: From Protein Abnormalities to Patient Impact. Protein J. 2021, 40, 466–488. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, K.; Goto, K.; Nishino, I.; Angelini, C.; Hayashi, Y.K. Dysferlin mutation analysis in a group of Italian patients with limb-girdle muscular dystrophy and Miyoshi myopathy. Eur. J. Neurol. 2004, 11, 657–661. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 7, D439–D444. [Google Scholar] [CrossRef]
- Urtizberea, J.; Bassez, G.; Leturcq, F.; Nguyen, K.; Krahn, M.; Levy, N. Dysferlinopathies. Neurol. India 2008, 56, 289–297. [Google Scholar] [CrossRef]
- Han, R.; Campbell, K.P. Dysferlin and muscle membrane repair. Curr. Opin. Cell Biol. 2007, 19, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Bansal, D.; Campbell, K.P. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004, 14, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Gayathrì, N.; Alefia, R.; Nalini, A.; Yasha, T.C.; Anita, M.; Santosh, V.; Shankar, S.K. Dysferlinopathy: Spectrum of pathological changes in skeletal muscle tissue. Indian J. Pathol. Microbiol. 2011, 54, 350–354. [Google Scholar] [CrossRef] [PubMed]
- McDade, J.R.; Archambeau, A.; Michele, D.E. Rapid actin-cytoskeleton-dependent recruitment of plasma membrane-derived dysferlin at wounds is critical for muscle membrane repair. FASEB J. 2014, 28, 3660–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tidball, J.G. Mechanisms of muscle injury, repair, and regeneration. Compr. Physiol. 2011, 1, 2029–2062. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, C.; Miyake, K.; Kameyama, K.; Keduka, E.; Takeshima, H.; Imamura, T.; Araki, N.; Nishino, I.; Hayashi, Y. The C2A domain in dysferlin is important for association with MG53 (TRIM72). PLoS Curr. 2012, 4, e5035add8caff4. [Google Scholar] [CrossRef] [PubMed]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef]
- McNeil, P. Membrane repair redux: Redox of MG53. Nat. Cell Biol. 2009, 11, 7–9. [Google Scholar] [CrossRef]
- Matsuda, C.; Hayashi, Y.K.; Ogawa, M.; Aoki, M.; Murayama, K.; Nishino, I.; Nonaka, I.; Arahata, K.; Brown, R.H., Jr. The sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal muscle. Hum. Mol. Genet. 2001, 10, 1761–1766. [Google Scholar] [CrossRef]
- Hogarth, M.W.; Defour, A.; Lazarski, C.; Gallardo, E.; Diaz Manera, J.; Partridge, T.A.; Nagaraju, K.; Jaiswal, J.K. Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nat. Commun. 2019, 10, 2430. [Google Scholar] [CrossRef] [Green Version]
- Angelini, C.; Giaretta, L.; Marozzo, R. An update on diagnostic options and considerations in limb-girdle dystrophies. Expert Rev. Neurother. 2018, 18, 693–703. [Google Scholar] [CrossRef]
- Harris, E.; Topf, A.; Barresi, R.; Hudson, J.; Powell, H.; Tellez, J.; Hicks, D.; Porter, A.; Bertoli, M.; Evangelista, T.; et al. Exome sequences versus sequential gene testing in the UK highly specialised Service for Limb Girdle Muscular Dystrophy. Orphanet J. Rare Dis. 2017, 12, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat. Rev. Genet. 2011, 13, 36–46, Erratum in Nat. Rev. Genet. 2012, 13, 146. [Google Scholar] [CrossRef] [PubMed]
- Nigro, V.; Piluso, G. Next generation sequencing (NGS) strategies for the genetic testing of myopathies. Acta Myol. 2012, 31, 196–200. [Google Scholar] [PubMed]
- Özyilmaz, B.; Kirbiyik, Ö.; Özdemir, T.R.; Kaya Özer, Ö.; Kutbay, Y.B.; Erdogan, K.M.; Güvenç, M.S.; Kale, M.Y.; Gazeteci, H.; Kiliç, B.; et al. Impact of next-generation sequencing panels in the evaluation of limb-girdle muscular dystrophies. Ann. Hum. Genet. 2019, 83, 331–347. [Google Scholar] [CrossRef]
- Fichna, J.P.; Macias, A.; Piechota, M.; Korostyński, M.; Potulska-Chromik, A.; Redowicz, M.J.; Zekanowski, C. Whole-exome sequencing identifies novel pathogenic mutations and putative phenotype-influencing variants in Polish limb-girdle muscular dystrophy patients. Hum. Genom. 2018, 12, 34. [Google Scholar] [CrossRef] [Green Version]
- Lemmers, R.J.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012, 44, 1370–1374. [Google Scholar] [CrossRef] [Green Version]
- Simeoni, S.; Russo, V.; Gigli, G.L.; Scalise, A. Facioscapulohumeral muscular dystrophy and limb-girdle muscular dystrophy: “double trouble” overlapping syndrome? J. Neurol. Sci. 2015, 348, 292–293. [Google Scholar] [CrossRef]
- Lam, C.W.; Wong, K.S.; Leung, H.W.; Law, C.Y. Limb girdle myasthenia with digenic RAPSN and a novel disease gene AK9 mutations. Eur. J. Hum. Genet. 2017, 25, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Saenz, A.; Lopez de Munain, A. Dominant LGMD2A: Alternative diagnosis or hidden digenism? Brain 2017, 140 Pt 2, e7. [Google Scholar] [CrossRef]
- Cagliani, R.; Magri, F.; Toscano, A.; Merlini, L.; Fortunato, F.; Lamperti, C.; Rodolico, C.; Prelle, A.; Sironi, M.; Aguennouz, M.; et al. Mutation finding in patients with dysferlin deficiency and role of the dysferlin interacting proteins annexin A1 and A2 in muscular dystrophies. Hum. Mutat. 2005, 26, 283. [Google Scholar] [CrossRef]
- Spadafora, P.; Liguori, M.; Andreoli, V.; Quattrone, A.; Gambardella, A. CAV3 T78M mutation as polymorphic variant in South Italy. Neuromuscul. Disord. 2012, 22, 669–670. [Google Scholar] [CrossRef] [PubMed]
- Xu, I.; Pallikkuth, S.; Hou, Z.; Mignery, G.A.; Robia, S.L.; Han, R. Dysferlin Forms a Dimer Mediated by the C2 Domains and the Transmembrane Domain in Vitro and in Living Cells. PLoS ONE 2011, 6, e27884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, L.V.; Harrison, R.M.; Pogue, R.; Vafiadaki, E.; Pollitt, C.; Davison, K.; Moss, J.A.; Keers, S.; Pyle, A.; Shaw, P.J.; et al. Secondary reduction in calpain 3 expression in patients with limb girdle muscular dystrophy type 2B and Miyoshi myopathy (primary dysferlinopathies). Neuromuscul. Disord. 2000, 10, 553–559. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Bean, L.J.; Tinker, S.W.; da Silva, C.; Hegde, M.R. Free the data: One laboratory’s approach to knowledge-based genomic variant classification and preparation for EMR integration of genomic data. Hum. Mutat. 2013, 34, 1183–1188. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | WT | nt Change | Func. Refgenze | Gene | ExonicFunc. Refgene | AAChange.Refgene | dbSNP | Inher |
|---|---|---|---|---|---|---|---|---|
| chr1 | T | C | exonic | POMGNT1 | nonsynonymous SNV | POMGNT1:NM_001290129:exon20:c.A1801G:p.M601V | rs6659553 | hom |
| chr2 | - | AGGCGG | exonic | DYSF | nonframeshift insertion | DYSF:NM_001130455:exon30:c.3184_3185insAGGCGG: p.Q1062delinsQAE | rs398123779 | hom |
| chr2 | G | A | exonic | DYSF | nonsynonymous SNV | DYSF:NM_003494:exon45:c.G5033A:p.C1678Y | . | hom |
| chr3 | C | G | exonic | DAG1 | nonsynonymous SNV | DAG1:NM_001177639:exon2:c.C41G:p.S14WW | rs2131107 | hom |
| chr3 | T | C | exonic | GMPPB | nonsynonymous SNV | GMPPB:NM_013334:exon5:c.A551G:p.Q184R | rs1466685 | hom |
| chr5 | A | C | exonic | MYOT | nonsynonymous SNV | MYOT:NM_006790:exon2:c.A220C:p.K74Q | rs6890689 | hom |
| chr17 | A | G | exonic | GAA | nonsynonymous SNV | GAA:NM_000152:exon3:c.A596G:p.H199R | rs1042393 | hom |
| chr17 | G | A | exonic | GAA | nonsynonymous SNV | GAA:NM_000152:exon3:c.G668A:p.R223H | rs1042395 | hom |
| chr17 | G | A | exonic | GAA | nonsynonymous SNV | GAA:NM_000152:exon17:c.G2338A:p.V780I | rs1126690 | hom |
| chrX | C | T | exonic | DMD | nonsynonymous SNV | DMD:NM_004014:exon5:c.G623A:p.R208Q | rs1800280 | hom |
| Tool | Effect | Score/ Reliability Index | Link |
|---|---|---|---|
| MutPred2 | Deleterious | 0.90 | http://mutpred.mutdb.org/ (accessed on 21 July 2021) |
| PhD-SNP | Deleterious | 10 | https://snps.biofold.org/phd-snp/phd-snp.html (accessed on 21 July 2021) |
| PolyPhen2 | Deleterious | 1 | http://genetics.bwh.harvard.edu/pph2/ (accessed on 21 July 2021) |
| CADD | Deleterious | 4.19 (PHRED 29.5) | https://cadd.gs.washington.edu/ (accessed on 21 July 2021) |
| SNAP2 | Deleterious | 66 | https://www.rostlab.org/services/snap/ (accessed on 21 July 2021) |
| Pmut | Deleterious | 0.87 | https://wwwmmb.irbbarcelona.org/PMut/ (accessed on 21 July 2021) |
| Panther | Deleterious | 0.86 | http://www.pantherdb.org/tools/ (accessed on 21 July 2021) |
| SIFT | Deleterious | 0.04 | https://sift.bii.a-star.edu.sg/ (accessed on 21 July 2021) |
| PROVEN | Deleterious | −10.41 | http://provean.jcvi.org/index.php (accessed on 21 July 2021) |
| Mutation Taster | Deleterious | 0.99 | https://www.mutationtaster.org/ (accessed on 21 July 2021) |
| Bonds | Angles | Torsion | Improper | NonBonded | Electrostatic | Constraint// | TOTAL | |
|---|---|---|---|---|---|---|---|---|
| Wt dysferlin | 16,540.810 | 18,356.452 | 14,731.280 | 1306.953 | −65,364.79 | −60,468.37 | 0.0000// | E = −74,897.672 |
| Mutated | 16,542.059 | 18,368.218 | 14,725.421 | 1308.030 | 842,455.48 | −60,547.98 | 0.0000// | E = 832,851.313 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spadafora, P.; Qualtieri, A.; Cavalcanti, F.; Di Palma, G.; Gallo, O.; De Benedittis, S.; Cerantonio, A.; Citrigno, L. A Novel Homozygous Variant in DYSF Gene Is Associated with Autosomal Recessive Limb Girdle Muscular Dystrophy R2/2B. Int. J. Mol. Sci. 2022, 23, 8932. https://doi.org/10.3390/ijms23168932
Spadafora P, Qualtieri A, Cavalcanti F, Di Palma G, Gallo O, De Benedittis S, Cerantonio A, Citrigno L. A Novel Homozygous Variant in DYSF Gene Is Associated with Autosomal Recessive Limb Girdle Muscular Dystrophy R2/2B. International Journal of Molecular Sciences. 2022; 23(16):8932. https://doi.org/10.3390/ijms23168932
Chicago/Turabian StyleSpadafora, Patrizia, Antonio Qualtieri, Francesca Cavalcanti, Gemma Di Palma, Olivier Gallo, Selene De Benedittis, Annamaria Cerantonio, and Luigi Citrigno. 2022. "A Novel Homozygous Variant in DYSF Gene Is Associated with Autosomal Recessive Limb Girdle Muscular Dystrophy R2/2B" International Journal of Molecular Sciences 23, no. 16: 8932. https://doi.org/10.3390/ijms23168932
APA StyleSpadafora, P., Qualtieri, A., Cavalcanti, F., Di Palma, G., Gallo, O., De Benedittis, S., Cerantonio, A., & Citrigno, L. (2022). A Novel Homozygous Variant in DYSF Gene Is Associated with Autosomal Recessive Limb Girdle Muscular Dystrophy R2/2B. International Journal of Molecular Sciences, 23(16), 8932. https://doi.org/10.3390/ijms23168932