
Inhibition of the Phospholipase Cε–c-Jun N-Terminal Kinase Axis Suppresses Glioma Stem Cell Properties

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The Knockdown of PLCε Suppresses the Viability of GSCs
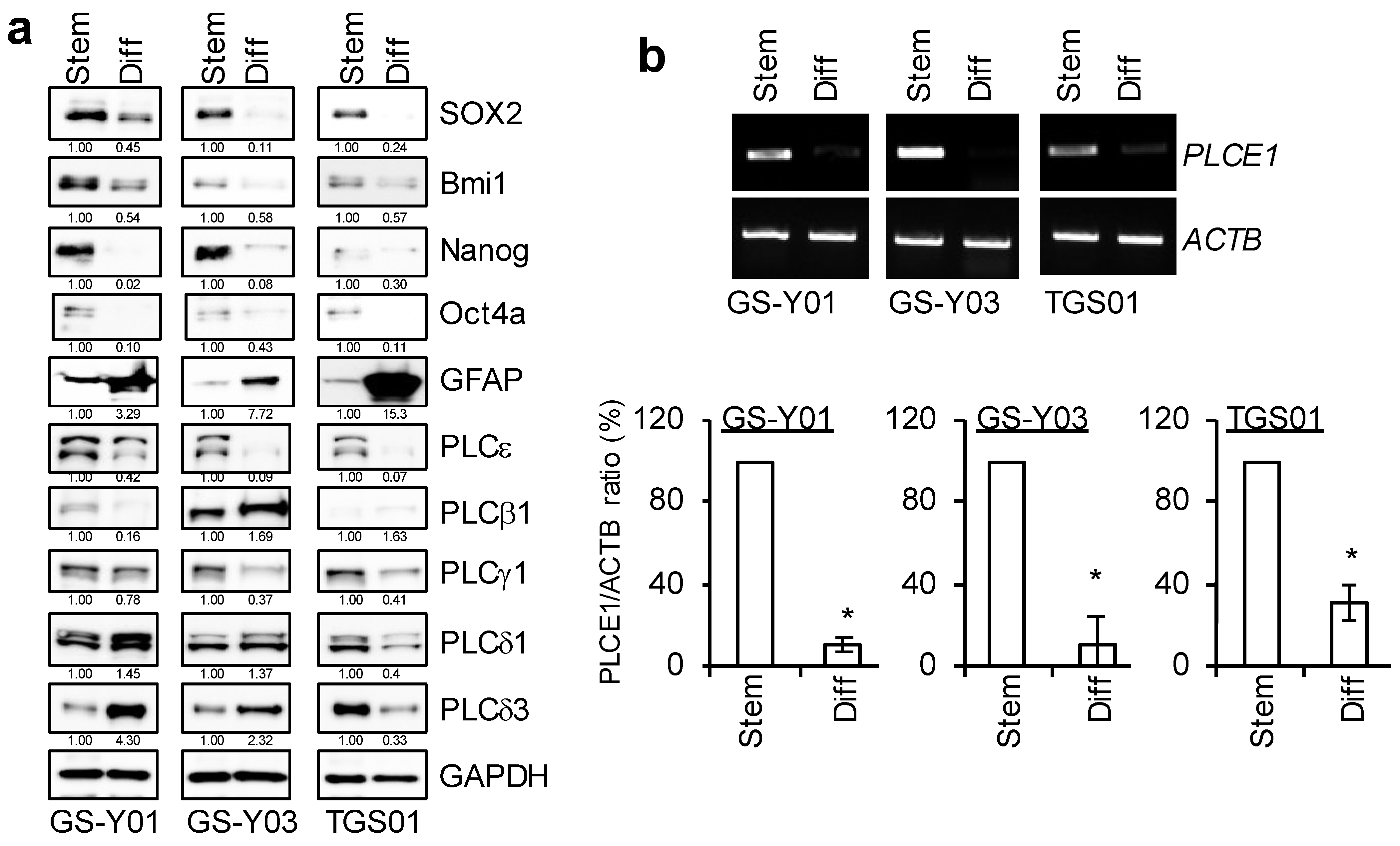
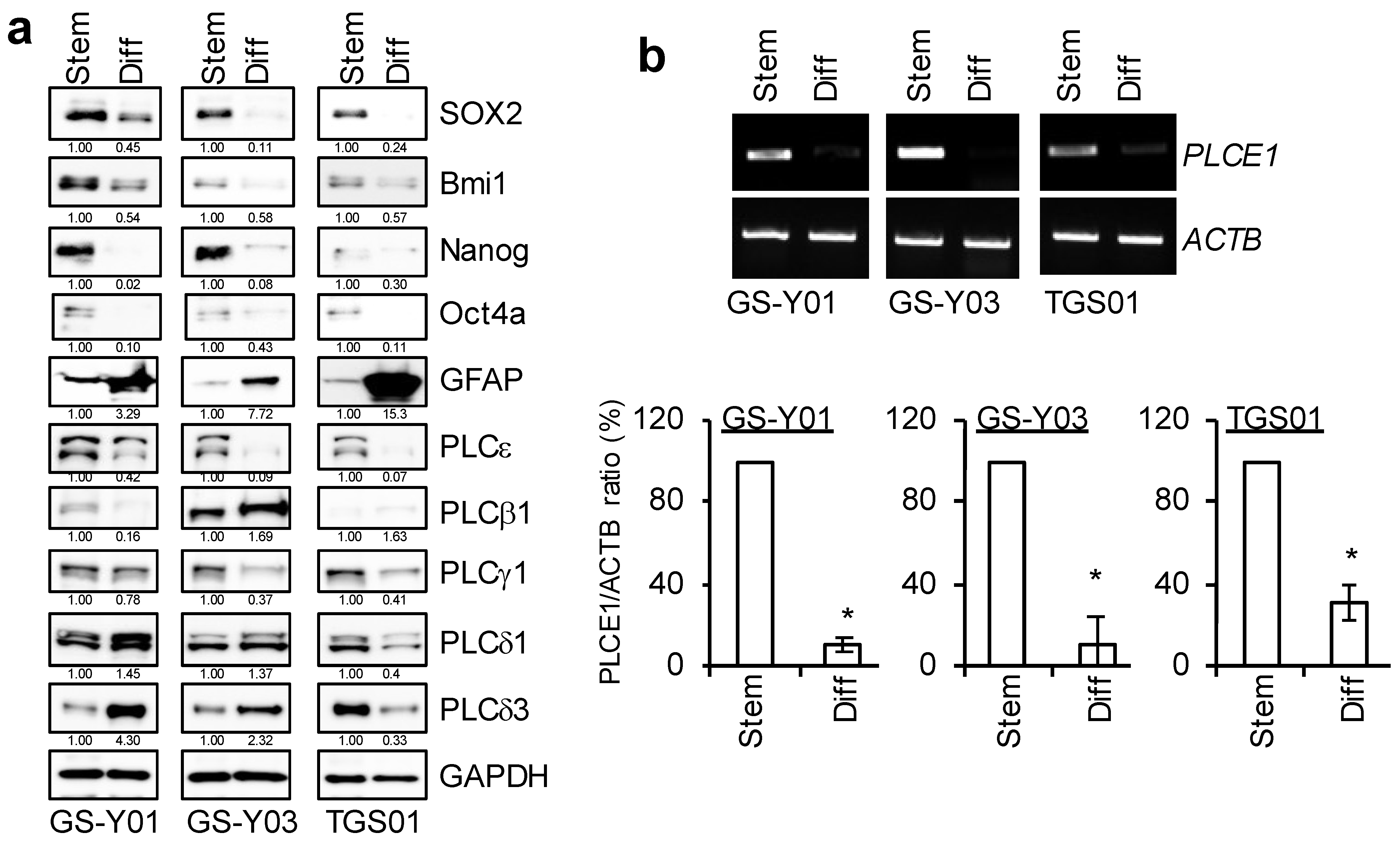
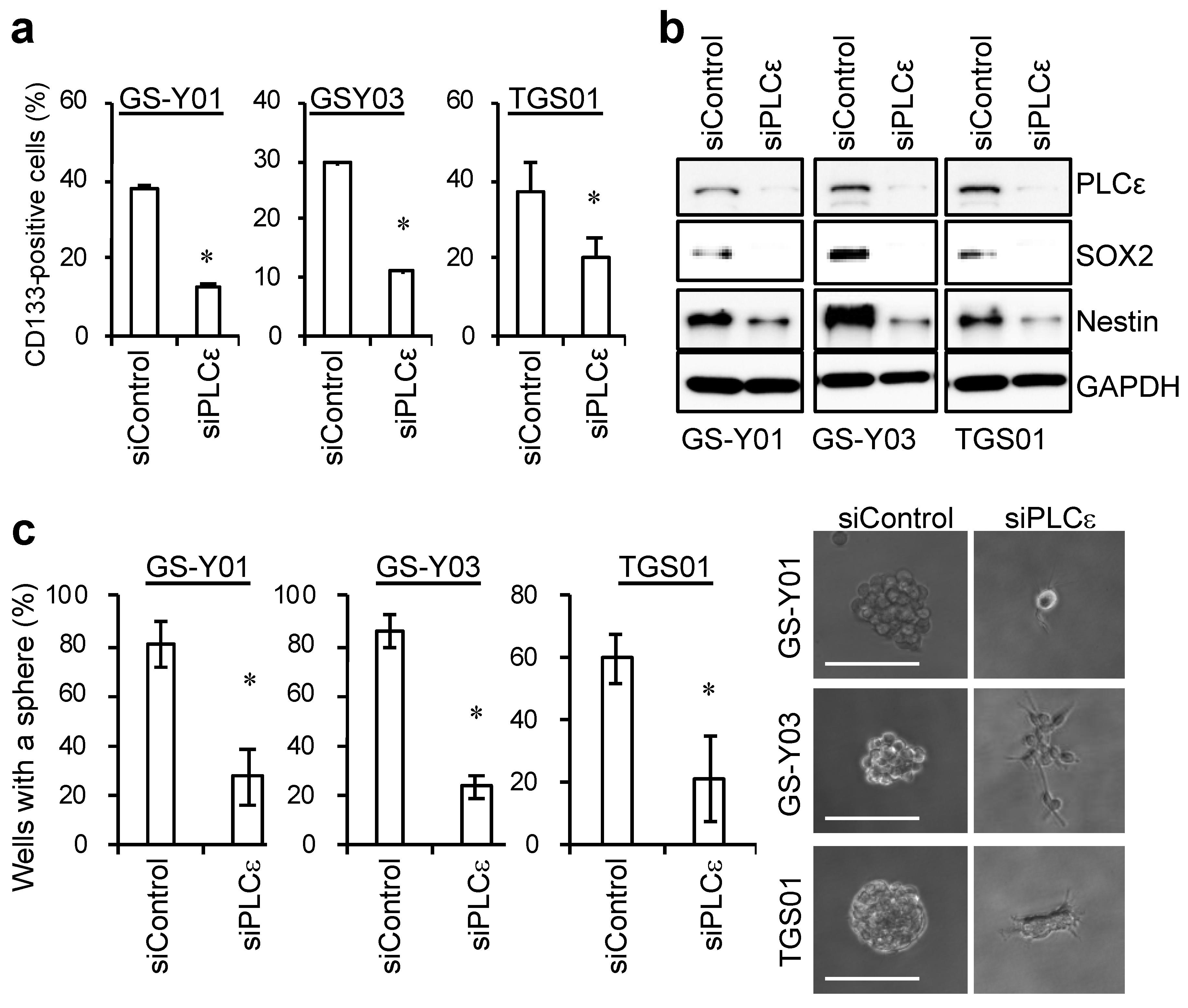
2.2. PLCε Contributes to the Maintenance of GSC Stemness
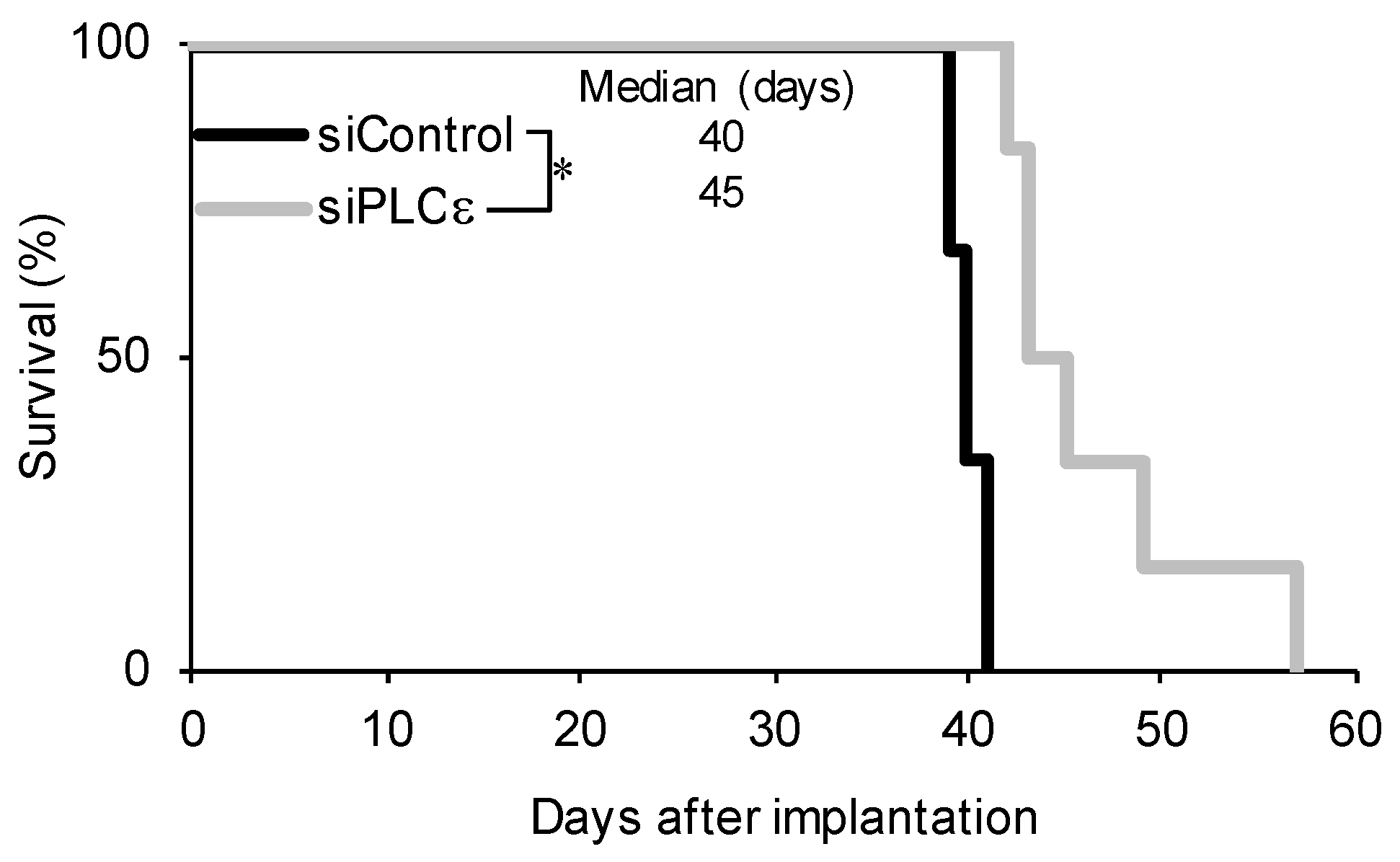
2.3. PLCε Contributes to the Tumor-Initiating Capacity of GSCs
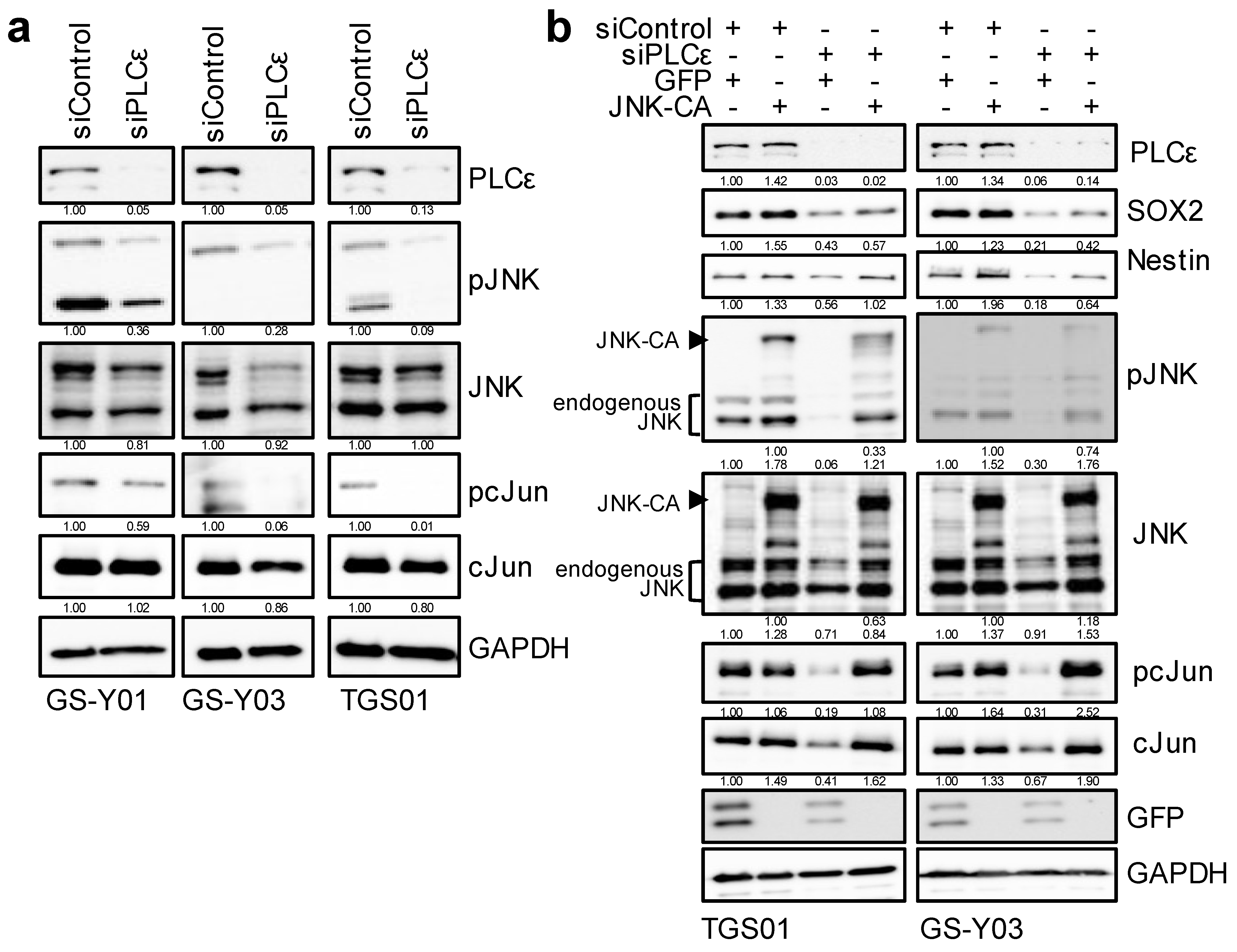
2.4. Down-Regulation of PLCε Suppresses the Stemness of GSCs by Inhibiting the JNK Axis
3. Discussion
4. Materials and Methods
4.1. Antibodies and Chemicals
4.2. Cell Culture
4.3. WST-8 Assay
4.4. Propidium Iodide (PI) Uptake Assay
4.5. Western Blotting
4.6. Flow Cytometric Analysis
4.7. Reverse Transcription–PCR Analysis
4.8. Sphere-Forming Analysis
4.9. Animal Study
4.10. Gene Silencing by siRNA
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matsuda, K.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, A.; Okada, M.; Shibuya, K.; Watanabe, E.; Seino, S.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; Kitanaka, C. Resveratrol promotes proteasome-dependent degradation of Nanog via p53 activation and induces differentiation of glioma stem cells. Stem Cell Res. 2013, 11, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef]
- Shibuya, K.; Okada, M.; Suzuki, S.; Seino, M.; Seino, S.; Takeda, H.; Kitanaka, C. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 2015, 6, 651–661. [Google Scholar] [CrossRef] [Green Version]
- Sunayama, J.; Sato, A.; Matsuda, K.; Tachibana, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; Kayama, T.; Tomiyama, A.; et al. Dual blocking of mTor and PI3K elicits a prodifferentiation effect on glioblastoma stem-like cells. Neuro-Oncology 2010, 12, 1205–1219. [Google Scholar] [CrossRef] [Green Version]
- Owusu Obeng, E.; Rusciano, I.; Marvi, M.V.; Fazio, A.; Ratti, S.; Follo, M.Y.; Xian, J.; Manzoli, L.; Billi, A.M.; Mongiorgi, S.; et al. Phosphoinositide-Dependent Signaling in Cancer: A Focus on Phospholipase C Isozymes. Int. J. Mol. Sci. 2020, 21, 2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratti, S.; Follo, M.Y.; Ramazzotti, G.; Faenza, I.; Fiume, R.; Suh, P.G.; McCubrey, J.A.; Manzoli, L.; Cocco, L. Nuclear phospholipase C isoenzyme imbalance leads to pathologies in brain, hematologic, neuromuscular, and fertility disorders. J. Lipid Res. 2019, 60, 312–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocco, L.; Follo, M.Y.; Manzoli, L.; Suh, P.G. Phosphoinositide-specific phospholipase C in health and disease. J. Lipid Res. 2015, 56, 1853–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusciano, I.; Marvi, M.V.; Owusu Obeng, E.; Mongiorgi, S.; Ramazzotti, G.; Follo, M.Y.; Zoli, M.; Morandi, L.; Asioli, S.; Fabbri, V.P.; et al. Location-dependent role of phospholipase C signaling in the brain: Physiology and pathology. Adv. Biol. Regul. 2021, 79, 100771. [Google Scholar] [CrossRef]
- Katan, M.; Cockcroft, S. Phospholipase C families: Common themes and versatility in physiology and pathology. Prog. Lipid Res. 2020, 80, 101065. [Google Scholar] [CrossRef]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C.; Okada, M. Verteporfin inhibits oxidative phosphorylation and induces cell death specifically in glioma stem cells. FEBS J. 2020, 287, 2023–2036. [Google Scholar] [CrossRef]
- Okada, M.; Suzuki, S.; Togashi, K.; Sugai, A.; Yamamoto, M.; Kitanaka, C. Targeting Folate Metabolism Is Selectively Cytotoxic to Glioma Stem Cells and Effectively Cooperates with Differentiation Therapy to Eliminate Tumor-Initiating Cells in Glioma Xenografts. Int. J. Mol. Sci. 2021, 22, 11633. [Google Scholar] [CrossRef]
- Sengelaub, C.A.; Navrazhina, K.; Ross, J.B.; Halberg, N.; Tavazoie, S.F. PTPRN2 and PLCβ1 promote metastatic breast cancer cell migration through PI(4,5)P2-dependent actin remodeling. EMBO J. 2016, 35, 62–76. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Johnson, M.D.; Todderud, G.; Coffey, R.J.; Carpenter, G.; Page, D.L. Elevated content of the tyrosine kinase substrate phospholipase C-gamma 1 in primary human breast carcinomas. Proc. Natl. Acad. Sci. USA 1991, 88, 10435–10439. [Google Scholar] [CrossRef] [Green Version]
- Nomoto, K.; Tomita, N.; Miyake, M.; Xhu, D.B.; LoGerfo, P.R.; Weinstein, I.B. Expression of phospholipases gamma 1, beta 1, and delta 1 in primary human colon carcinomas and colon carcinoma cell lines. Mol. Carcinog. 1995, 12, 146–152. [Google Scholar] [CrossRef]
- Koss, H.; Bunney, T.D.; Behjati, S.; Katan, M. Dysfunction of phospholipase Cγ in immune disorders and cancer. Trends Biochem. Sci. 2014, 39, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Qin, Y.R.; Xie, D.; Hu, L.; Kwong, D.L.; Srivastava, G.; Tsao, S.W.; Guan, X.Y. Characterization of a novel tumor-suppressor gene PLC delta 1 at 3p22 in esophageal squamous cell carcinoma. Cancer Res. 2007, 67, 10720–10726. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, W.; Zhang, T.; Ji, J.; Qian, Q.; Lu, L.; Fu, H.; Jin, W.; Cui, D. Differential expression of phospholipase C epsilon 1 is associated with chronic atrophic gastritis and gastric cancer. PLoS ONE 2012, 7, e47563. [Google Scholar] [CrossRef]
- Ratti, S.; Marvi, M.V.; Mongiorgi, S.; Obeng, E.O.; Rusciano, I.; Ramazzotti, G.; Morandi, L.; Asioli, S.; Zoli, M.; Mazzatenta, D.; et al. Impact of phospholipase C β1 in glioblastoma: A study on the main mechanisms of tumor aggressiveness. Cell. Mol. Life Sci. 2022, 79, 195. [Google Scholar] [CrossRef]
- Okada, M.; Kuramoto, K.; Takeda, H.; Watarai, H.; Sakaki, H.; Seino, S.; Seino, M.; Suzuki, S.; Kitanaka, C. The novel JNK inhibitor AS602801 inhibits cancer stem cells in vitro and in vivo. Oncotarget 2016, 7, 27021–27032. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Takeda, H.; Sakaki, H.; Kuramoto, K.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C. Repositioning CEP-1347, a chemical agent originally developed for the treatment of Parkinson’s disease, as an anti-cancer stem cell drug. Oncotarget 2017, 8, 94872–94882. [Google Scholar] [CrossRef] [Green Version]
- Bunney, T.D.; Katan, M. PLC regulation: Emerging pictures for molecular mechanisms. Trends Biochem. Sci. 2011, 36, 88–96. [Google Scholar] [CrossRef]
- Lopez, I.; Mak, E.C.; Ding, J.; Hamm, H.E.; Lomasney, J.W. A novel bifunctional phospholipase c that is regulated by Galpha 12 and stimulates the Ras/mitogen-activated protein kinase pathway. J. Biol. Chem. 2001, 276, 2758–2765. [Google Scholar] [CrossRef] [Green Version]
- Tyutyunnykova, A.; Telegeev, G.; Dubrovska, A. The controversial role of phospholipase C epsilon (PLCε) in cancer development and progression. J. Cancer 2017, 8, 716–729. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.B.; Li, S.; Li, T.T.; Peng, H.; Jin, T.T.; Zhang, S.M.; Liu, C.X.; Yang, L.; Shen, Y.Y.; Li, S.G.; et al. Targeting oncogenic PLCE1 by miR-145 impairs tumor proliferation and metastasis of esophageal squamous cell carcinoma. Oncotarget 2016, 7, 1777–1795. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Luo, C.; Wu, X.; Zhang, Y.; He, Y.; Wu, Q.; Xia, Y.; Zhang, J. shRNA targeting PLCε inhibits bladder cancer cell growth in vitro and in vivo. Urology 2011, 78, 474.e7–474.e11. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, L.; Zhao, Y.; Ou, L.; Wu, X.; Luo, C. Knockdown of phospholipase C-epsilon by short-hairpin RNA-mediated gene silencing induces apoptosis in human bladder cancer cell lines. Cancer Biother. Radiopharm. 2013, 28, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xin, H.; Peng, H.; Shi, Q.; Li, M.; Yu, J.; Tian, Y.; Han, X.; Chen, X.; Zheng, Y.; et al. Hypomethylation-Linked Activation of PLCE1 Impedes Autophagy and Promotes Tumorigenesis through MDM2-Mediated Ubiquitination and Destabilization of p53. Cancer Res. 2020, 80, 2175–2189. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Hu, J.; Huang, Z.; Yang, L.; Chen, M. MicroRNA-1976 functions as a tumor suppressor and serves as a prognostic indicator in non-small cell lung cancer by directly targeting PLCE1. Biochem. Biophys. Res. Commun. 2016, 473, 1144–1151. [Google Scholar] [CrossRef]
- Bai, Y.; Edamatsu, H.; Maeda, S.; Saito, H.; Suzuki, N.; Satoh, T.; Kataoka, T. Crucial role of phospholipase Cepsilon in chemical carcinogen-induced skin tumor development. Cancer Res. 2004, 64, 8808–8810. [Google Scholar] [CrossRef] [Green Version]
- Ikuta, S.; Edamatsu, H.; Li, M.; Hu, L.; Kataoka, T. Crucial role of phospholipase C epsilon in skin inflammation induced by tumor-promoting phorbol ester. Cancer Res. 2008, 68, 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, Y.; Edamatsu, H.; Kataoka, T. PLCε cooperates with the NF-κB pathway to augment TNFα-stimulated CCL2/MCP1 expression in human keratinocyte. Biochem. Biophys. Res. Commun. 2011, 414, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, N.; Edamatsu, H.; Suzuki, N.; Saito, H.; Inoue, Y.; Oka, M.; Hu, L.; Kataoka, T. Overexpression of phospholipase Cε in keratinocytes upregulates cytokine expression and causes dermatitis with acanthosis and T-cell infiltration. Eur. J. Immunol. 2011, 41, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Desbaillets, I.; Tada, M.; de Tribolet, N.; Diserens, A.C.; Hamou, M.F.; Van Meir, E.G. Human astrocytomas and glioblastomas express monocyte chemoattractant protein-1 (MCP-1) in vivo and in vitro. Int. J. Cancer 1994, 58, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.; da Hora, C.C.; Kantar, R.S.; Nakano, I.; Wakimoto, H.; Batchelor, T.T.; Chiocca, E.A.; Badr, C.E.; Tannous, B.A. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro-Oncology 2017, 19, 820–832. [Google Scholar] [CrossRef] [Green Version]
- Semba, T.; Sammons, R.; Wang, X.; Xie, X.; Dalby, K.N.; Ueno, N.T. JNK Signaling in Stem Cell Self-Renewal and Differentiation. Int. J. Mol. Sci. 2020, 21, 2613. [Google Scholar] [CrossRef] [Green Version]
- Tam, S.Y.; Law, H.K. JNK in Tumor Microenvironment: Present Findings and Challenges in Clinical Translation. Cancers 2021, 13, 2196. [Google Scholar] [CrossRef]
- de Los Reyes Corrales, T.; Losada-Pérez, M.; Casas-Tintó, S. JNK Pathway in CNS Pathologies. Int. J. Mol. Sci. 2021, 22, 3883. [Google Scholar] [CrossRef]
- Han, M.S.; Barrett, T.; Brehm, M.A.; Davis, R.J. Inflammation Mediated by JNK in Myeloid Cells Promotes the Development of Hepatitis and Hepatocellular Carcinoma. Cell Rep. 2016, 15, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Fan, Y.; Wang, X.; Niu, L.; Duan, L.; Yang, J.; Li, L.; Gao, Y.; Wu, X.; Luo, C. PLCε regulates prostate cancer mitochondrial oxidative metabolism and migration via upregulation of Twist1. J. Exp. Clin. Cancer Res. 2019, 38, 337. [Google Scholar] [CrossRef] [Green Version]
- Martins, M.; McCarthy, A.; Baxendale, R.; Guichard, S.; Magno, L.; Kessaris, N.; El-Bahrawy, M.; Yu, P.; Katan, M. Tumor suppressor role of phospholipase C epsilon in Ras-triggered cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 4239–4244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, S.; Kitanaka, C.; Noguchi, K.; Mochizuki, T.; Nagashima, Y.; Shirouzu, M.; Fujita, H.; Yoshida, M.; Chen, W.; Asai, A.; et al. Oncogenic Ras triggers cell suicide through the activation of a caspase-independent cell death program in human cancer cells. Oncogene 1999, 18, 2281–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gömöri, E.; Dóczi, T.; Pajor, L.; Matolcsy, A. Sporadic p53 mutations and absence of ras mutations in glioblastomas. Acta Neurochir. 1999, 141, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Khoshyomn, S.; Penar, P.L.; Rossi, J.; Wells, A.; Abramson, D.L.; Bhushan, A. Inhibition of phospholipase C-gamma1 activation blocks glioma cell motility and invasion of fetal rat brain aggregates. Neurosurgery 1999, 44, 568–577; discussion 577–568. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, H.; Howell, G.; Suzuki, S.; Zhang, Q.; Qi, Y.; Klein-Seetharaman, J.; Wells, A.; Grandis, J.R.; Thomas, S.M. Combined inhibition of PLCγ-1 and c-Src abrogates epidermal growth factor receptor-mediated head and neck squamous cell carcinoma invasion. Clin. Cancer Res. 2008, 14, 4336–4344. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.; Hadjiyanni, I.; Wei, H.C.; Polevoy, G.; McBride, R.; Sem, K.P.; Brill, J.A. PIP2 hydrolysis and calcium release are required for cytokinesis in Drosophila spermatocytes. Curr. Biol. 2005, 15, 1401–1406. [Google Scholar] [CrossRef] [Green Version]
- Naito, Y.; Okada, M.; Yagisawa, H. Phospholipase C isoforms are localized at the cleavage furrow during cytokinesis. J. Biochem. 2006, 140, 785–791. [Google Scholar] [CrossRef]
- Thapa, N.; Anderson, R.A. PIP2 signaling, an integrator of cell polarity and vesicle trafficking in directionally migrating cells. Cell Adh. Migr. 2012, 6, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.; Pentassuglia, L.; Sawyer, D.B. Emerging anticancer therapeutic targets and the cardiovascular system: Is there cause for concern? Circ. Res. 2010, 106, 1022–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, A.; Sunayama, J.; Matsuda, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Tachibana, K.; Tomiyama, A.; Kayama, T.; Kitanaka, C. MEK-ERK signaling dictates DNA-repair gene MGMT expression and temozolomide resistance of stem-like glioblastoma cells via the MDM2-p53 axis. Stem Cells 2011, 29, 1942–1951. [Google Scholar] [CrossRef]
- Sunayama, J.; Matsuda, K.; Sato, A.; Tachibana, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; Kayama, T.; Tomiyama, A.; et al. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells 2010, 28, 1930–1939. [Google Scholar] [CrossRef]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Miyazawa, K.; Miyazono, K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 2009, 5, 504–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Sato, A.; Shibuya, K.; Watanabe, E.; Seino, S.; Suzuki, S.; Seino, M.; Narita, Y.; Shibui, S.; Kayama, T.; et al. JNK contributes to temozolomide resistance of stem-like glioblastoma cells via regulation of MGMT expression. Int. J. Oncol. 2014, 44, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Okada, M.; Sanomachi, T.; Togashi, K.; Seino, S.; Sato, A.; Yamamoto, M.; Kitanaka, C. Therapeutic targeting of pancreatic cancer stem cells by dexamethasone modulation of the MKP-1-JNK axis. J. Biol. Chem. 2020, 295, 18328–18342. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Togashi, K.; Sanomachi, T.; Kitanaka, C.; Okada, M. Inhibition of the Lipid Droplet-Peroxisome Proliferator-Activated Receptor α Axis Suppresses Cancer Stem Cell Properties. Genes 2021, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Proserpio, C.; Galardi, S.; Desimio, M.G.; Michienzi, A.; Doria, M.; Minutolo, A.; Matteucci, C.; Ciafrè, S.A. MEOX2 Regulates the Growth and Survival of Glioblastoma Stem Cells by Modulating Genes of the Glycolytic Pathway and Response to Hypoxia. Cancers 2022, 14, 2304. [Google Scholar] [CrossRef] [PubMed]
- Hägerstrand, D.; He, X.; Bradic Lindh, M.; Hoefs, S.; Hesselager, G.; Ostman, A.; Nistér, M. Identification of a SOX2-dependent subset of tumor- and sphere-forming glioblastoma cells with a distinct tyrosine kinase inhibitor sensitivity profile. Neuro-Oncology 2011, 13, 1178–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okada, M.; Nakagawa-Saito, Y.; Mitobe, Y.; Sugai, A.; Togashi, K.; Suzuki, S.; Kitanaka, C. Inhibition of the Phospholipase Cε–c-Jun N-Terminal Kinase Axis Suppresses Glioma Stem Cell Properties. Int. J. Mol. Sci. 2022, 23, 8785. https://doi.org/10.3390/ijms23158785
Okada M, Nakagawa-Saito Y, Mitobe Y, Sugai A, Togashi K, Suzuki S, Kitanaka C. Inhibition of the Phospholipase Cε–c-Jun N-Terminal Kinase Axis Suppresses Glioma Stem Cell Properties. International Journal of Molecular Sciences. 2022; 23(15):8785. https://doi.org/10.3390/ijms23158785
Chicago/Turabian StyleOkada, Masashi, Yurika Nakagawa-Saito, Yuta Mitobe, Asuka Sugai, Keita Togashi, Shuhei Suzuki, and Chifumi Kitanaka. 2022. "Inhibition of the Phospholipase Cε–c-Jun N-Terminal Kinase Axis Suppresses Glioma Stem Cell Properties" International Journal of Molecular Sciences 23, no. 15: 8785. https://doi.org/10.3390/ijms23158785
APA StyleOkada, M., Nakagawa-Saito, Y., Mitobe, Y., Sugai, A., Togashi, K., Suzuki, S., & Kitanaka, C. (2022). Inhibition of the Phospholipase Cε–c-Jun N-Terminal Kinase Axis Suppresses Glioma Stem Cell Properties. International Journal of Molecular Sciences, 23(15), 8785. https://doi.org/10.3390/ijms23158785