
Activation Markers on B and T Cells and Immune Checkpoints in Autoimmune Rheumatic Diseases


Abstract
1. Introduction
2. B- and T-Cell Activation in Autoimmune Rheumatic Diseases
3. Prognostic Role of Surface Molecules on B and T Cells in Autoimmune Rheumatic Diseases
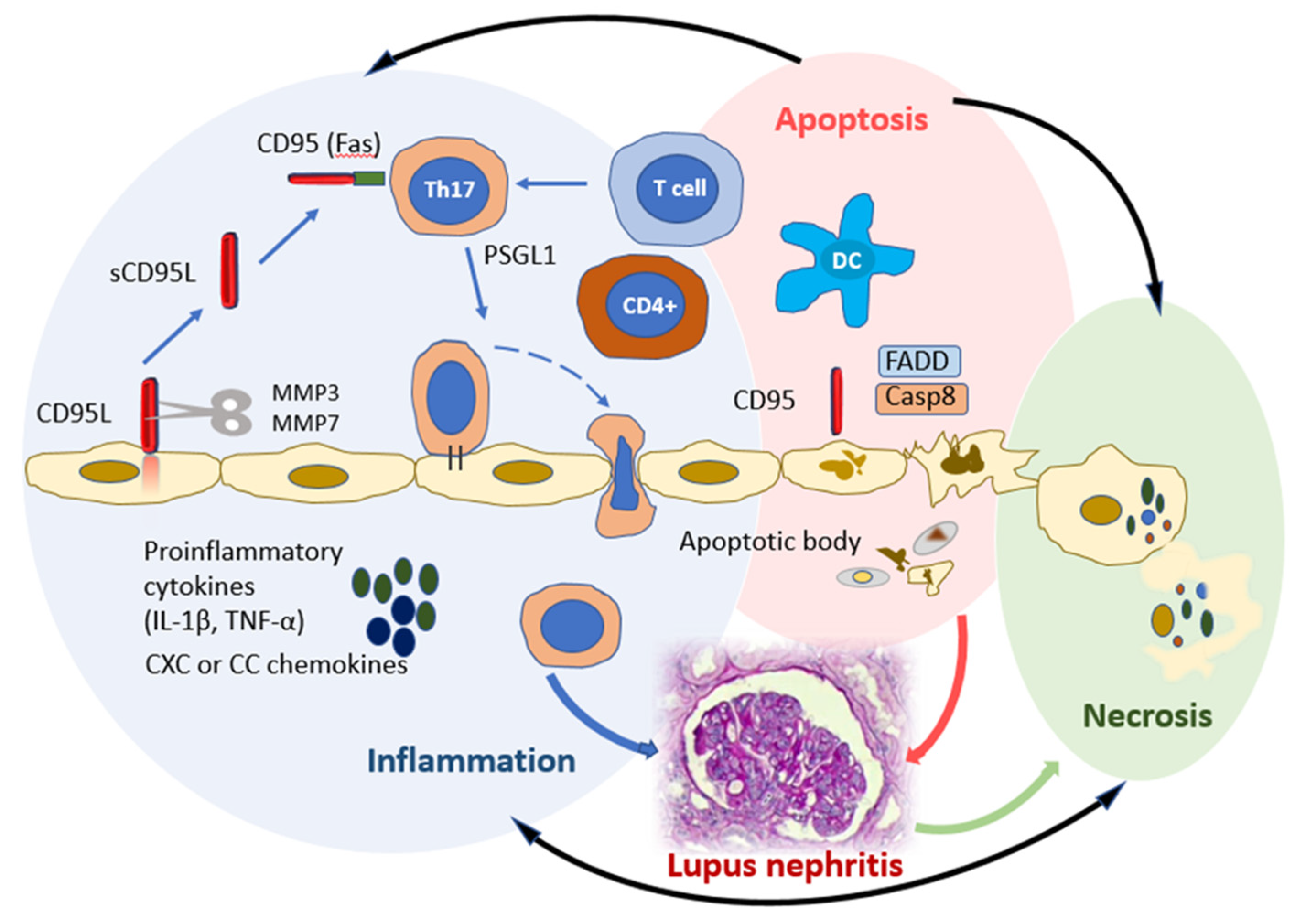
3.1. CD95
3.2. HLA-DR
3.3. CD25
4. Immune Checkpoints and Autoimmune Rheumatic Diseases
5. Immune Checkpoints Are Promising Targets for Targeted Immunotherapy of Autoimmune Rheumatic Diseases
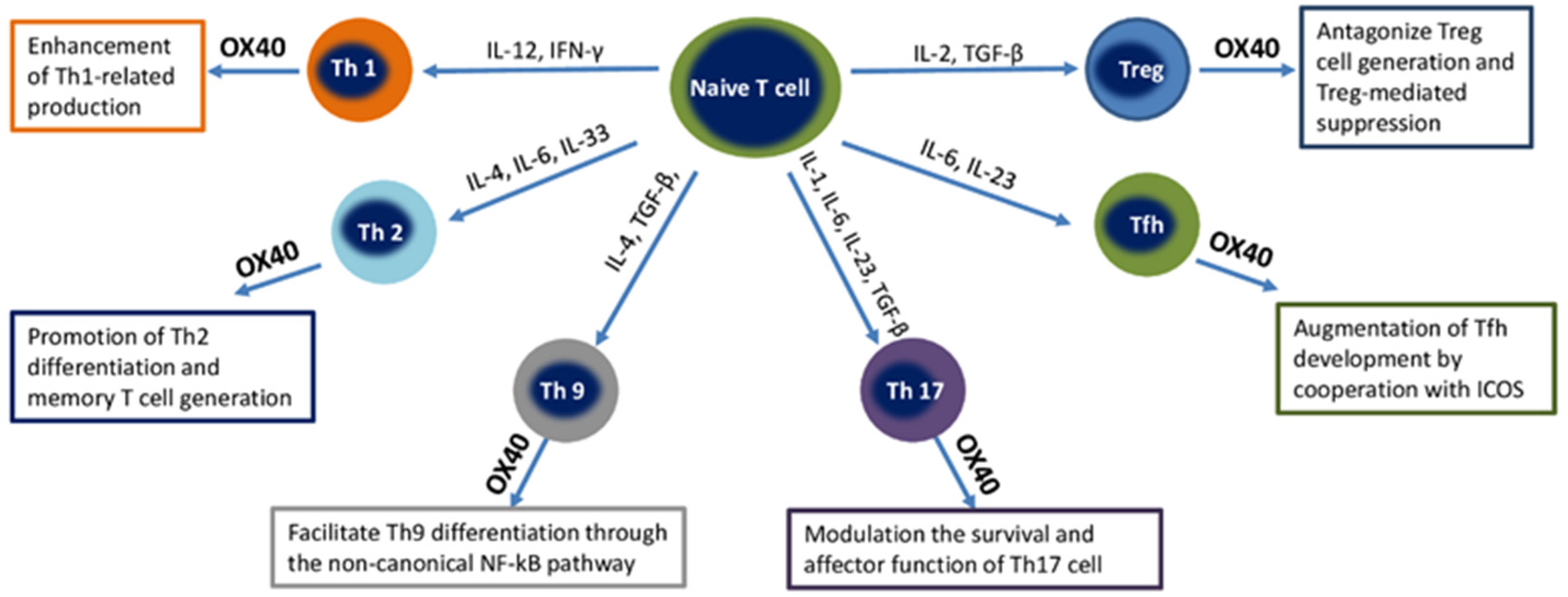
5.1. OX40
5.2. GITR
5.3. ICOS
5.4. PD-1
5.5. CTLA-4
5.6. CD40
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APCs | Antigen-presenting cells |
| ARDs | Autoimmune rheumatic diseases |
| CIA | Collagen-induced arthritis |
| CRP | C-reactive protein |
| DCs | Dendritic cells |
| DM | Diabetes mellitus |
| FADD | Fas-associated death domain |
| ICPs | Immune checkpoints |
| Ig | Immunoglobulin |
| IL | Interleukin |
| MAP | Mitogen-activated protein kinase |
| MHC | Major histocompatibility complex |
| MMP | Metalloproteases |
| NFAT | Nuclear factor of activated T cells |
| NF-κB | Nuclear factor kappa B |
| NK cells | Natural killer cells |
| NKT cells | Natural killer T cells |
| RA | Rheumatoid arthritis |
| RF | Rheumatoid factor |
| SLE | Systemic lupus erythematosus |
| TCR | T-cell receptor |
| Tfh | T follicular helper cells |
| TGF-β | Transforming growth factor beta |
| Th | T helper |
| TLR | Toll-like receptor |
| TNF | Tumoral necrosis factor |
| Tregs | T regulatory cells |
References
- Serra, P.; Santamaria, P. Antigen-specific therapeutic approaches for autoimmunity. Nat. Biotechnol. 2019, 37, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Selmi, C. Autoimmunity in 2018. Clin. Rev. Allergy Immunol. 2019, 56, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.X.; Miller, J.S.; Zheng, S.G. An updated advance of autoantibodies in autoimmune diseases. Autoimmun. Rev. 2021, 20, 102743. [Google Scholar] [CrossRef] [PubMed]
- Edner, N.M.; Carlesso, G.; Rush, J.S.; Walker, L.S.K. Targeting co-stimulatory molecules in autoimmune disease. Nat. Rev. Drug Discov. 2020, 19, 860–883. [Google Scholar] [CrossRef] [PubMed]
- Bachelet, I.; Levi-Schaffer, F. Mast cells as effector cells: A co-stimulating question. Trends Immunol. 2007, 28, 360–365. [Google Scholar] [CrossRef]
- Tak, P.P.; Doorenspleet, M.E.; de Hair, M.J.H.; Klarenbeek, P.L.; van Beers-Tas, M.E.; van Kampen, A.H.C.; van Schaardenburg, D.; Gerlag, D.M.; Baas, F.; de Vries, N. Dominant B cell receptor clones in peripheral blood predict onset of arthritis in individuals at risk for rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 1924–1930. [Google Scholar] [CrossRef]
- Tu, J.; Huang, W.; Zhang, W.; Mei, J.; Zhu, C. A Tale of Two Immune Cells in Rheumatoid Arthritis: The crosstalk between macrophages and T cells in the synovium. Front. Immunol. 2021, 12, 655477. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. The immunology of rheumatoid arthritis. Nat. Immunol. 2021, 22, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Bader, L.; Gullaksen, S.E.; Blaser, N.; Brun, M.; Bringeland, G.H.; Sulen, A.; Gjesdal, G.; Vedeler, C.; Gavasso, S. Candidate Markers for Stratification and Classification in Rheumatoid Arthritis. Front. Immunol. 2019, 10, 1488. [Google Scholar] [CrossRef]
- Coit, P.; Dozmorov, M.G.; Merrill, J.T.; McCune, W.J.; Maksimowicz-McKinnon, K.; Wren, J.D.; Sawalha, A.H. Epigenetic Reprogramming in Naive CD4 + T Cells Favoring T Cell Activation and Non-Th1 Effector T Cell Immune Response as an Early Event in Lupus Flares. Arthritis Rheumatol. 2016, 68, 2200–2209. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Yang, G.; Liu, Q.; Wang, S.; Cui, D. Function and Role of Regulatory T Cells in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 626193. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Benoist, C.; Bluestone, J.A.; Campbell, D.J.; Ghosh, S.; Hori, S.; Jiang, S.; Kuchroo, V.K.; Mathis, D.; Roncarolo, M.G.; et al. Regulatory T cells: Recommendations to simplify the nomenclature. Nat. Immunol. 2013, 14, 300–308. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Zhuang, L.; Xu, C.; Li, T.; Zhang, G.; Liu, Y. Decreased regulatory T-cell frequency and interleukin-35 levels in patients with rheumatoid arthritis. Exp. Ther. Med. 2018, 16, 5366–5372. [Google Scholar] [CrossRef]
- Rosenblum, M.D.; Way, S.S.; Abbas, A.K. Regulatory T cell memory. Nat. Rev. Immunol. 2016, 16, 90–101. [Google Scholar] [CrossRef]
- Hammaker, D.; Firestein, G.S. Epigenetics of inflammatory arthritis. Curr. Opin. Rheumatol. 2018, 30, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Adlowitz, D.G.; Barnard, J.; Biear, J.N.; Cistrone, C.; Owen, T.; Wang, W.; Palanichamy, A.; Ezealah, E.; Campbell, D.; Chungwen, W.; et al. Expansion of Activated Peripheral Blood Memory B Cells in Rheumatoid Arthritis, Impact of B Cell Depletion Therapy, and Biomarkers of Response. PLoS ONE 2015, 10, e0128269. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, A.M.; Mei, H.; Hoyer, B.F.; Mumtaz, I.M.; Thiele, K.; Radbruch, A.; Burmester, G.-R.; Hiepe, F.; Dörner, T. HLA-DRhigh/CD27high plasmablasts indicate active disease in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2010, 69, 305–308. [Google Scholar] [CrossRef]
- Flores-Mendoza, G.; Rodríguez-Rodríguez, N.; Rubio, R.M.; Madera-Salcedo, I.K.; Rosetti, F.; Crispín, J.C. Fas/FasL Signaling Regulates CD8 Expression During Exposure to Self-Antigens. Front. Immunol. 2021, 12, 635862. [Google Scholar] [CrossRef] [PubMed]
- Matson, D.R.; Yang, D.T. Autoimmune Lymphoproliferative Syndrome: An Overview. Arch. Pathol. Lab. Med. 2020, 144, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Galli, G.; Vacher, P.; Ryffel, B.; Blanco, P.; Legembre, P. Fas/CD95 Signaling Pathway in Damage-Associated Molecular Pattern (DAMP)-Sensing Receptors. Cells 2022, 11, 1438. [Google Scholar] [CrossRef]
- Guégan, J.P.; Legembre, P. Nonapoptotic functions of Fas/CD95 in the immune response. FEBS J. 2018, 285, 809–827. [Google Scholar] [CrossRef]
- Han, M.; Hu, R.; Ma, J.; Zhang, B.; Chen, C.; Li, H.; Yang, J.; Huang, G. Fas signaling in Dendritic Cells mediates Th2 polarization in HDM-induced allergic pulmonary inflammation. Front. Immunol. 2018, 9, 3045. [Google Scholar] [CrossRef] [PubMed]
- Mabrouk, I.; Buart, S.; Hasmim, M.; Michiels, C.; Connault, E.; Opolon, P.; Chiocchia, G.; Lévi-Strauss, M.; Chouaib, S.; Karrayet, S. Prevention of autoimmunity and control of recall response to exogenous antigen by Fas death receptor ligand expression on T cells. Immunity 2008, 29, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Poissonnier, A.; Guégan, J.P.; Nguyen, H.T.; Best, D.; Levoin, N.; Kozlov, G.; Gehring, K.; Pineau, R.; Jouan, F.; Morere, L.; et al. Disrupting the CD95-PLCγ1 interaction prevents Th17-driven inflammation. Nat. Chem. Biol. 2018, 14, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Zu Horste, M.G.; Przybylski, D.; Schramm, M.A.; Wang, C.; Schnell, A.; Lee, Y.; Sobel, R.; Regev, A.; Kuchroo, V.K. Fas Promotes T helper 17 cell differentiation and inhibits T helper 1 Cell development by binding and Sequestering Transcription Factor STAT1. Immunity 2018, 48, 556–569.e7. [Google Scholar] [CrossRef]
- Fleischer, S.J.; Giesecke, C.; Mei, H.E.; Lipsky, P.E.; Daridon, C.; Dörner, T. Increased frequency of a unique spleen tyrosine kinase bright memory B cell population in systemic lupus erythematosus. Arthritis Rheumatol. 2014, 66, 3424–3435. [Google Scholar] [CrossRef]
- Jacobi, A.M.; Reiter, K.; Mackay, M.; Aranow, C.; Hiepe, F.; Radbruch, A.; Hansen, A.; Burmester, R.-G.; Diamond, F.; Lipsky, P.E.; et al. Activated memory B cell subsets correlate with disease activity in systemic lupus erythematosus: Delineation by expression of CD27, IgD, and CD95. Arthritis Rheum. 2008, 58, 1762–1773. [Google Scholar] [CrossRef]
- Zazzeroni, F.; Papa, S.; Algeciras-Schimnich, A.; Alvarez, K.; Melis, T.; Bubici, C.; Majewski, N.; Hay, N.; De Smaele, E.; Peter, M.E.; et al. Gadd45 beta mediates the protective effects of CD40 costimulation against Fas-induced apoptosis. Blood 2003, 102, 3270–3279. [Google Scholar] [CrossRef]
- Chodorge, M.; Züger, S.; Stirnimann, C.; Briand, C.; Jermutus, L.; Grütter, M.G.; Minter, R.R. A series of Fas receptor agonist antibodies that demonstrate an inverse correlation between affinity and potency. Cell Death Differ. 2012, 19, 1187–1195. [Google Scholar] [CrossRef]
- Clauder, A.K.; Kordowski, A.; Bartsch, Y.C.; Köhl, G.; Lilienthal, G.M.; Almeida, L.N.; Lindemann, T.; Petry, J.; Rau, C.N.; Gramalla-Schmitz, A.; et al. IgG Fc N-Glycosylation Translates MHCII Haplotype into Autoimmune Skin Disease. J. Investig. Dermatol. 2021, 141, 285–294. [Google Scholar] [CrossRef]
- Viallard, J.F.; Bloch-Michel, C.; Neau-Cransac, M.; Taupin, J.L.; Garrigue, S.; Miossec, V.; Mercie, P.; Pellegrin, J.L.; Moreau, F. HLA-DR expression on lymphocyte subsets as a marker of disease activity in patients with systemic lupus erythematosus. Clin. Exp. Immunol. 2001, 125, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Daca, A.; Czuszyńska, Z.; Smoleńska, Z.; Zdrojewski, Z.; Witkowski, J.M.; Bryl, E. Two systemic lupus erythematosus (SLE) global disease activity indexes--the SLE Disease Activity Index and the Systemic Lupus Activity Measure--demonstrate different correlations with activation of peripheral blood CD4 + T cells. Hum. Immunol. 2011, 72, 1160–1167. [Google Scholar] [CrossRef]
- Zhou, H.; Li, B.; Li, J.; Wu, T.; Jin, X.; Yuan, R.; Shi, P.; Zhou, Y.; Li, L.; Yu, F. Dysregulated T Cell activation and aberrant cytokine expression profile in Systemic Lupus Erythematosus. Mediat. Inflamm. 2019, 2019, 8450947. [Google Scholar] [CrossRef]
- Perry, D.J.; Titov, A.A.; Sobel, E.S.; Brusko, T.M.; Morel, L. Immunophenotyping reveals distinct subgroups of lupus patients based on their activated T cell subsets. Clin. Immunol. 2020, 221, 108602. [Google Scholar] [CrossRef] [PubMed]
- Niu, Q.; Huang, Z.C.; Cai, B.; Wang, L.L.; Feng, W.H. Analysis of frequency of peripheral blood CD4+; CD25(high);Tregs and CD4+; CD25(low);T cells and expression of PD-1 in SLE and RA patients. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2011, 27, 23–25. [Google Scholar] [PubMed]
- Zhu, Y.; Huang, Y.; Ming, B.; Wu, X.; Chen, Y.; Dong, L. Regulatory T-cell levels in systemic lupus erythematosus patients: A meta-analysis. Lupus 2019, 28, 445–454. [Google Scholar] [CrossRef]
- Sonawale, A.; Bohara, V.; Bichile, L.S. Evaluation of the Association between CD4, CD8 and CD25 Cell Counts and SLE in Active Disease and in Remission. J. Assoc. Physicians India 2017, 65, 37–42. [Google Scholar]
- Darlan, D.M.; Munir, D.; Putra, A.; Jusuf, N.K. MSCs-released TGFβ1 generate CD4+CD25+Foxp3+ in T-reg cells of human SLE PBMC. J. Formos Med. Assoc. 2021, 120, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-related adverse events associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Lidar, M.; Giat, E.; Garelick, D.; Horowitz, Y.; Amital, H.; Steinberg-Silman, Y.; Schachter, J.; Shapira-Frommer, R.; Markel, G. Rheumatic manifestations among cancer patients treated with immune checkpoint inhibitors. Autoimmun. Rev. 2018, 17, 284–289. [Google Scholar] [CrossRef]
- Zhai, Y.; Moosavi, R.; Chen, M. Immune Checkpoints, a Novel Class of Therapeutic Targets for Autoimmune Diseases. Front. Immunol. 2021, 12, 645699. [Google Scholar] [CrossRef]
- Farres, M.N.; Al-Zifzaf, D.S.; Aly, A.A.; Abd Raboh, N.M. OX40/OX40L in systemic lupus erythematosus: Association with disease activity and lupus nephritis. Ann. Saudi Med. 2011, 31, 29–34. [Google Scholar] [CrossRef][Green Version]
- Webb, G.J.; Hirschfield, G.M.; Lane, P.J. OX40, OX40L and Autoimmunity: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 50, 312–332. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, C.; Schmitt, N.; Contin-Bordes, C.; Liu, Y.; Narayanan, P.; Seneschal, J.; Maurouard, T.; Dougall, D.; Davizon, E.S.; Dumortier, H. OX40 Ligand Contributes to Human Lupus Pathogenesis by Promoting T Follicular Helper Response. Immunity 2015, 42, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Kshirsagar, S.; Binder, E.; Riedl, M.; Wechselberger, G.; Steichen, E.; Edelbauer, M. Enhanced activity of Akt in Teff cells from children with lupus nephritis is associated with reduced induction of tumor necrosis factor receptor-associated factor 6 and increased OX40 expression. Arthritis Rheum. 2013, 65, 2996–3006. [Google Scholar] [CrossRef] [PubMed]
- Börnsen, L.; Christensen, J.R.; Ratzer, R.; Oturai, A.B.; Sørensen, P.S.; Søndergaard, H.B.; Sellebjerget, F. Effect of natalizumab on circulating CD4+ T-cells in multiple sclerosis. PLoS ONE 2012, 7, e47578. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, C.; Augusto, J.F.; Scherlinger, M.; Gensous, N.; Forcade, E.; Douchet, I.; Levionnois, E.; Richez, C.; Lazaro, E.; Duffau, P.; et al. OX40L/OX40 axis impairs follicular and natural Treg function in human SLE. JCI Insight 2018, 3, e122167. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, C.; Liu, M.; Shen, Y.; Hu, X.; Wang, Q.; Wu, J.; Wu, M.; Fang, Q.; Zhang, X. OX40 signaling is involved in the autoactivation of CD4 + CD28- T cells and contributes to the pathogenesis of autoimmune arthritis. Arthritis Res. Ther. 2017, 19, 67. [Google Scholar] [CrossRef]
- Wang, R.; Gao, C.; Raymond, M.; Dito, G.; Kabbabe, D.; Shao, X.; Hilt, E.; Sun, Y.; Pak, I.; Gutierrez, M.; et al. An Integrative Approach to Inform Optimal Administration of OX40 Agonist Antibodies in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 6709–6720. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Pavel, A.B.; Zhou, L.; Estrada, Y.D.; Zhang, N.; Xu, H.; Peng, X.; Wen, H.C.; Govas, P.; Gudi, G.; et al. GBR 830, an anti-OX40, improves skin gene signatures and clinical scores in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 144, 482–493.e7. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chau, B.; West, S.M.; Kimberlin, C.R.; Cao, F.; Schwarz, F.; Aguilar, B.; Han, M.; Morishige, W.; Bee, C.; et al. Structures of mouse and human GITR-GITRL complexes reveal unique TNF superfamily interactions. Nat. Commun. 2021, 12, 1378. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.; Kim, W.J.; Kang, Y.M.; Suk, K.; Koh, E.-M.; Cha, H.-S.; Ahn, K.-S.; Huh, T.-L.; Lee, W.-H. Glucocorticoid-induced tumour necrosis factor receptor-related protein-mediated macrophage stimulation may induce cellular adhesion and cytokine expression in rheumatoid arthritis. Clin. Exp. Immunol. 2007, 148, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Yu, N.; Li, X.; Wang, L.; Pan, Y.; Li, X.; Tao, J.; Chen, C.; Wang, G. Aberrant GITR expression on different T cell subsets and the regulation by glucocorticoid in systemic lupus erythematosus. Int. J. Rheum. Dis. 2016, 19, 199–204. [Google Scholar] [CrossRef]
- Lee, J.H.; Wang, L.C.; Lin, Y.T.; Yang, Y.H.; Lin, D.T.; Chiang, B.L. Inverse correlation between CD4+ regulatory T-cell population and autoantibody levels in paediatric patients with systemic lupus erythematosus. Immunology 2006, 117, 280–286. [Google Scholar] [CrossRef]
- Nocentini, G.; Alunno, A.; Petrillo, M.G.; Bistoni, O.; Bartoloni, E.; Caterbi, S.; Ronchetti, S.; Migliorati, G.; Riccardi, C.; Gerli, R. Expansion of regulatory GITR + CD25 low/-CD4 + T cells in systemic lupus erythematosus patients. Arthritis Res. Ther. 2014, 16, 444. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Zhang, B.; Rui, K.; Wang, S. The Role of GITR/GITRL Interaction in Autoimmune Diseases. Front. Immunol. 2020, 11, 588682. [Google Scholar] [CrossRef]
- Hilaire, M.; Aubert, N. La stimulation des lymphocytes Treg via le TNFR2 et GITR comme nouvelle approche thérapeutique dans les maladies auto-immunes [Boosting Treg activity by TNFR2 and GITR agonists: New therapeutic approaches for autoimmune diseases]. Med. Sci. 2019, 35, 702–705. (In French) [Google Scholar] [CrossRef]
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczeket, R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999, 397, 263–266. [Google Scholar] [CrossRef]
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J. Neurosci. Res. 2006, 84, 370–378. [Google Scholar] [CrossRef]
- Kuol, N.; Stojanovska, L.; Nurgali, K.; Apostolopoulos, V. PD-1/PD-L1 in disease. Immunotherapy 2018, 10, 149–160. [Google Scholar] [CrossRef]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Ledbetter, J.A.; Shu, G.; Gallagher, M.; Clark, E.A. Augmentation of normal and malignant B cell proliferation by monoclonal antibody to the B cell-specific antigen BP50 (CDW40). J. Immunol. 1987, 138, 788–794. [Google Scholar] [PubMed]
- Madissoon, E.; Wilbrey-Clark, A.; Miragaia, R.J.; Saeb-Parsy, K.; Mahbubani, K.; Georgakopoulos, N.; Harding, P.; Polanski, K.; Huang, N.; Nowicki-Osuch, K.; et al. scRNA-seq assessment of the human lung, spleen, and esophagus tissue stability after cold preservation. Genome Biol. 2019, 21, 1. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Cheng, X.; Truong, B.; Sun, L.; Yang, X.; Wang, H. Molecular basis and therapeutic implications of CD40/CD40L immune checkpoint. Pharmacol. Ther. 2021, 219, 107709. [Google Scholar] [CrossRef]
- Lu, X. OX40 and OX40L Interaction in Cancer. Curr. Med. Chem. 2021, 28, 5659–5673. [Google Scholar] [CrossRef]
- Willoughby, J.; Griffiths, J.; Tews, I.; Cragg, M.S. OX40: Structure and function—What questions remain? Mol. Immunol. 2017, 83, 13–22. [Google Scholar] [CrossRef]
- Kumari, R.; Chakraborty, S.; Jain, R.; Mitra, S.; Mohan, A.; Guleria, R.; Pandey, S.; Chaudhury, U.; Mitra, D.K. Inhibiting OX40 restores regulatory T-cell function and suppresses inflammation in pulmonary sarcoidosis. Chest 2021, 160, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Lin, Q.; Zhang, Z.; Zhang, L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm. Sin. B 2020, 10, 414–433. [Google Scholar] [CrossRef]
- Cui, D.; Lv, Y.; Yuan, X.; Ruan, G.; Zhang, Y.; Yan, C.; Xu, D.; Lv, M.; Mao, Y.; Cao, J.; et al. Increased Expressions of OX40 and OX40 Ligand in Patients with Primary Immune Thrombocytopenia. J. Immunol. Res. 2019, 2019, 6804806. [Google Scholar] [CrossRef]
- Cunninghame Graham, D.S.; Graham, R.R.; Manku, H.; Wong, A.K.; Whittaker, J.C.; Gaffney, P.M.; Moser, K.L.; Rioux, J.D.; Altshuler, D.; Behrens, T.W.; et al. Polymorphism at the TNF superfamily gene TNFSF4 confers susceptibility to systemic lupus erythematosus. Nat. Genet. 2008, 40, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Zhu, P. Functional niche of inflamed synovium for Th17-cell expansion and activation in rheumatoid arthritis: Implication to clinical therapeutics. Autoimmun. Rev. 2012, 11, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Bhattacharya, P.; Prabhakar, B.S. A comprehensive review on the role of co-signaling receptors and Treg homeostasis in autoimmunity and tumor immunity. J. Autoimmun. 2018, 95, 77–99. [Google Scholar] [CrossRef]
- Riccardi, C.; Ronchetti, S.; Nocentini, G. Glucocorticoid-induced TNFR-related gene (GITR) as a therapeutic target for immunotherapy. Expert Opin. Ther. Targets 2018, 22, 783–797. [Google Scholar] [CrossRef]
- Ronchetti, S.; Nocentini, G.; Bianchini, R.; Krausz, L.T.; Migliorati, G.; Riccardi, C. Glucocorticoid-induced TNFR-related protein lowers the threshold of CD28 costimulation in CD8+ T cells. J. Immunol. 2007, 179, 5916–5926. [Google Scholar] [CrossRef]
- Kohm, A.P.; Podojil, J.R.; Williams, J.S.; McMahon, J.S.; Miller, S.D. CD28 regulates glucocorticoid-induced TNF receptor family-related gene expression on CD4+ T cells via IL-2-dependent mechanisms. Cell Immunol. 2005, 235, 56–64. [Google Scholar] [CrossRef]
- Wang, S.; Shi, Y.; Yang, M.; Ma, J.; Tian, J.; Chen, J.; Mao, C.; Jiao, Z.; Ko, K.-H.; Baidooet, S.E.; et al. Glucocorticoid-induced tumor necrosis factor receptor family-related protein exacerbates collagen-induced arthritis by enhancing the expansion of Th17 cells. Am. J. Pathol. 2012, 180, 1059–1067. [Google Scholar] [CrossRef]
- Tang, X.; Tian, J.; Ma, J.; Wang, J.; Qi, C.; Rui, K.; Wang, Y.; Xu, H.; Lu, L.; Wang, S. GITRL modulates the activities of p38 MAPK and STAT3 to promote Th17 cell differentiation in autoimmune arthritis. Oncotarget 2016, 7, 8590–8600. [Google Scholar] [CrossRef]
- Ma, J.; Feng, D.; Wei, Y.; Tian, J.; Tang, X.; Rui, K.; Lu, L.; Xu, H.; Wang, S. Blockade of Glucocorticoid-Induced Tumor Necrosis Factor-Receptor-Related Protein Signaling Ameliorates Murine Collagen-Induced Arthritis by Modulating Follicular Helper T Cells. Am. J. Pathol. 2016, 186, 1559–1567. [Google Scholar] [CrossRef]
- Li, L.; Wen, W.; Jia, R.; Li, Y.; Liu, X.; Sun, X.; Li, Z. GITRL is associated with increased autoantibody production in patients with rheumatoid arthritis. Clin. Rheumatol. 2016, 35, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, G.; Cari, L.; Migliorati, G.; Riccardi, C. Treatment of autoimmune diseases and prevention of transplant rejection and graft-versus-host disease by regulatory T cells: The state of the art and perspectives. In The Epigenetics of Autoimmunity; Zhang, R., Tollefsbol, T., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 321–357. [Google Scholar] [CrossRef]
- Mages, H.W.; Hutloff, A.; Heuck, C.; Büchner, K.; Himmelbauer, H.; Oliveri, F.; Kroczeket, R.-A. Molecular cloning and characterization of murine ICOS and identification of B7h as ICOS ligand. Eur. J. Immunol. 2000, 30, 1040–1047. [Google Scholar] [CrossRef]
- Wikenheiser, D.J.; Stumhofer, J.S. ICOS Co-Stimulation: Friend or Foe? Front. Immunol. 2016, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Amatore, F.; Gorvel, L.; Olive, D. Role of Inducible Co-Stimulator (ICOS) in cancer immunotherapy. Expert. Opin. Biol. Ther. 2020, 20, 141–150. [Google Scholar] [CrossRef]
- Kunicki, M.A.; Hernandez, L.C.; Davis, K.L.; Bacchetta, R.; Roncarolo, M.G. Identity and Diversity of Human Peripheral Th and T Regulatory Cells Defined by Single-Cell Mass Cytometry. J. Immunol. 2018, 200, 336–346. [Google Scholar] [CrossRef]
- Fonseca, V.R.; Ribeiro, F.; Graca, L. T follicular regulatory (Tfr) cells: Dissecting the complexity of Tfr-cell compartments. Immunol. Rev. 2019, 288, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Y.; Xiong, X.Z. ICOS+ Tregs: A Functional Subset of Tregs in Immune Diseases. Front. Immunol. 2020, 11, 2104. [Google Scholar] [CrossRef]
- Yong, P.F.; Salzer, U.; Grimbacher, B. The role of costimulation in antibody deficiencies: ICOS and common variable immunodeficiency. Immunol. Rev. 2009, 229, 101–113. [Google Scholar] [CrossRef]
- Chen, Q.; Mo, L.; Cai, X.; Wei, L.; Xie, Z.; Li, H.; Li, J.; Hu, Z. ICOS signal facilitates Foxp3 transcription to favor suppressive function of regulatory T cells. Int. J. Med. Sci. 2018, 15, 666–673. [Google Scholar] [CrossRef]
- Kornete, M.; Sgouroudis, E.; Piccirillo, C.A. ICOS-dependent homeostasis and function of Foxp3+ regulatory T cells in islets of nonobese diabetic mice. J. Immunol. 2012, 188, 1064–1074. [Google Scholar] [CrossRef]
- Ito, T.; Hanabuchi, S.; Wang, Y.H.; Park, W.R.; Arima, K.; Bover, L.; Qin, F.H.-F.; Gilliet, M.; Liuet, Y.-J. Two functional subsets of FOXP3+ regulatory T cells in human thymus and periphery. Immunity 2008, 28, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, H.; Yan, C.; Mei, Y.; Lin, C.; Hong, Y.; Lin, X.; Zhang, Q.; Yu, J. Plasmacytoid Dendritic Cells and ICOS+ Regulatory T Cells Predict Poor Prognosis in Gastric Cancer: A Pilot Study. J. Cancer 2019, 10, 6711–6715. [Google Scholar] [CrossRef] [PubMed]
- Montes-Casado, M.; Ojeda, G.; Aragoneses-Fenoll, L.; López, D.; de Andrés, B.; Gaspar, M.L.; Dianzani, U.; Rojo, J.M.; Portolés, P. ICOS deficiency hampers the homeostasis, development and function of NK cells. PLoS ONE 2019, 14, e0219449. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, T.; Cai, G.; Qin, Y.; Wang, W.; Tang, G.; Zhao, D.; Shenet, Q. Elevated circulating CD4 + ICOS + Foxp3 + T cells contribute to overproduction of IL-10 and are correlated with disease severity in patients with systemic lupus erythematosus. Lupus 2011, 20, 620–627. [Google Scholar] [CrossRef]
- Kälble, F.; Wu, L.; Lorenz, H.M.; Zeier, M.; Schaier, M.; Steinborn, A. Impaired differentiation of highly proliferative ICOS+-Tregs is involved in the transition from low to high disease activity in Systemic Lupus Erythematosus (SLE) patients. Int. J. Mol. Sci. 2021, 22, 9501. [Google Scholar] [CrossRef]
- Sakthivel, P.; Grunewald, J.; Eklund, A.; Bruder, D.; Wahlström, J. Pulmonary sarcoidosis is associated with high-level inducible co-stimulator (ICOS) expression on lung regulatory T cells--possible implications for the ICOS/ICOS-ligand axis in disease course and resolution. Clin. Exp. Immunol. 2016, 183, 294–306. [Google Scholar] [CrossRef]
- Slauenwhite, D.; McAlpine, S.M.; Hanly, J.G.; Malik, A.; Haidl, I.D.; Marshall, J.S.; Issekutz, T.B. Association of a Type 2-Polarized T Cell Phenotype with Methotrexate Nonresponse in Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2020, 72, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.A.; Tsuji, W.; Kivitz, A.; Peng, J.; Arnold, G.E.; Boedigheimer, M.J.; Chiu, K.; Green, C.L.; Kaliyaperumal, A.; Wang, C.; et al. Inducible T-cell co-stimulator ligand (ICOSL) blockade leads to selective inhibition of anti-KLH IgG responses in subjects with systemic lupus erythematosus. Lupus Sci. Med. 2016, 3, e000146. [Google Scholar] [CrossRef]
- Cheng, L.E.; Amoura, Z.; Cheah, B.; Hiepe, F.; Sullivan, B.A.; Zhou, L.; Arnold, G.E.; Tsuji, W.H.; Merrill, J.T.; Chung, J.B. Brief Report: A Randomized, Double-Blind, Parallel-Group, Placebo-Controlled, Multiple-Dose Study to Evaluate AMG 557 in Patients with Systemic Lupus Erythematosus and Active Lupus Arthritis. Arthritis Rheumatol. 2018, 70, 1071–1076. [Google Scholar] [CrossRef]
- Mariette, X.; Bombardieri, M.; Alevizos, I.; Moate, R.; Sullivan, B.; Noaiseh, G.; Kvarnström, M.; Rees, W.; Wang, L.; Illei, G. A Phase 2a Study of MEDI5872 (AMG557), a Fully Human Anti-ICOS Ligand Monoclonal Antibody in Patients with Primary Sjögren’s Syndrome [bstract]. Arthritis Rheumatol. 2019, 71, 2417. Available online: https://acrabstracts.org/abstract/a-phase-2a-study-of-medi5872-amg557-a-fully-human-anti-icos-ligand-monoclonal-antibody-in-patients-with-primary-sjogrens-syndrome/ (accessed on 1 January 2020).
- Shi, J.; Hou, S.; Fang, Q.; Liu, X.; Liu, X.; Qi, H. PD-1 Controls Follicular T Helper Cell Positioning and Function. Immunity 2018, 49, 264–274.e4. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.J.I.; Salama, A.D.; Chitnis, C.T.; Smith, R.N.; Yagita, H.; Akiba, H.; Yamazaki, T.; Azuma, M.; Iwai, H.; Khoury, S.J.; et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J. Exp. Med. 2003, 198, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptordeficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef]
- Suarez-Gestal, M.; Ferreiros-Vidal, I.; Ortiz, J.A.; Gomez-Reino, J.J.; Gonzalez, A. Analysis of the functional relevance of a putative regulatory SNP of PDCD1, PD1.3, associated with systemic lupus erythematosus. Genes Immun. 2008, 9, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Kong, E.K.; Prokunina-Olsson, L.; Wong, W.H.; Lau, C.S.; Chan, T.M.; Alarcon-Riquelme, M.; Lau, Y.L. A new haplotype of PDCD1 is associated with rheumatoid arthritis in Hong Kong Chinese. Arthritis Rheum. 2005, 52, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Shang, Y.; Zhu, G.; Bao, X.; Xu, S.; Chen, Y. The expression and distribution of immunomodulatory proteins B7-H1, B7-DC, B7-H3, and B7-H4 in rheumatoid synovium. Clin. Rheumatol. 2012, 31, 271–281. [Google Scholar] [CrossRef]
- Matsuda, K.; Miyoshi, H.; Hiraoka, K.; Hamada, T.; Yoshida, S.; Ishibashi, Y.; Haraguchi, T.; Shiba, N.; Ohshima, K. Clinicopathological value of programmed cell death 1 (PD-1) and programmed cell death ligand 1 (PD-L1) expression in synovium of patients with rheumatoid arthritis. Clin. Exp. Med. 2018, 18, 487–494. [Google Scholar] [CrossRef]
- Ajam, F.; Aghaei, M.; Mohammadi, S.; Samiei, H.; Behnampour, N.; Memarian, A. PD-1 Expression on CD8+CD28- T cells within inflammatory synovium is associated with Relapse: A cohort of Rheumatoid Arthritis. Immunol. Lett. 2020, 228, 76–82. [Google Scholar] [CrossRef]
- Huang, Y.; Pan, C.; Liu, Y.; Lin, S.; Zhan, Y.; Zhang, Y.; Zhan, F. Immune Function and Mechanism of Costimulating Molecules PD-1 and OX40 in Rheumatoid Arthritis. J. Interferon Cytokine Res. 2020, 40, 530–539. [Google Scholar] [CrossRef]
- Wang, S.; Wang, J.; Kumar, V.; Karnell, J.L.; Naiman, B.; Gross, P.S.; Rahman, S.; Zerrouki, K.; Hanna, R.; Morehouse, C.; et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11c(hi)T-bet(+) B cells in SLE. Nat. Commun. 2018, 9, 1758. [Google Scholar] [CrossRef]
- Luo, Q.; Huang, Z.; Ye, J.; Deng, Y.; Fang, L.; Li, X.; Guo, Y.; Jiang, H.; Ju, B.; Huang, Q.; et al. PD-L1-expressing neutrophils as a novel indicator to assess disease activity and severity of systemic lupus erythematosus. Arthritis Res. Ther. 2016, 18, 47. [Google Scholar] [CrossRef]
- Mozaffarian, N.; Wiedeman, A.E.; Stevens, A.M. Active systemic lupus erythematosus is associated with failure of antigen-presenting cells to express programmed death ligand-1. Rheumatology 2008, 47, 1335–1341. [Google Scholar] [CrossRef]
- Curran, C.S.; Gupta, S.; Sanz, I.; Sharon, E. PD-1 immunobiology in systemic lupus erythematosus. J. Autoimmun. 2019, 97, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Ye, J.; Teng, J.; Yin, Y.; Hu, Q.; Wu, X.; Liu, H.; Cheng, X.; Su, Y.; Liu, M.; et al. Elevated serum autoantibodies against co-inhibitory PD-1 facilitate T cell proliferation and correlate with disease activity in new-onset systemic lupus erythematosus patients. Arthritis Res. Ther. 2017, 19, 52. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, V.; Bouttier, M.; Boukhaled, G.; Salehi-Tabar, R.; Avramescu, R.G.; Memari, B.; Hasaj, B.; Lukacs, G.; Krawczyk, K.M.; White, J.H. Hormonal vitamin D up-regulates tissue-specific PD-L1 and PD-L2 surface glycoprotein expression in humans but not mice. J. Biol. Chem. 2017, 292, 20657–20668. [Google Scholar] [CrossRef]
- Wanchoo, R.; Karam, S.; Uppal, N.N.; Barta, V.S.; Deray, G.; Devoe, C.; Launay-Vacher, V.; Jhaveri, K.D. Adverse renal effects of immune checkpoint inhibitors: A narrative review. Am. J. Nephrol. 2017, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Usui, Y.; Hattori, T.; Takeuchi, M.; Takayama, K.; Karasawa, Y.; Nishio, Y.; Yamakawa, N.; Saitoh, D.; Goto, H.; et al. Programmed Cell Death-1 Pathway Deficiency Enhances Autoimmunity Leading to Dacryoadenitis of Mice. Am. J. Pathol. 2021, 191, 1077–1093. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kawano, S.; Hatachi, S.; Kurimoto, C.; Okazaki, T.; Iwai, Y.; Honjo, T.; Tanaka, Y.; Minato, N.; Komoet, T.; et al. Enhanced expression of programmed death-1 (PD-1)/PD-L1 in salivary glands of patients with Sjögren’s syndrome. J. Rheumatol. 2005, 32, 2156–2163. [Google Scholar]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Yamasaki, K.; Kira, J.I.; Kawano, Y.; Kobayashi, T.; Kanai, T.; Nishimura, Y.; Matsushita, S.; Hasuo, K.; Tobimatsu, S. Western versus asian types of multiple sclerosis: Immunogenetically and clinically distinct disorders. Ann. Neurol. 1996, 40, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhou, X.; Zhou, X.; Wang, H.; Gu, L.; Ke, Y.; Zhang, M.; Ji, J.; Yang, X. Low expressions of PD-L1 and CTLA-4 by induced CD4+ CD25+ Foxp3+ Tregs in patients with SLE and their correlation with the disease activity. Cytokine 2020, 133, 155119. [Google Scholar] [CrossRef] [PubMed]
- Cha, E.; Klinger, M.; Hou, Y.; Hou, Y.; Cummings, C.; Ribas, A.; Faham, M.; Fong, L. Improved Survival with T Cell Clonotype Stability After Anti–CTLA-4 Treatment in Cancer Patients. Sci. Transl. Med. 2014, 6, 238ra270. [Google Scholar] [CrossRef]
- Liu, M.; Yu, Y.; Hu, S. A review on applications of abatacept in systemic rheumatic diseases. Int. Immunopharmacol. 2021, 96, 107612. [Google Scholar] [CrossRef] [PubMed]
- Verstappen, G.M.; Meiners, P.M.; Corneth, O.; Visser, A.; Arends, S.; Abdulahad, W.H.; Hendriks, R.W.; Vissink, A.; Kroese, F.; Bootsma, H. Attenuation of follicular helper T cell–dependent B cell hyperactivity by abatacept treatment in primary Sjögren’s syndrome. Arthritis Rheumatol. 2017, 69, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, A.; Gharibi, T.; Marofi, F.; Babaloo, Z.; Baradaran, B. CTLA-4: From mechanism to autoimmune therapy. Int. Immunopharmacol. 2020, 80, 106221. [Google Scholar] [CrossRef]
- Karnell, J.L.; Rieder, S.A.; Ettinger, R.; Kolbeck, R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv. Drug Deliv. Rev. 2019, 141, 92–103. [Google Scholar] [CrossRef]
- Berner, B.; Wolf, G.; Hummel, K.M.; Müller, G.A.; Reuss-Borst, M.A. Increased expression of CD40 ligand (CD154) on CD4+ T cells as a marker of disease activity in rheumatoid arthritis. Ann. Rheum. Dis. 2000, 59, 190–195. [Google Scholar] [CrossRef]
- Harigai, M.; Hara, M.; Nakazawa, S.; Fukasawa, C.; Ohta, S.; Sugiura, T.; Inoue, K.; Kashiwazaki, S. Ligation of CD40 induced tumor necrosis factor-alpha in rheumatoid arthritis: A novel mechanism of activation of synoviocytes. J. Rheumatol. 1999, 26, 1035–1043. [Google Scholar]
- Dimitriou, I.D.; Kapsogeorgou, E.K.; Moutsopoulos, H.M.; Manoussakis, M.N. CD40 on salivary gland epithelial cells: High constitutive expression by cultured cells from Sjögren’s syndrome patients indicating their intrinsic activation. Clin. Exp. Immunol. 2002, 127, 386–392. [Google Scholar] [CrossRef]
- Chen, D.; Ireland, S.J.; Remington, G.; Alvarez, E.; Racke, M.K.; Greenberg, B.; Frohman, E.M.; Monson, N.L. CD40-Mediated NF-κB Activation in B Cells Is Increased in Multiple Sclerosis and Modulated by Therapeutics. J. Immunol. 2016, 197, 4257–4265. [Google Scholar] [CrossRef] [PubMed]
- Boumpas, D.T.; Furie, R.; Manzi, S.; Illei, G.G.; Wallace, D.J.; Balow, J.E.; Vaishnaw, A. BG9588 Lupus Nephritis Trial Group. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003, 48, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, C.; Colman, P.J.; Ranger, A.M.; Burkly, L.C.; Johnston, G.I.; Otoul, C.; Stach, C.; Zamacona, M.; Dörner, T.; Urowitz, M.; et al. Repeated administration of dapirolizumab pegol in a randomised phase I study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann. Rheum. Dis. 2017, 76, 1837–1844. [Google Scholar] [CrossRef]
- Karnell, J.L.; Albulescu, M.; Drabic, S.; Wang, L.; Moate, R.; Baca, M.; Oganesyan, V.; Gunsior, M.; Thisted, T.; Yan, L.; et al. A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci. Transl. Med. 2019, 11, eaar6584. [Google Scholar] [CrossRef] [PubMed]
- Gueiros, L.A.; France, K.; Posey, R.; Mays, J.W.; Carey, B.; Sollecito, T.P.; Setterfield, J.; Woo, S.B.; Culton, D.; Payne, A.S.; et al. World Workshop on Oral Medicine VII: Immunobiologics for salivary gland disease in Sjogren’s syndrome: A systematic review. Oral Dis. 2019, 25, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Espié, P.; He, Y.; Koo, P.; Sickert, D.; Dupuy, C.; Chokoté, E.; Schuler, R.; Mergentaler, H.; Ristov, J.; Milojevic, J.; et al. First-in-human clinical trial to assess pharmacokinetics, pharmacodynamics, safety, and tolerability of iscalimab, an anti-CD40 monoclonal antibody. Am. J. Transplant. 2020, 20, 463–473. [Google Scholar] [CrossRef]
- Visvanathan, S.; Daniluk, S.; Ptaszynski, R.; Muller-Ladner, U.; Ramanujam, M.; Rosenstock, B.; Eleftheraki, A.G.; Vinisko, R.; Petrikova, A.; Kellner, H.; et al. Effects of BI 655064, an antagonistic anti-CD40 antibody, on clinical and biomarker variables in patients with active rheumatoid arthritis: A randomised, double-blind, placebo-controlled, phase IIa study. Ann. Rheum. Dis. 2019, 78, 754–760. [Google Scholar] [CrossRef]
- Xiao, Z.X.; Olsen, N.; Zheng, S.G. The essential role of costimulatory molecules in systemic lupus erythematosus. Lupus 2019, 28, 575–582. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Immune Checkpoints | Expression and Functions/Properties | References |
|---|---|---|
| OX40 (CD134) | Mainly expressed on activated APCs | [34] |
| Expressed on hematopoietic cells such as activated NK cells, NKT cells, mast cells or the responding CD4+ T cells, and non-hematopoietic cells, such as endothelial cells or smooth muscle cells | [35,36] | |
| An increase in the survival of T cells and their transition to memory T cells during the immune response | [37] | |
| GITR | Inhibition of suppressive activity of Tregs and prolongation of T-effector-cell survival | [45,46] |
| Present on the membrane of NK cells and, to a lesser extent, B lymphocytes, macrophages and DCs | [47] | |
| Increases survival, activation, and stimulation of T-cell proliferation by increasing TCR-induced proliferation, cytokine production, and blocking anti-CD3-induced T-cell apoptosis | [48,49] | |
| Differentiation of naive CD4+ T cells into Th17 cells | [50] | |
| Activation of the p38 MAP signaling pathway and the STAT3 signaling pathway influencing the development of Th17 cells | [51] | |
| ICOS | Expressed on activated CD4+ and CD8+ T cells, presumably regulates adaptive T-cell response | [52,53] |
| Expressed on Th1, Th2, Th17, Tfh, Tfr, Treg, Tr1, and innate lymphoid cells | [54,55] | |
| Participation in differentiation, proliferation, and survival of Tregs | [56,57,58] | |
| Regulation of the humoral immune response, namely in the formation of germinal centers and the generation of Ig class switching | [55] | |
| PD-1/PDL1 | PD-1 is expressed on activated T cells, B cells, and monocytes | [59] |
| PD-L1 expression on proliferation and activation of T cells by interacting with PD-1. | [60] | |
| PD-1 negatively regulates the TCR signal by recruiting SHP-2 to the phosphorylated tyrosine residue in the cytoplasmic region | [61] | |
| CTLA-4 | Inhibition of T-cell proliferation, cell cycle progression, IL-2 production, and differentiation of T cells | [62] |
| CD40/CD40L | CD-40 is expressed mainly on B cells and regulation of B-cell proliferation | [63] |
| CD40L expressed mainly on T cells | [64] | |
| T-cell-dependent B-cell differentiation and activation | [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerasimova, E.V.; Tabakov, D.V.; Gerasimova, D.A.; Popkova, T.V. Activation Markers on B and T Cells and Immune Checkpoints in Autoimmune Rheumatic Diseases. Int. J. Mol. Sci. 2022, 23, 8656. https://doi.org/10.3390/ijms23158656
Gerasimova EV, Tabakov DV, Gerasimova DA, Popkova TV. Activation Markers on B and T Cells and Immune Checkpoints in Autoimmune Rheumatic Diseases. International Journal of Molecular Sciences. 2022; 23(15):8656. https://doi.org/10.3390/ijms23158656
Chicago/Turabian StyleGerasimova, Elena V., Dmitry V. Tabakov, Daria A. Gerasimova, and Tatiana V. Popkova. 2022. "Activation Markers on B and T Cells and Immune Checkpoints in Autoimmune Rheumatic Diseases" International Journal of Molecular Sciences 23, no. 15: 8656. https://doi.org/10.3390/ijms23158656
APA StyleGerasimova, E. V., Tabakov, D. V., Gerasimova, D. A., & Popkova, T. V. (2022). Activation Markers on B and T Cells and Immune Checkpoints in Autoimmune Rheumatic Diseases. International Journal of Molecular Sciences, 23(15), 8656. https://doi.org/10.3390/ijms23158656