
HDAC8-Selective Inhibition by PCI-34051 Enhances the Anticancer Effects of ACY-241 in Ovarian Cancer Cells

 ,
,  and
and 
Abstract
:1. Introduction
2. Results
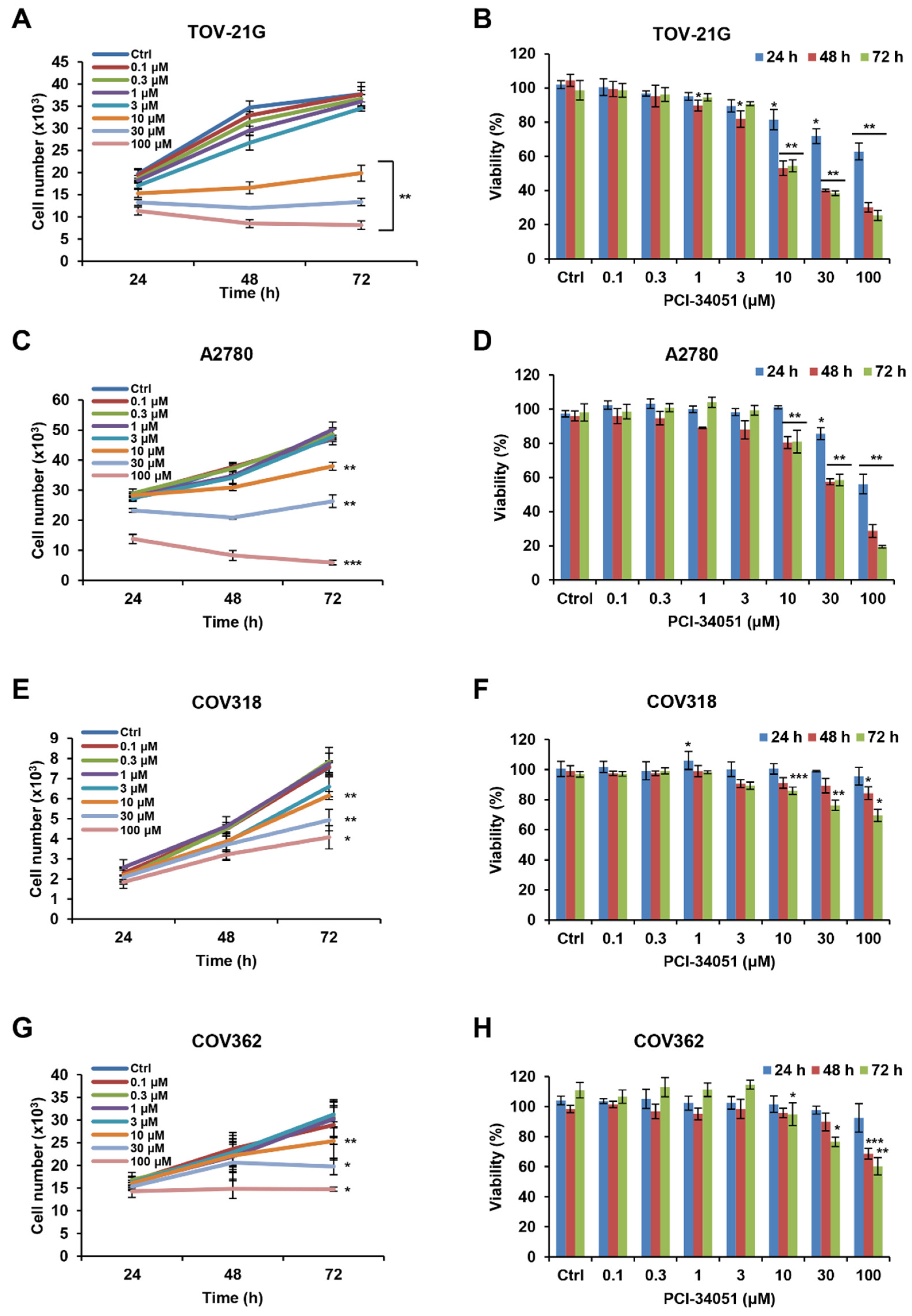
2.1. HDAC8 Inhibitor, PCI-34051, Suppresses Cell Growth and Reduces Cell Viability in p53 Wild-Type Ovarian Cancer Cells
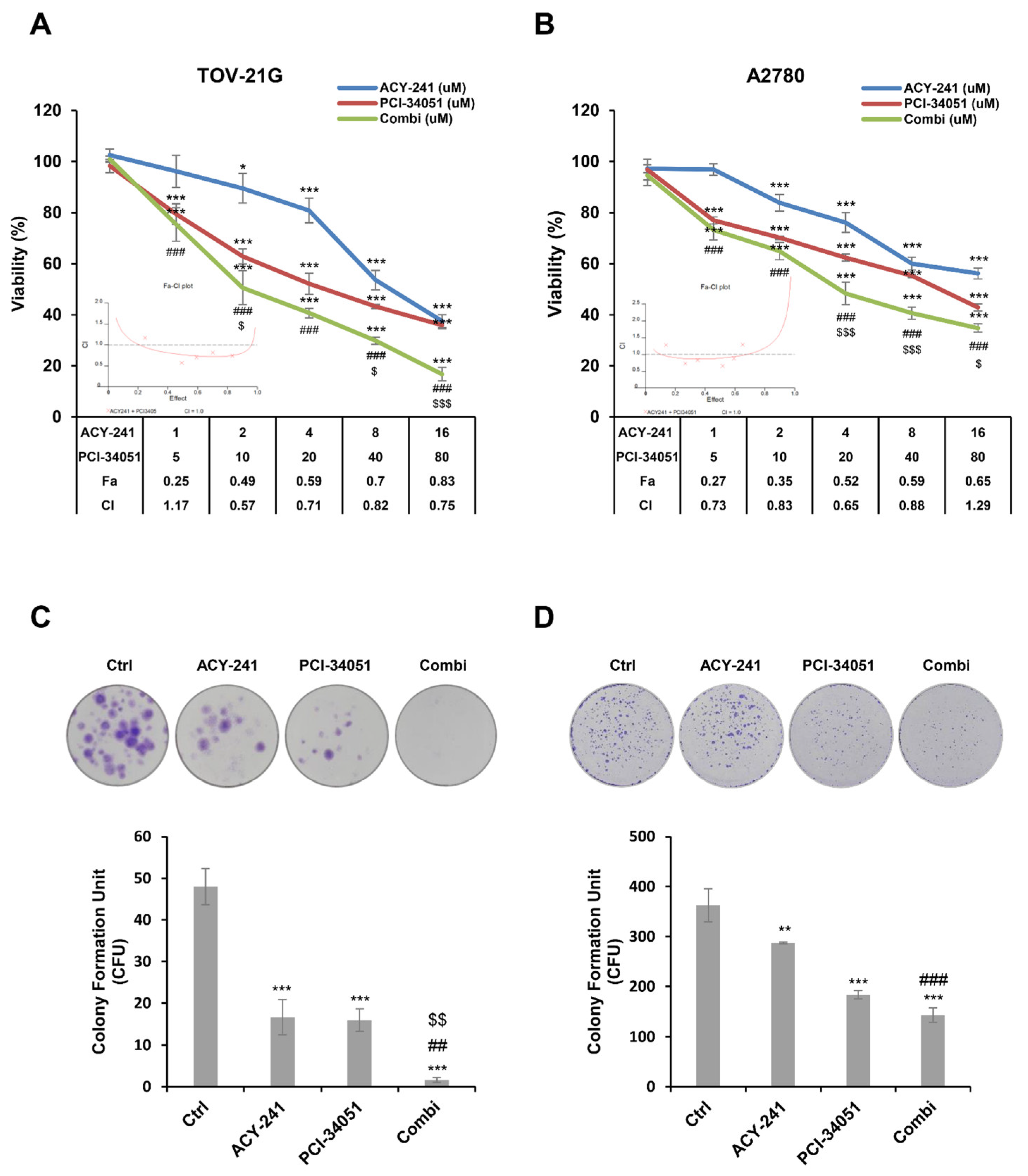
2.2. ACY-241 and PCI-34051 Treatment Synergistically Suppresses Cell Growth and Reduces Cell Viability in p53 Wild-Type Ovarian Cancer Cells
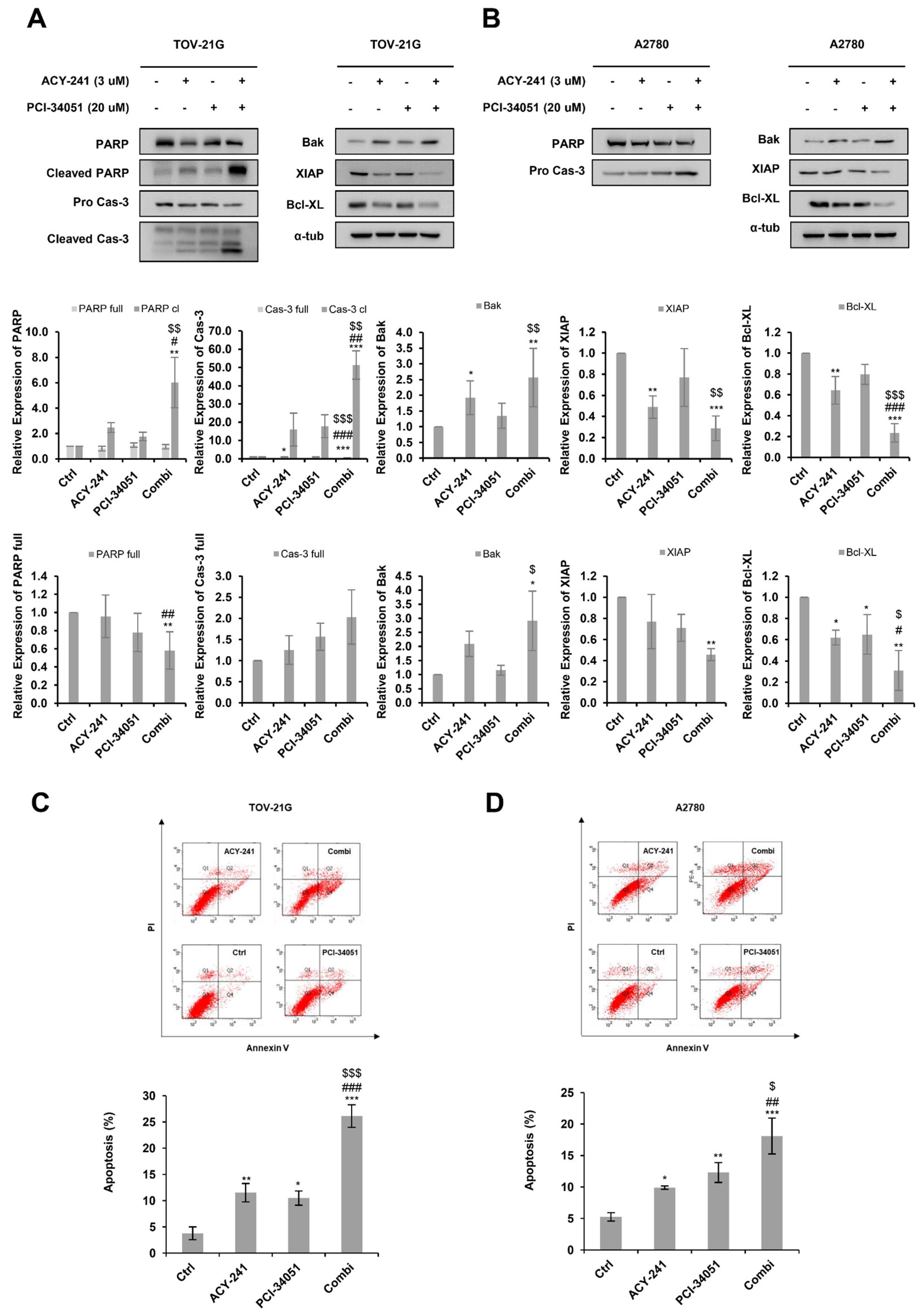
2.3. ACY-241 and PCI-34051 Treatment Synergistically Induces Apoptosis in p53 Wild-Type Ovarian Cancer Cells
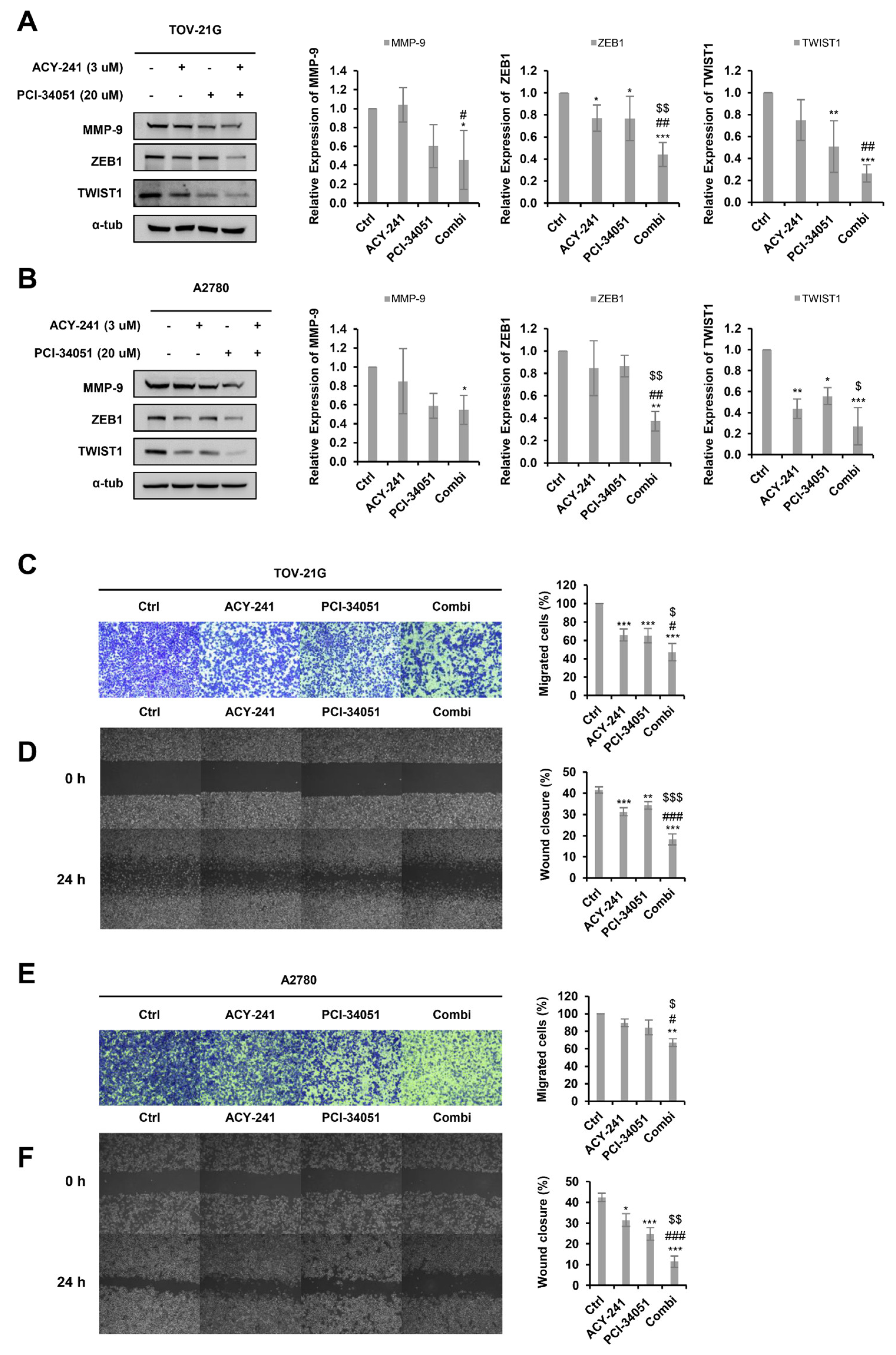
2.4. ACY-241 and PCI-34051 Treatment Synergistically Inhibits Cell Migration in p53 Wild-Type Ovarian Cancer Cells
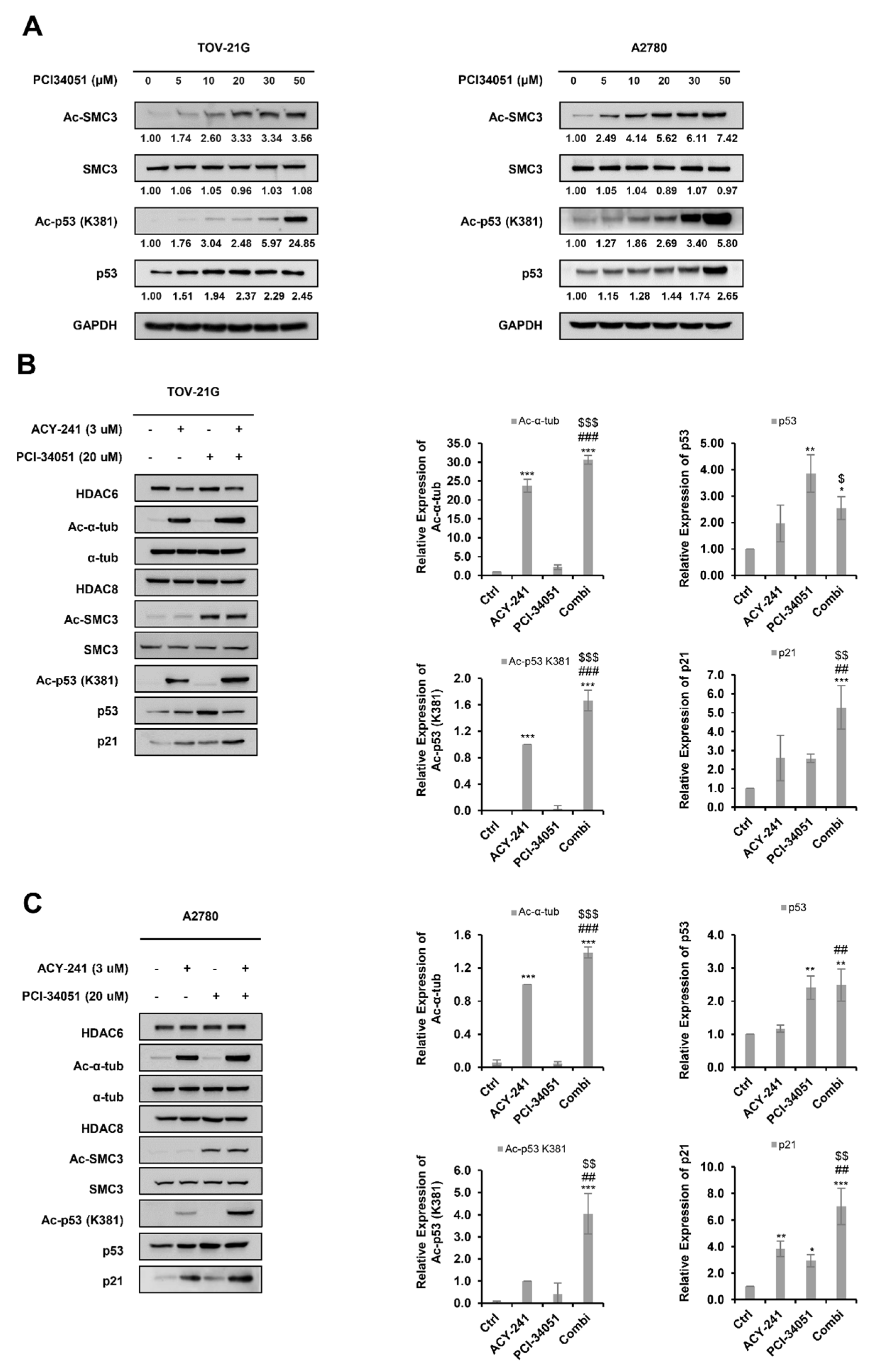
2.5. ACY-241 and PCI-34051 Treatment Synergistically Enhances p53 Stability in p53 Wild-Type Ovarian Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Ovarian Cancer Cell Lines and CELL Culture
4.3. Cell Growth and Viability Assay
4.4. Drug Combination Analysis
4.5. Apoptosis Assay
4.6. Colony Formation Assay
4.7. Wound Healing Assay
4.8. Transwell Migration Assay
4.9. Western Blot
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef] [Green Version]
- Kurnit, K.C.; Fleming, G.F.; Lengyel, E. Updates and New Options in Advanced Epithelial Ovarian Cancer Treatment. Obstet. Gynecol. 2021, 137, 108. [Google Scholar] [CrossRef]
- Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.; Niesporek, S.; Denkert, C.; Dietel, M. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef] [Green Version]
- Weichert, W.; Röske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.-C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef] [Green Version]
- Krusche, C.A.; Wülfing, P.; Kersting, C.; Vloet, A.; Böcker, W.; Kiesel, L.; Beier, H.M.; Alfer, J. Histone deacetylase-1 and-3 protein expression in human breast cancer: A tissue microarray analysis. Breast Cancer Res. Treat. 2005, 90, 15–23. [Google Scholar] [CrossRef]
- Minamiya, Y.; Ono, T.; Saito, H.; Takahashi, N.; Ito, M.; Mitsui, M.; Motoyama, S.; Ogawa, J. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer 2011, 74, 300–304. [Google Scholar] [CrossRef]
- Pulya, S.; Amin, S.A.; Adhikari, N.; Biswas, S.; Jha, T.; Ghosh, B. HDAC6 as privileged target in drug discovery: A perspective. Pharmacol. Res. 2021, 163, 105274. [Google Scholar] [CrossRef] [PubMed]
- Selenica, M.-L.; Benner, L.; Housley, S.B.; Manchec, B.; Lee, D.C.; Nash, K.R.; Kalin, J.; Bergman, J.A.; Kozikowski, A.; Gordon, M.N. Histone deacetylase 6 inhibition improves memory and reduces total tau levels in a mouse model of tau deposition. Alzheimers Res. Ther. 2014, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- McLendon, P.M.; Ferguson, B.S.; Osinska, H.; Bhuiyan, M.S.; James, J.; McKinsey, T.A.; Robbins, J. Tubulin hyperacetylation is adaptive in cardiac proteotoxicity by promoting autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, E5178–E5186. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Omoto, Y.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hayashi, S.-i.; Iwase, H. HDAC6 expression is correlated with better survival in breast cancer. Clin. Cancer Res. 2004, 10, 6962–6968. [Google Scholar] [CrossRef] [Green Version]
- Bazzaro, M.; Lin, Z.; Santillan, A.; Lee, M.K.; Wang, M.-C.; Chan, K.C.; Bristow, R.E.; Mazitschek, R.; Bradner, J.; Roden, R.B. Ubiquitin proteasome system stress underlies synergistic killing of ovarian cancer cells by bortezomib and a novel HDAC6 inhibitor. Clin. Cancer Res. 2008, 14, 7340–7347. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Q.; Wu, W.; Li, X.; Zhao, L.; Chen, W. HDAC6 and SIRT2 promote bladder cancer cell migration and invasion by targeting cortactin. Oncol. Rep. 2012, 27, 819–824. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Tang, F.; Hu, P.; Wang, Y.; Gong, J.; Sun, S.; Xie, C. HDAC6 promotes cell proliferation and confers resistance to gefitinib in lung adenocarcinoma. Oncol. Rep. 2016, 36, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.-L.; Zhu, H.-Y.; Zhou, B.-Y.; Chu, Y.; Huo, J.-R.; Tan, Y.-Y.; Liu, D.-L. Histone deacetylase 6 is overexpressed and promotes tumor growth of colon cancer through regulation of the MAPK/ERK signal pathway. Oncol. Targets Ther. 2019, 12, 2409. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Zhang, F.; Maguire, A.; Byrne, T.; Weiner-Gorzel, K.; Bridgett, S.; O’Toole, S.; O’Leary, J.; Beggan, C.; Fitzpatrick, P.; et al. HDAC6 degradation inhibits the growth of high-grade serous ovarian cancer cells. Cancers 2020, 12, 3734. [Google Scholar] [CrossRef]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef]
- Huang, P.; Almeciga-Pinto, I.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Yang, M.; Jones, S.S.; Quayle, S.N. Selective HDAC inhibition by ACY-241 enhances the activity of paclitaxel in solid tumor models. Oncotarget 2017, 8, 2694. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Das, D.S.; Song, Y.; Hideshima, T.; Tai, Y.-T.; Chauhan, D.; Anderson, K.C. Combination of a novel HDAC6 inhibitor ACY-241 and anti-PD-L1 antibody enhances anti-tumor immunity and cytotoxicity in multiple myeloma. Leukemia 2018, 32, 843–846. [Google Scholar] [CrossRef]
- North, B.J.; Almeciga-Pinto, I.; Tamang, D.; Yang, M.; Jones, S.S.; Quayle, S.N. Enhancement of pomalidomide anti-tumor response with ACY-241, a selective HDAC6 inhibitor. PLoS ONE 2017, 12, e0173507. [Google Scholar] [CrossRef] [Green Version]
- García-Guerrero, E.; Götz, R.; Doose, S.; Sauer, M.; Rodríguez-Gil, A.; Nerreter, T.; Kortüm, K.M.; Pérez-Simón, J.A.; Einsele, H.; Hudecek, M. Upregulation of CD38 expression on multiple myeloma cells by novel HDAC6 inhibitors is a class effect and augments the efficacy of daratumumab. Leukemia 2021, 35, 201–214. [Google Scholar] [CrossRef]
- Awad, M.M.; Le Bruchec, Y.; Lu, B.; Ye, J.; Miller, J.; Lizotte, P.H.; Cavanaugh, M.E.; Rode, A.J.; Dumitru, C.D.; Spira, A. Selective Histone Deacetylase Inhibitor ACY-241 (Citarinostat) Plus Nivolumab in Advanced Non-Small Cell Lung Cancer: Results From a Phase Ib Study. Front. Oncol. 2021, 11, 696512. [Google Scholar] [CrossRef]
- Park, S.-J.; Joo, S.H.; Lee, N.; Jang, W.-J.; Seo, J.H.; Jeong, C.-H. ACY-241, an HDAC6 inhibitor, overcomes erlotinib resistance in human pancreatic cancer cells by inducing autophagy. Arch. Pharm. Res. 2021, 44, 1062–1075. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. HDAC8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Jun, J.; Jeong, K.J.; Heo, H.J.; Sohn, J.S.; Lee, H.Y.; Park, C.G.; Kang, J. Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncol. Rep. 2011, 25, 1677–1681. [Google Scholar] [CrossRef] [Green Version]
- Niegisch, G.; Knievel, J.; Koch, A.; Hader, C.; Fischer, U.; Albers, P.; Schulz, W.A. Changes in histone deacetylase (HDAC) expression patterns and activity of HDAC inhibitors in urothelial cancers. Urol. Oncol. 2013, 31, 1770–1779. [Google Scholar] [CrossRef]
- Wu, J.; Du, C.; Lv, Z.; Ding, C.; Cheng, J.; Xie, H.; Zhou, L.; Zheng, S. The up-regulation of histone deacetylase 8 promotes proliferation and inhibits apoptosis in hepatocellular carcinoma. Dig. Dis. Sci. 2013, 58, 3545–3553. [Google Scholar] [CrossRef]
- Gruhn, B.; Naumann, T.; Gruner, D.; Walther, M.; Wittig, S.; Becker, S.; Beck, J.F.; Sonnemann, J. The expression of histone deacetylase 4 is associated with prednisone poor-response in childhood acute lymphoblastic leukemia. Leuk. Res. 2013, 37, 1200–1207. [Google Scholar] [CrossRef]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.-P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; Von Deimling, A. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Marek, M.; Shaik, T.B.; Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Ramos-Morales, E.; Da Veiga, C.; Kalinin, D.; Melesina, J.; Robaa, D. Characterization of histone deacetylase 8 (HDAC8) selective inhibition reveals specific active site structural and functional determinants. J. Med. Chem. 2018, 61, 10000–10016. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef]
- Bae, J.; Hideshima, T.; Tai, Y.-T.; Song, Y.; Richardson, P.; Raje, N.; Munshi, N.C.; Anderson, K.C. Histone deacetylase (HDAC) inhibitor ACY241 enhances anti-tumor activities of antigen-specific central memory cytotoxic T lymphocytes against multiple myeloma and solid tumors. Leukemia 2018, 32, 1932–1947. [Google Scholar] [CrossRef]
- Yoo, J.; Jeon, Y.H.; Lee, D.H.; Kim, G.W.; Lee, S.W.; Kim, S.Y.; Park, J.; Kwon, S.H. HDAC6-selective inhibitors enhance anticancer effects of paclitaxel in ovarian cancer cells. Oncol. Lett. 2021, 21, 201. [Google Scholar] [CrossRef]
- Chou, T.-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Hsieh, Y.-L.; Tu, H.-J.; Pan, S.-L.; Liou, J.-P.; Yang, C.-R. Anti-metastatic activity of MPT0G211, a novel HDAC6 inhibitor, in human breast cancer cells in vitro and in vivo. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 992–1003. [Google Scholar] [CrossRef]
- Tang, X.; Li, G.; Su, F.; Cai, Y.; Shi, L.; Meng, Y.; Liu, Z.; Sun, J.; Wang, M.; Qian, M. HDAC8 cooperates with SMAD3/4 complex to suppress SIRT7 and promote cell survival and migration. Nucleic Acids Res. 2020, 48, 2912–2923. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.-W.; Shin, D.-H.; Lee, D.H.; Choi, J.; Han, G.; Lee, K.Y.; Kwon, S.H. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 2017, 391, 162–171. [Google Scholar] [CrossRef]
- Chen, Y.; Pan, C.; Lu, Y.; Miao, Y.; Xiong, B. HDAC8 drives spindle organization during meiotic maturation of porcine oocytes. Cell Prolif. 2021, 54, e13119. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609. [Google Scholar] [CrossRef]
- Aylon, Y.; Oren, M. New plays in the p53 theater. Curr. Opin. Genet. Dev. 2011, 21, 86–92. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Hua, W.-K.; Qi, J.; Cai, Q.; Carnahan, E.; Ayala Ramirez, M.; Li, L.; Marcucci, G.; Kuo, Y.-H. HDAC8 regulates long-term hematopoietic stem-cell maintenance under stress by modulating p53 activity. Blood 2017, 130, 2619–2630. [Google Scholar] [CrossRef]
- Qi, J.; Singh, S.; Hua, W.-K.; Cai, Q.; Chao, S.-W.; Li, L.; Liu, H.; Ho, Y.; McDonald, T.; Lin, A. HDAC8 inhibition specifically targets Inv (16) acute myeloid leukemic stem cells by restoring p53 acetylation. Cell Stem Cell 2015, 17, 597–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, C.M.; Menko, A.S. Microtubules: Evolving roles and critical cellular interactions. Exp. Biol. Med. 2019, 244, 1240–1254. [Google Scholar] [CrossRef]
- Lee, C.-C.; Cheng, Y.-C.; Chang, C.-Y.; Lin, C.-M.; Chang, J.-Y. Alpha-tubulin acetyltransferase/MEC-17 regulates cancer cell migration and invasion through epithelial–mesenchymal transition suppression and cell polarity disruption. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boggs, A.E.; Vitolo, M.I.; Whipple, R.A.; Charpentier, M.S.; Goloubeva, O.G.; Ioffe, O.B.; Tuttle, K.C.; Slovic, J.; Lu, Y.; Mills, G.B. α-Tubulin acetylation elevated in metastatic and basal-like breast cancer cells promotes microtentacle formation, adhesion, and invasive migration. Cancer Res. 2015, 75, 203–215. [Google Scholar] [CrossRef] [Green Version]
- Nekooki-Machida, Y.; Hagiwara, H. Role of tubulin acetylation in cellular functions and diseases. Med. Mol. Morphol. 2020, 53, 191–197. [Google Scholar] [CrossRef]
- Vanaja, G.; Ramulu, H.G.; Kalle, A.M. Overexpressed HDAC8 in cervical cancer cells shows functional redundancy of tubulin deacetylation with HDAC6. Cell Commun. Signal. 2018, 16, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Chan, Y.-T.; Tan, H.-Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022. [Google Scholar] [CrossRef] [Green Version]
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; De Álava, E.; Hajji, N.; García-Domínguez, D.J. Synergistic enhancement of cancer therapy using HDAC inhibitors: Opportunity for clinical trials. Front. Genet. 2020, 11, 578011. [Google Scholar] [CrossRef]
- Cho, H.Y.; Lee, S.W.; Jeon, Y.H.; Lee, D.H.; Kim, G.W.; Yoo, J.; Kim, S.Y.; Kwon, S.H. Combination of ACY-241 and JQ1 Synergistically Suppresses Metastasis of HNSCC via Regulation of MMP-2 and MMP-9. Int. J. Mol. Sci. 2020, 21, 6873. [Google Scholar] [CrossRef]
- Zhang, P.; Brinton, L.T.; Williams, K.; Sher, S.; Orwick, S.; Tzung-Huei, L.; Mims, A.S.; Coss, C.C.; Kulp, S.K.; Youssef, Y. Targeting DNA damage repair functions of two histone deacetylases, HDAC8 and SIRT6, sensitizes acute myeloid leukemia to NAMPT inhibition. Clin. Cancer Res. 2021, 27, 2352–2366. [Google Scholar] [CrossRef]
- Spreafico, M.; Gruszka, A.M.; Valli, D.; Mazzola, M.; Deflorian, G.; Quintè, A.; Totaro, M.G.; Battaglia, C.; Alcalay, M.; Marozzi, A. HDAC8: A Promising Therapeutic Target for Acute Myeloid Leukemia. Front. Cell Dev. Biol. 2020, 8, 844. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Najafi, S.; Stäble, S.; Fabian, J.; Koeneke, E.; Kolbinger, F.R.; Wrobel, J.K.; Meder, B.; Distel, M.; Heimburg, T.; et al. A kinome-wide RNAi screen identifies ALK as a target to sensitize neuroblastoma cells for HDAC8-inhibitor treatment. Cell Death Differ. 2018, 25, 2053–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015, 6, e1657. [Google Scholar] [CrossRef]
- Konze, K.D.; Ma, A.; Li, F.; Barsyte-Lovejoy, D.; Parton, T.; Macnevin, C.J.; Liu, F.; Gao, C.; Huang, X.P.; Kuznetsova, E.; et al. An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- José-Enériz, S.; Agirre, X.; Rabal, O.; Vilas-Zornoza, A.; Sanchez-Arias, J.A.; Miranda, E.; Ugarte, A.; Roa, S.; Paiva, B.; Estella-Hermoso de Mendoza, A. Discovery of first-in-class reversible dual small molecule inhibitors against G9a and DNMTs in hematological malignancies. Nat. Commun. 2017, 8, 15424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalin, J.H.; Wu, M.; Gomez, A.V.; Song, Y.; Das, J.; Hayward, D.; Adejola, N.; Wu, M.; Panova, I.; Chung, H.J. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat. Commun. 2018, 9, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolbinger, F.R.; Koeneke, E.; Ridinger, J.; Heimburg, T.; Müller, M.; Bayer, T.; Sippl, W.; Jung, M.; Gunkel, N.; Miller, A.K. The HDAC6/8/10 inhibitor TH34 induces DNA damage-mediated cell death in human high-grade neuroblastoma cell lines. Arch. Toxicol. 2018, 92, 2649–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Yang, W.; Zeng, H.; Hu, C.; Zhang, Y.; Ding, N.; Fan, G.; Shao, L.; Kuang, B. Droxinostat sensitizes human colon cancer cells to apoptotic cell death via induction of oxidative stress. Cell. Mol. Biol. Lett. 2018, 23, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time | 72 h | |||
|---|---|---|---|---|
| Cell line | TOV-21G | A2780 | COV318 | COV362 |
| 1 IC50 (µM) | 9.7 | 28.3 | 127.6 | 120.4 |
| 2 GI50 (µM) | 9.3 | 25.9 | 127.2 | 81.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.Y.; Han, S.Y.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Park, J.; Kwon, S.H. HDAC8-Selective Inhibition by PCI-34051 Enhances the Anticancer Effects of ACY-241 in Ovarian Cancer Cells. Int. J. Mol. Sci. 2022, 23, 8645. https://doi.org/10.3390/ijms23158645
Kim JY, Han SY, Yoo J, Kim GW, Jeon YH, Lee SW, Park J, Kwon SH. HDAC8-Selective Inhibition by PCI-34051 Enhances the Anticancer Effects of ACY-241 in Ovarian Cancer Cells. International Journal of Molecular Sciences. 2022; 23(15):8645. https://doi.org/10.3390/ijms23158645
Chicago/Turabian StyleKim, Ji Yoon, Seung Yoon Han, Jung Yoo, Go Woon Kim, Yu Hyun Jeon, Sang Wu Lee, Jongsun Park, and So Hee Kwon. 2022. "HDAC8-Selective Inhibition by PCI-34051 Enhances the Anticancer Effects of ACY-241 in Ovarian Cancer Cells" International Journal of Molecular Sciences 23, no. 15: 8645. https://doi.org/10.3390/ijms23158645
APA StyleKim, J. Y., Han, S. Y., Yoo, J., Kim, G. W., Jeon, Y. H., Lee, S. W., Park, J., & Kwon, S. H. (2022). HDAC8-Selective Inhibition by PCI-34051 Enhances the Anticancer Effects of ACY-241 in Ovarian Cancer Cells. International Journal of Molecular Sciences, 23(15), 8645. https://doi.org/10.3390/ijms23158645