
Validated Growth Rate-Dependent Regulation of Lipid Metabolism in Yarrowia lipolytica


Abstract
:1. Introduction
2. Results and Discussion
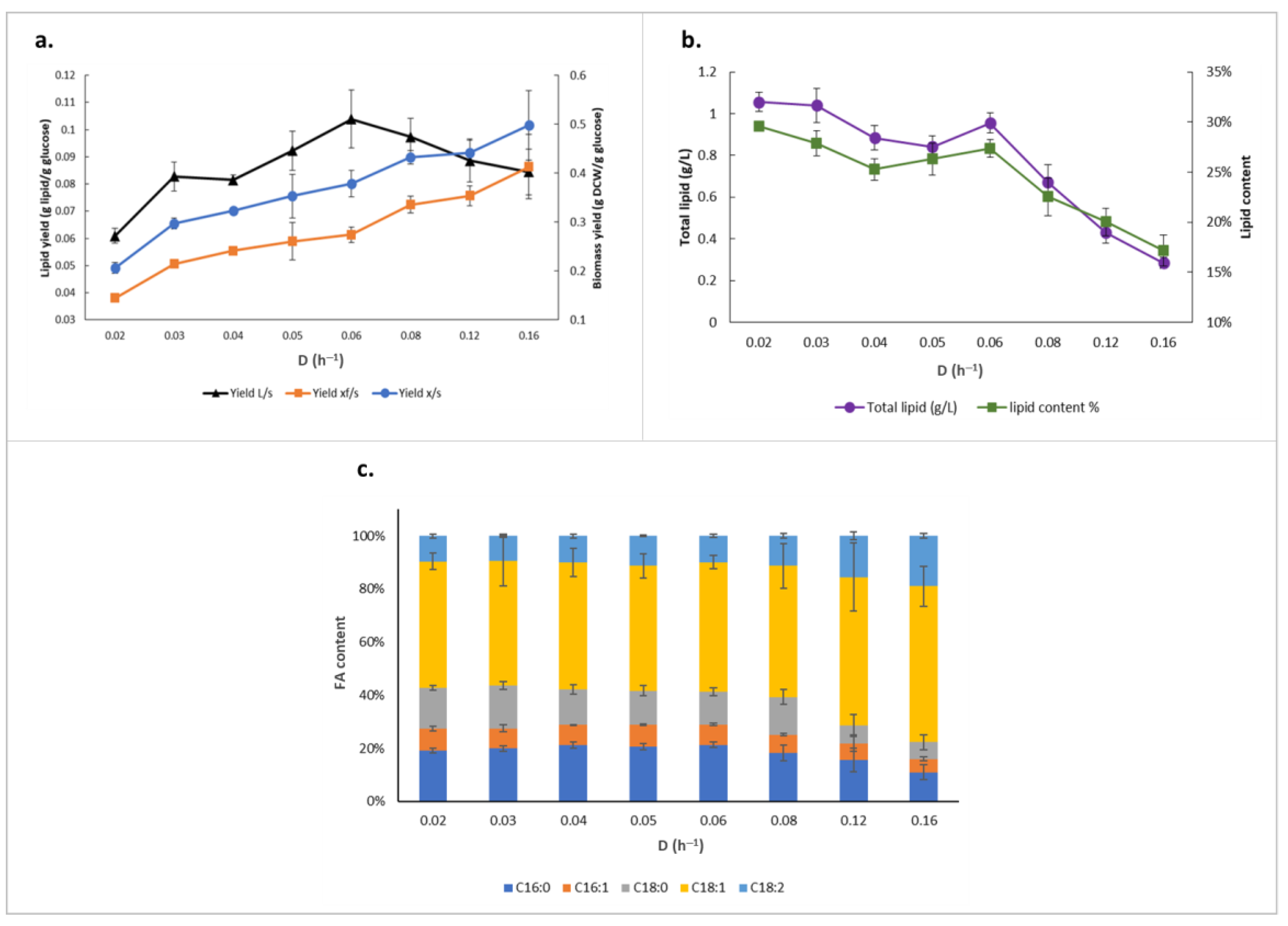
2.1. Relationship between Growth and Lipid Accumulation in Y. lipolytica
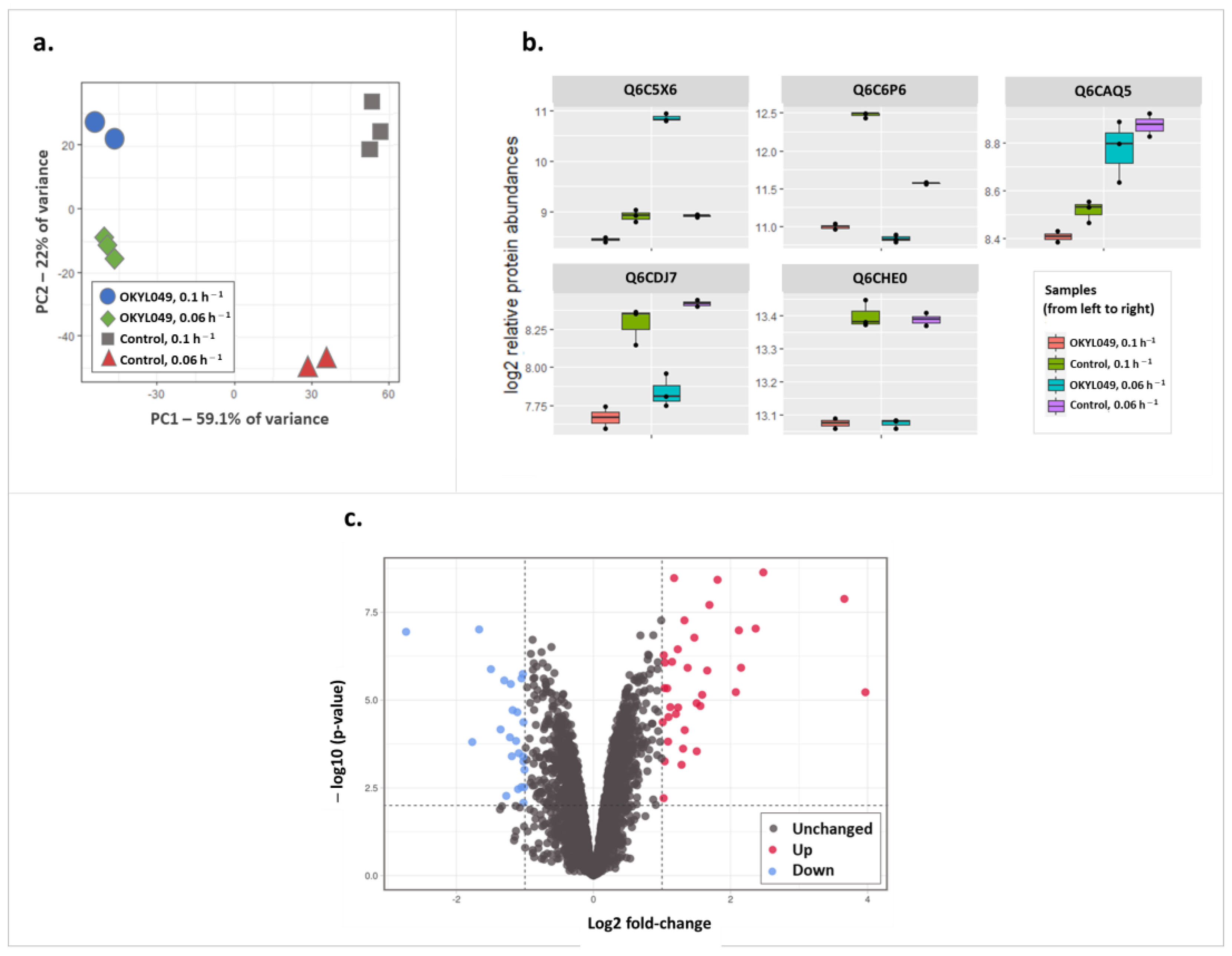
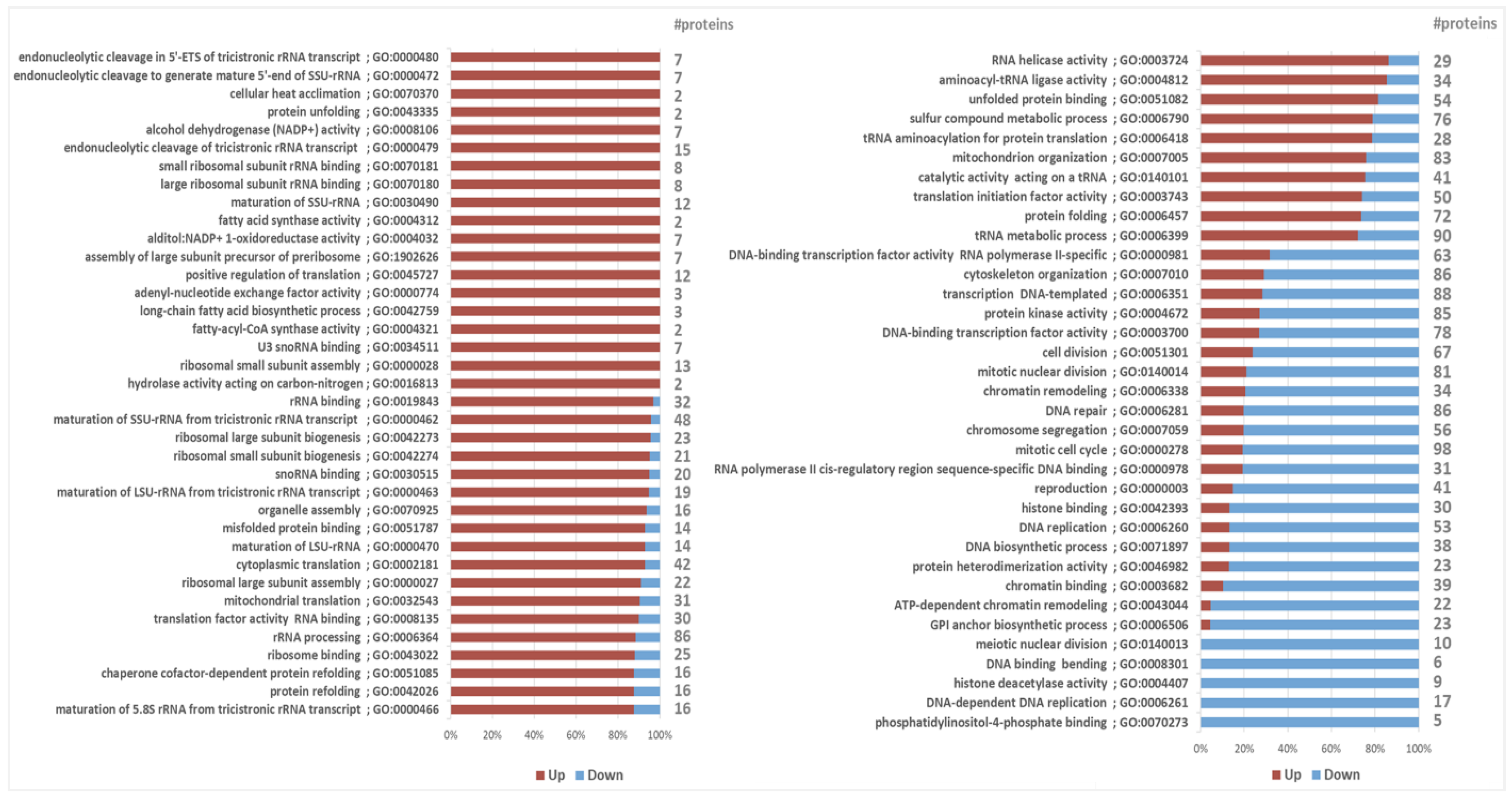
2.2. Effect of Growth Rate on Proteome Composition of Y. lipolytica during Lipogenic Conditions
2.2.1. Alternative NAPDH Suppliers for Lipid Accumulation
2.2.2. Boosting Precursor Supply
2.2.3. The Link between Unfolded Protein Response (UPR) and Lipid Accumulation
2.2.4. Sulfur Amino Acid (SAA) Metabolism May Affect Lipid Accumulation
2.2.5. Other Differentially Expressed Proteins and Protein Sets
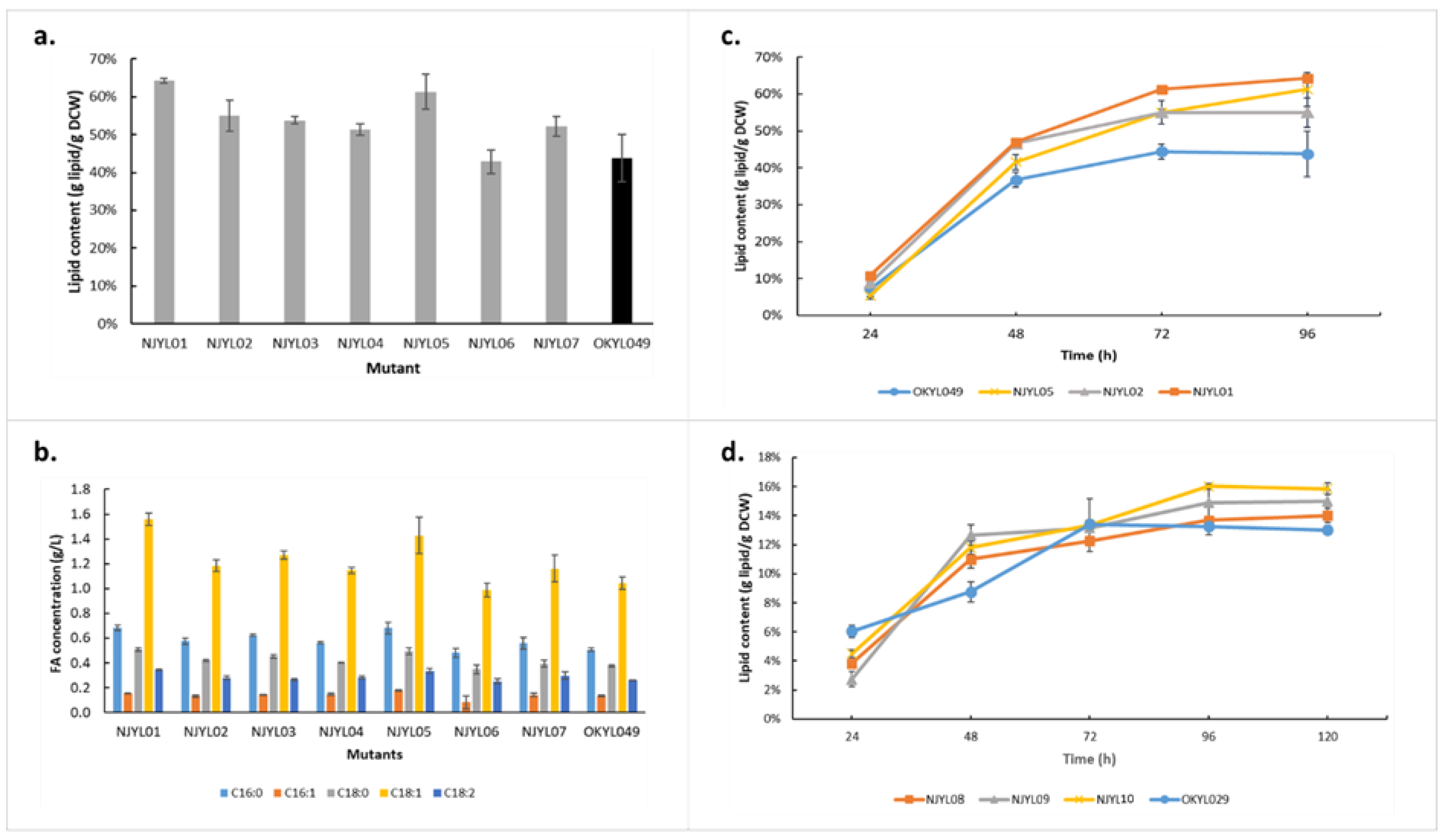
2.3. Strain Engineering
3. Materials and Methods
3.1. Yeast Strains, Cultivation, and Sample Collection
3.2. Strain Construction
3.3. Exometabolic Analysis
3.4. Lipid Extraction and Quantification
3.5. Proteomic Analysis
3.5.1. Protein Extraction
3.5.2. Tryptic Digestion and Tandem Mass Tag (TMT) Labeling
3.5.3. nLC-MS/MS
3.5.4. Proteomic Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Markham, K.A.; Alper, H.S. Synthetic Biology Expands the Industrial Potential of Yarrowia Lipolytica. Trends Biotechnol. 2018, 36, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Poorinmohammad, N.; Kerkhoven, E.J. Systems-level Approaches for Understanding and Engineering of the Oleaginous Cell Factory Yarrowia Lipolytica. Biotechnol. Bioeng. 2021, 118, 3640–3654. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Shen, H.; Zhou, W.; Wang, Y.; Yang, X.; Zhao, Z.K. Efficient Conversion of Acetate into Lipids by the Oleaginous Yeast Cryptococcus Curvatus. Biotechnol. Biofuels 2015, 8, 189. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Gong, Z.; Yang, X.; Jin, G.; Bai, F.; Zhao, Z.K. Kinetics of Continuous Cultivation of the Oleaginous Yeast Rhodosporidium Toruloides. J. Biotechnol. 2013, 168, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, S.; Aggelis, G. Lipid Production by Yarrowia Lipolytica Growing on Industrial Glycerol in a Single-Stage Continuous Culture. Bioresour. Technol. 2002, 82, 43–49. [Google Scholar] [CrossRef]
- Brown, B.D.; Hsu, K.H.; Hammond, E.G.; Glatz, B.A. A Relationship between Growth and Lipid Accumulation in Candida Curvata D. J. Ferment. Bioeng. 1989, 68, 344–352. [Google Scholar] [CrossRef]
- Konzock, O.; Norbeck, J. Deletion of MHY1 Abolishes Hyphae Formation in Yarrowia Lipolytica without Negative Effects on Stress Tolerance. PLoS ONE 2020, 15, e0231161. [Google Scholar] [CrossRef] [PubMed]
- Gajdoš, P.; Nicaud, J.-M.; Rossignol, T.; Čertík, M. Single Cell Oil Production on Molasses by Yarrowia Lipolytica Strains Overexpressing DGA2 in Multicopy. Appl. Microbiol. Biotechnol. 2015, 99, 8065–8074. [Google Scholar] [CrossRef] [PubMed]
- Beopoulos, A.; Haddouche, R.; Kabran, P.; Dulermo, T.; Chardot, T.; Nicaud, J.-M. Identification and Characterization of DGA2, an Acyltransferase of the DGAT1 Acyl-CoA: Diacylglycerol Acyltransferase Family in the Oleaginous Yeast Yarrowia Lipolytica. New Insights into the Storage Lipid Metabolism of Oleaginous Yeasts. Appl. Microbiol. Biotechnol. 2012, 93, 1523–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajdoš, P.; Ledesma-Amaro, R.; Nicaud, J.-M.; Čertík, M.; Rossignol, T. Overexpression of Diacylglycerol Acyltransferase in Yarrowia Lipolytica Affects Lipid Body Size, Number and Distribution. FEMS Yeast Res. 2016, 16, fow062. [Google Scholar] [CrossRef] [Green Version]
- Napora-Wijata, K.; Strohmeier, G.A.; Sonavane, M.N.; Avi, M.; Robins, K.; Winkler, M. Enantiocomplementary Yarrowia Lipolytica Oxidoreductases: Alcohol Dehydrogenase 2 and Short Chain Dehydrogenase/Reductase. Biomolecules 2013, 3, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Chen, H.; Tang, X.; Zhang, H.; Hao, G.; Chen, W.; Chen, Y.Q. The Role of Glyceraldehyde-3-Phosphate Dehydrogenases in NADPH Supply in the Oleaginous Filamentous Fungus Mortierella Alpina. Front. Microbiol. 2020, 11, 818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; He, Y.; Sen, B.; Wang, G. Reactive Oxygen Species and Their Applications toward Enhanced Lipid Accumulation in Oleaginous Microorganisms. Bioresour. Technol. 2020, 307, 123234. [Google Scholar] [CrossRef] [PubMed]
- Kieliszek, M.; Dourou, M. Effect of Selenium on the Growth and Lipid Accumulation of Yarrowia Lipolytica Yeast. Biol. Trace Elem. Res. 2021, 199, 1611–1622. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.; Holkenbrink, C.; Marella, E.R.; Wang, G.; Liebal, U.; Lieven, C.; Weber, D.; McCloskey, D.; Wang, H.-L.; Ebert, B.E. Corrigendum: Multi-Omics Analysis of Fatty Alcohol Production in Engineered Yeasts Saccharomyces Cerevisiae and Yarrowia Lipolytica. Front. Genet. 2021, 11, 637738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Jagtap, S.S.; Deewan, A.; Rao, C. V PH Selectively Regulates Citric Acid and Lipid Production in Yarrowia Lipolytica W29 during Nitrogen-Limited Growth on Glucose. J. Biotechnol. 2019, 290, 10–15. [Google Scholar] [CrossRef]
- Wasylenko, T.M.; Ahn, W.S.; Stephanopoulos, G. The Oxidative Pentose Phosphate Pathway Is the Primary Source of NADPH for Lipid Overproduction from Glucose in Yarrowia Lipolytica. Metab. Eng. 2015, 30, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Dulermo, T.; Lazar, Z.; Dulermo, R.; Rakicka, M.; Haddouche, R.; Nicaud, J.-M. Analysis of ATP-Citrate Lyase and Malic Enzyme Mutants of Yarrowia Lipolytica Points out the Importance of Mannitol Metabolism in Fatty Acid Synthesis. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2015, 1851, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Matthew, D. Truppo Cofactor Recycling for Enzyme Catalyzed Processes. In Comprehensive Chirality; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; pp. 46–70. [Google Scholar]
- Van Rossum, H.M.; Kozak, B.U.; Pronk, J.T.; van Maris, A.J.A. Engineering Cytosolic Acetyl-Coenzyme A Supply in Saccharomyces Cerevisiae: Pathway Stoichiometry, Free-Energy Conservation and Redox-Cofactor Balancing. Metab. Eng. 2016, 36, 99–115. [Google Scholar] [CrossRef]
- Kerkhoven, E.J.; Pomraning, K.R.; Baker, S.E.; Nielsen, J. Regulation of Amino-Acid Metabolism Controls Flux to Lipid Accumulation in Yarrowia Lipolytica. Npj Syst. Biol. Appl. 2016, 2, 16005. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Zhang, S.; Liu, H.; Shen, H.; Lin, X.; Yang, F.; Zhou, Y.J.; Jin, G.; Ye, M.; Zou, H. A Multi-Omic Map of the Lipid-Producing Yeast Rhodosporidium Toruloides. Nat. Commun. 2012, 3, 1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, M.F. Vitamin Formation from Fatty Acid Precursors. In Biogenesis of Fatty Acids, Lipids and Membranes; Springer International Publishing: Cham, Switzerland, 2019; pp. 259–271. [Google Scholar]
- Shi, K.; Gao, Z.; Shi, T.-Q.; Song, P.; Ren, L.-J.; Huang, H.; Ji, X.-J. Reactive Oxygen Species-Mediated Cellular Stress Response and Lipid Accumulation in Oleaginous Microorganisms: The State of the Art and Future Perspectives. Front. Microbiol. 2017, 8, 793. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Qiao, K.; Stephanopoulos, G. Engineering Oxidative Stress Defense Pathways to Build a Robust Lipid Production Platform in Yarrowia Lipolytica. Biotechnol. Bioeng. 2017, 114, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Meng, T.; Huang, H.; Zhuang, H.; He, Z.; Yang, H.; Feng, D. Molecular Machineries and Physiological Relevance of ER-Mediated Membrane Contacts. Theranostics 2021, 11, 974. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.J.; Liao, P.-C.; Tan, G.; Vevea, J.D.; Sing, C.N.; Tsang, C.A.; McCaffery, J.M.; Boldogh, I.R.; Pon, L.A. Membrane Dynamics and Protein Targets of Lipid Droplet Microautophagy during ER Stress-Induced Proteostasis in the Budding Yeast, Saccharomyces Cerevisiae. Autophagy 2021, 17, 2363–2383. [Google Scholar] [CrossRef]
- Ruiz-Lopez, N.; Pérez-Sancho, J.; Del Valle, A.E.; Haslam, R.P.; Vanneste, S.; Catalá, R.; Perea-Resa, C.; Van Damme, D.; García-Hernández, S.; Albert, A. Synaptotagmins at the Endoplasmic Reticulum–Plasma Membrane Contact Sites Maintain Diacylglycerol Homeostasis during Abiotic Stress. Plant Cell 2021, 33, 2431–2453. [Google Scholar] [CrossRef]
- Manford, A.G.; Stefan, C.J.; Yuan, H.L.; MacGurn, J.A.; Emr, S.D. ER-to-Plasma Membrane Tethering Proteins Regulate Cell Signaling and ER Morphology. Dev. Cell 2012, 23, 1129–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quon, E.; Nenadic, A.; Zaman, M.F.; Johansen, J.; Beh, C.T. ER-PM Membrane Contact Site Regulation by Yeast ORPs and Membrane Stress Pathways. PLoS Genet. 2022, 18, e1010106. [Google Scholar] [CrossRef]
- Jacquier, N.; Choudhary, V.; Mari, M.; Toulmay, A.; Reggiori, F.; Schneiter, R. Lipid Droplets Are Functionally Connected to the Endoplasmic Reticulum in Saccharomyces Cerevisiae. J. Cell Sci. 2011, 124, 2424–2437. [Google Scholar] [CrossRef] [Green Version]
- Rennert, C.; Heil, T.; Schicht, G.; Stilkerich, A.; Seidemann, L.; Kegel-Hübner, V.; Seehofer, D.; Damm, G. Prolonged Lipid Accumulation in Cultured Primary Human Hepatocytes Rather Leads to ER Stress than Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 7097. [Google Scholar] [CrossRef]
- Song, M.J.; Malhi, H. The Unfolded Protein Response and Hepatic Lipid Metabolism in Non Alcoholic Fatty Liver Disease. Pharmacol. Ther. 2019, 203, 107401. [Google Scholar] [CrossRef] [PubMed]
- Moncan, M.; Mnich, K.; Blomme, A.; Almanza, A.; Samali, A.; Gorman, A.M. Regulation of Lipid Metabolism by the Unfolded Protein Response. J. Cell. Mol. Med. 2021, 25, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kaufman, R.J. The Role of ER Stress in Lipid Metabolism and Lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yang, Q.; Mao, Z. Signaling and Induction of Chaperone-Mediated Autophagy by the Endoplasmic Reticulum under Stress Conditions. Autophagy 2018, 14, 1094–1096. [Google Scholar] [CrossRef] [PubMed]
- Kamisaka, Y.; Kimura, K.; Uemura, H.; Yamaoka, M. Addition of Methionine and Low Cultivation Temperatures Increase Palmitoleic Acid Production by Engineered Saccharomyces Cerevisiae. Appl. Microbiol. Biotechnol. 2015, 99, 201–210. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Yang, Y.; Yang, S.; Donohoe, B.S.; Van Wychen, S.; Zhang, M.; Himmel, M.E.; Knoshaug, E.P. Oleaginicity of the Yeast Strain Saccharomyces Cerevisiae D5A. Biotechnol. Biofuels 2018, 11, 258. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; Vowinckel, J.; Keller, M.A.; Ralser, M. Methionine Metabolism Alters Oxidative Stress Resistance via the Pentose Phosphate Pathway 2016. Antioxid. Redox Signal. 2016, 24, 543–547. [Google Scholar] [CrossRef] [Green Version]
- Werstuck, G.H.; Lentz, S.R.; Dayal, S.; Hossain, G.S.; Sood, S.K.; Shi, Y.Y.; Zhou, J.; Maeda, N.; Krisans, S.K.; Malinow, M.R. Homocysteine-Induced Endoplasmic Reticulum Stress Causes Dysregulation of the Cholesterol and Triglyceride Biosynthetic Pathways. J. Clin. Invest. 2001, 107, 1263–1273. [Google Scholar] [CrossRef] [Green Version]
- Higgins, R.; Gendron, J.M.; Rising, L.; Mak, R.; Webb, K.; Kaiser, S.E.; Zuzow, N.; Riviere, P.; Yang, B.; Fenech, E. The Unfolded Protein Response Triggers Site-Specific Regulatory Ubiquitylation of 40S Ribosomal Proteins. Mol. Cell 2015, 59, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Hamanaka, R.B.; Bobrovnikova-Marjon, E.; Gordan, J.D.; Dai, M.-S.; Lu, H.; Simon, M.C.; Diehl, J.A. Ribosomal Stress Couples the Unfolded Protein Response to P53-Dependent Cell Cycle Arrest. J. Biol. Chem. 2006, 281, 30036–30045. [Google Scholar] [CrossRef] [Green Version]
- Shcherbakov, D.; Teo, Y.; Boukari, H.; Cortes-Sanchon, A.; Mantovani, M.; Osinnii, I.; Moore, J.; Juskeviciene, R.; Brilkova, M.; Duscha, S. Ribosomal Mistranslation Leads to Silencing of the Unfolded Protein Response and Increased Mitochondrial Biogenesis. Commun. Biol. 2019, 2, 381. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Kim, H.; Kim, I. Ribosomal Protein L19 Overexpression Activates the Unfolded Protein Response and Sensitizes MCF7 Breast Cancer Cells to Endoplasmic Reticulum Stress-Induced Cell Death. Biochem. Biophys. Res. Commun. 2014, 450, 673–678. [Google Scholar] [CrossRef]
- Herrero-de-Dios, C.; Román, E.; Pla, J.; Alonso-Monge, R. Hog1 Controls Lipids Homeostasis upon Osmotic Stress in Candida Albicans. J. Fungi 2020, 6, 355. [Google Scholar] [CrossRef]
- Manzanares-Estreder, S.; Espí-Bardisa, J.; Alarcón, B.; Pascual-Ahuir, A.; Proft, M. Multilayered Control of Peroxisomal Activity upon Salt Stress in S Accharomyces Cerevisiae. Mol. Microbiol. 2017, 104, 851–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy Regulates Lipid Metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Marella, E.R.; Dahlin, J.; Dam, M.I.; Ter Horst, J.; Christensen, H.B.; Sudarsan, S.; Wang, G.; Holkenbrink, C.; Borodina, I. A Single-Host Fermentation Process for the Production of Flavor Lactones from Non-Hydroxylated Fatty Acids. Metab. Eng. 2020, 61, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Holkenbrink, C.; Dam, M.I.; Kildegaard, K.R.; Beder, J.; Dahlin, J.; Doménech Belda, D.; Borodina, I. EasyCloneYALI: CRISPR/Cas9-Based Synthetic Toolbox for Engineering of the Yeast Yarrowia Lipolytica. Biotechnol. J. 2018, 13, 1700543. [Google Scholar] [CrossRef] [Green Version]
- Konzock, O.; Zaghen, S.; Norbeck, J. Tolerance of Yarrowia Lipolytica to Inhibitors Commonly Found in Lignocellulosic Hydrolysates. BMC Microbiol. 2021, 21, 77. [Google Scholar] [CrossRef]
- Yu, R.; Vorontsov, E.; Sihlbom, C.; Nielsen, J. Quantifying Absolute Gene Expression Profiles Reveals Distinct Regulation of Central Carbon Metabolism Genes in Yeast. Elife 2021, 10, e65722. [Google Scholar] [CrossRef]
- Qiao, K.; Abidi, S.H.I.; Liu, H.; Zhang, H.; Chakraborty, S.; Watson, N.; Ajikumar, P.K.; Stephanopoulos, G. Engineering Lipid Overproduction in the Oleaginous Yeast Yarrowia Lipolytica. Metab. Eng. 2015, 29, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal Sample Preparation Method for Proteome Analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef]
- Zhu, Y.; Orre, L.M.; Tran, Y.Z.; Mermelekas, G.; Johansson, H.J.; Malyutina, A.; Anders, S.; Lehtiö, J. DEqMS: A Method for Accurate Variance Estimation in Differential Protein Expression Analysis. Mol. Cell. Proteom. 2020, 19, 1047–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Väremo, L.; Nielsen, J.; Nookaew, I. Enriching the Gene Set Analysis of Genome-Wide Data by Incorporating Directionality of Gene Expression and Combining Statistical Hypotheses and Methods. Nucleic Acids Res. 2013, 41, 4378–4391. [Google Scholar] [CrossRef]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S.; Hub, A.; Group, W.P.W. AmiGO: Online Access to Ontology and Annotation Data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A Universal Tool for Annotation, Visualization and Analysis in Functional Genomics Research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S. The InterPro Protein Families and Domains Database: 20 Years On. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: The Universal Protein Knowledgebase in 2021. Nuclein Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dilution Rate (h−1) | Residual Glucose (g/L) | q-Glucose (g/g/h) | Total Lipid (g/L) | Lipid Content (%) | Non-Lipid CDW (g/L) | CDW (g/L) |
|---|---|---|---|---|---|---|
| 0.02 | 10.03 ± 0.63 | 0.1 ± 0.005 | 1.06 ± 0.05 | 30 ± 0.46 | 2.51 ± 0.11 | 3.57 ± 0.16 |
| 0.03 | 11.94 ± 0.98 | 0.1 ± 0.004 | 1.04 ± 0.08 | 28 ± 1.27 | 2.68 ± 0.05 | 3.72 ± 0.12 |
| 0.04 | 15.05 ± 0.47 | 0.12 ± 0.003 | 0.88 ± 0.06 | 25 ± 1.09 | 2.61 ± 0.05 | 3.49 ± 0.10 |
| 0.05 | 16.58 ± 0.17 | 0.14 ± 0.019 | 0.84 ± 0.05 | 26 ± 1.61 | 2.36 ± 0.18 | 3.20 ± 0.20 |
| 0.06 | 16.44 ± 0.16 | 0.16 ± 0.011 | 0.95 ± 0.05 | 27 ± 0.89 | 2.53 ± 0.03 | 3.48 ± 0.07 |
| 0.08 | 18.68 ± 0.40 | 0.19 ± 0.006 | 0.67 ± 0.08 | 23 ± 1.90 | 2.30 ± 0.20 | 2.97 ± 0.26 |
| 0.12 | 19.61 ± 0.10 | 0.27 ± 0.015 | 0.43 ± 0.05 | 20 ± 1.39 | 1.71 ± 0.10 | 2.14 ± 0.14 |
| 0.16 | 20.74 ± 0.17 | 0.33 ± 0.054 | 0.28 ± 0.03 | 17 ± 1.55 | 1.37 ± 0.03 | 1.66 ± 0.03 |
| Protein Accession | Protein Annotation | GO (Molecular Function) | Change | log2 (FC) | −log10 (p-Value) |
|---|---|---|---|---|---|
| Q6C5X6 | Formate dehydrogenase | formate dehydrogenase (NAD+) activity [GO:0008863]; oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor [GO:0016616] | Up | 2.48 | 8.64 |
| Q6CAP1 | - | carbohydrate:proton symporter activity [GO:0005351] | Up | 3.66 | 7.88 |
| F2Z678 | Alcohol dehydrogenase 2 | alcohol dehydrogenase (NAD+) activity [GO:0004022]; zinc ion binding [GO:0008270] | Up | 1.81 | 8.42 |
| Q6CI12 | - | hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds, in linear amidines [GO:0016813]; peptidase activity [GO:0008233] | Up | 1.18 | 8.47 |
| Q6C676 | - | - | Up | 1.69 | 7.71 |
| Q6CBW3 | - | - | Down | 2.73 | 6.94 |
| Q92389 | Acid extracellular protease | aspartic-type endopeptidase activity [GO:0004190] | Up | 2.37 | 7.04 |
| Q6CBQ1 | Superoxide dismutase | manganese ion binding [GO:0030145]; superoxide dismutase activity [GO:0004784] | Up | 1.33 | 7.27 |
| Q6C395 | Triose phosphate isomerase | triose-phosphate isomerase activity [GO:0004807] | Up | 2.12 | 6.99 |
| Q6C8H3 | - | hydrolase activity, hydrolyzing O-glycosyl compounds [GO:0004553] | Down | 1.67 | 7.01 |
| Mutant (in OKYL049) | Mutant (in OKYL029) | Target Accession | Target Name | Biological Significance | FC Direction and p-Value | Mutation Strategy |
|---|---|---|---|---|---|---|
| NJYL01 | NJYL08 | Q6C903 | Biotin synthase | Biotin shares biosynthetic precursors with lipids | Down, 5.4 × 10−3 | Knockout |
| NJYL02 | NJYL09 | Q6C6P6 | Arginase | Puts burden on cell energetically, produces ammonia to alleviate nitrogen limitation | Down, 5.4 × 10−7 | Knockout |
| NJYL03 | Q6CA74 | PapD-like protein | ER-PM tether which maintains ER homeostasis | Down, 9.8 × 10−5 | Knockout | |
| NJYL04 | Q6C375 | Similar to MdM1 | Role in lipophagy | Down, 2 × 10−3 | Knockout | |
| NJYL05 | NJYL10 | Q6C4M9 | HOG1 | A MAPK known to affect lipogenesis during osmotic stress | Down, 1.5 × 10−3 | Knockout |
| NJYL06 and NJYL07 | Q6C5X6 | Formate dehydrogenase | NADPH is a key cofactor for lipid biogenesis | Up, 2.3 × 10−9 | Overexpression, different promoters |
| Y. lipolytica Strain | Genotype | Reference |
|---|---|---|
| OKYL049 | MATa, ∆ku70::Cas9::DsdA, lntE1::Tef1inp-DGA1PEX20t, Δare1Δmhy1 | [50] |
| OKYL029 | MATa, ∆ku70::Cas9::DsdA, Δmhy1 | [7] |
| JFYL007 (Q4) | MATa, ∆ku70::cas9∆mhy1∆ARE1∆LRO1∆DGA1∆DGA2 | Unpublished |
| NJYL01 | MATa, ∆ku70::Cas9::DsdA, lntE1::Tef1inp-DGA1PEX20t, Δare1Δmhy1ΔYALI0D15400g | This study |
| NJYL02 | MATa, ∆ku70::Cas9::DsdA, lntE1::Tef1inp-DGA1PEX20t, Δare1Δmhy1ΔYALI0E07535g | This study |
| NJYL03 | MATa, ∆ku70::Cas9::DsdA, lntE1::Tef1inp-DGA1PEX20t, Δare1Δmhy1ΔYALI0D05291g | This study |
| NJYL04 | MATa, ∆ku70::Cas9::DsdA, lntE1::Tef1inp-DGA1PEX20t, Δare1Δmhy1ΔYALI0F02035g | This study |
| NJYL05 | MATa, ∆ku70::Cas9::DsdA, lntE1::Tef1inp-DGA1PEX20t, Δare1Δmhy1ΔYALI0E25135g | This study |
| NJYL06 | MATa, ∆ku70::Cas9::DsdA, lntE1::Tef1inp-DGA1PEX20t, Δare1Δmhy1, lntD1::Tefp- YALI0E14256g-lip2t | This study |
| NJYL07 | MATa, ∆ku70::Cas9::DsdA, lntE1::Tef1inp-DGA1PEX20t, Δare1Δmhy1, lntD1::Tefinp-YALI0E14256g-lip2t | This study |
| NJYL08 | MATa, ∆ku70::Cas9::DsdA, Δmhy1 ΔYALI0D15400g | This study |
| NJYL09 | MATa, ∆ku70::Cas9::DsdA, Δmhy1 ΔYALI0E07535g | This study |
| NJYL10 | MATa, ∆ku70::Cas9::DsdA, Δmhy1 ΔYALI0E25135g | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poorinmohammad, N.; Fu, J.; Wabeke, B.; Kerkhoven, E.J. Validated Growth Rate-Dependent Regulation of Lipid Metabolism in Yarrowia lipolytica. Int. J. Mol. Sci. 2022, 23, 8517. https://doi.org/10.3390/ijms23158517
Poorinmohammad N, Fu J, Wabeke B, Kerkhoven EJ. Validated Growth Rate-Dependent Regulation of Lipid Metabolism in Yarrowia lipolytica. International Journal of Molecular Sciences. 2022; 23(15):8517. https://doi.org/10.3390/ijms23158517
Chicago/Turabian StylePoorinmohammad, Naghmeh, Jing Fu, Bob Wabeke, and Eduard J. Kerkhoven. 2022. "Validated Growth Rate-Dependent Regulation of Lipid Metabolism in Yarrowia lipolytica" International Journal of Molecular Sciences 23, no. 15: 8517. https://doi.org/10.3390/ijms23158517
APA StylePoorinmohammad, N., Fu, J., Wabeke, B., & Kerkhoven, E. J. (2022). Validated Growth Rate-Dependent Regulation of Lipid Metabolism in Yarrowia lipolytica. International Journal of Molecular Sciences, 23(15), 8517. https://doi.org/10.3390/ijms23158517