
Osteopathy in Complex Lymphatic Anomalies

 , ,
, , 
Abstract
:1. Introduction
2. Complex Lymphatic Anomalies (CLAs)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of CLA | Bone Phenotype | Defining Pathology | Somatic Mutations | References |
|---|---|---|---|---|
| Gorham Stout Disease (GSD) | Localized cortical and trabecular osteolysis | Massive bone destruction and resorption | KRAS | [53,59] |
| Generalized Lymphatic Anomaly (GLA) | Generalized trabecular osteolysis | Multi-organ, multicentric, proliferative lesions | PIK3CA, NRAS | [49,54,60] |
| Kaposiform Lymphangiomatosis (KLA) | Generalized trabecular osteolysis | Multi-organ, multicentric, proliferative lesions | NRAS, BAD, TSC1, CBL | [52,55,61,62] |
| Central Conducting Lymphatic Anomaly (CCLA) | Channel-like osseous lesions | Dilated central conducting channels | EPHB4, ARAF, MDFIC | [56,58,63] |
2.1. Gorham Stout Disease (GSD)
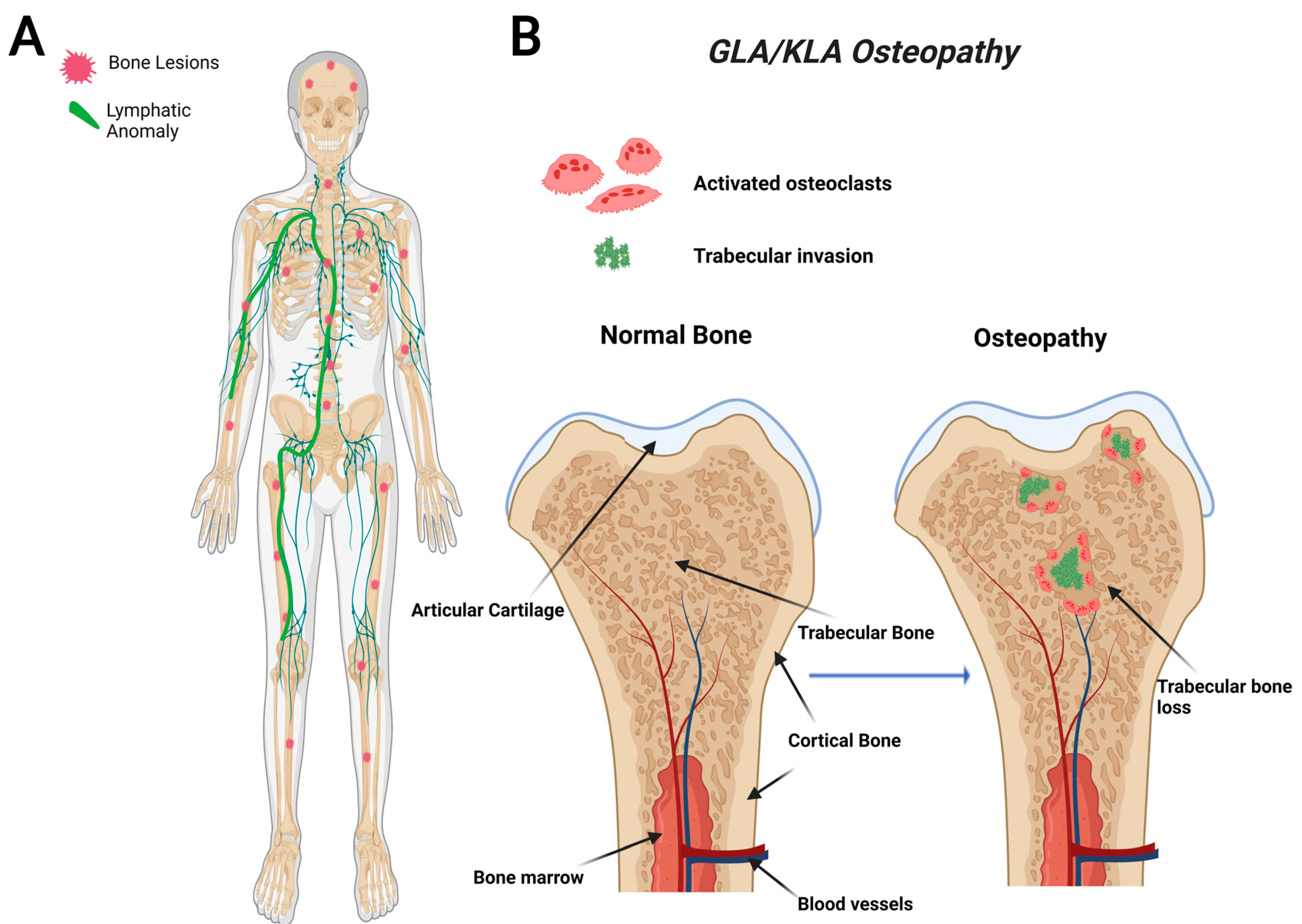
2.2. General Lymphatic Anomaly (GLA)
2.3. Kaposiform Lymphangiomatosis (KLA)
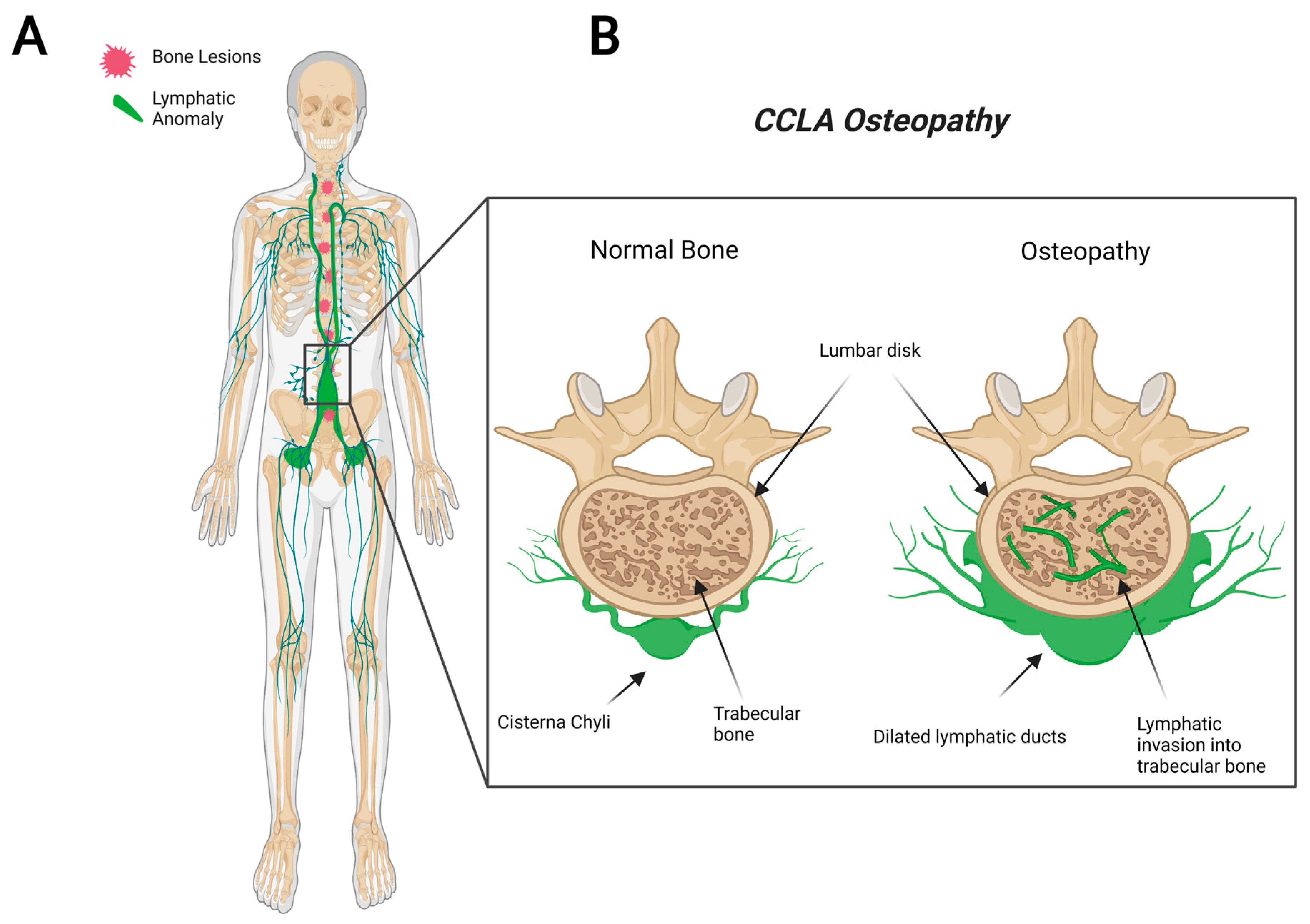
2.4. Central Conducting Lymphatic Anomaly (CCLA)
3. Lymphatic Bone Invasion
4. Current Knowledge of CLA Osteopathy
5. Proposed CLA Osteopathy
6. Osteocyte Osteolysis in GSD
7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Azzali, G. Transendothelial transport of lipids in the absorbing lymphatic vessel. Experientia 1982, 38, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Randolph, G.J.; Miller, N.E. Lymphatic transport of high-density lipoproteins and chylomicrons. J. Clin. Investig. 2014, 124, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Swartz, M.A.; Kaipainen, A.; Netti, P.; Brekken, C.; Boucher, Y.; Grodzinsky, A.J.; Jain, R.K. Mechanics of interstitial-lymphatic fluid transport: Theoretical foundation and experimental validation. J. Biomech. 1999, 32, 1297–1307. [Google Scholar] [CrossRef]
- Alderfer, L.; Wei, A.; Hanjaya-Putra, D. Lymphatic Tissue Engineering and Regeneration. J. Biol. Eng. 2018, 12, 32. [Google Scholar] [CrossRef]
- Petrova, T.V.; Koh, G.Y. Organ-specific lymphatic vasculature: From development to pathophysiology. J. Exp. Med. 2017, 215, 35–49. [Google Scholar] [CrossRef]
- Martinez-Corral, I.; Ulvmar, M.H.; Stanczuk, L.; Tatin, F.; Kizhatil, K.; John, S.W.; Alitalo, K.; Ortega, S.; Makinen, T. Nonvenous origin of dermal lymphatic vasculature. Circ. Res. 2015, 116, 1649–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camara, A.; Cordeiro, O.G.; Alloush, F.; Sponsel, J.; Chypre, M.; Onder, L.; Asano, K.; Tanaka, M.; Yagita, H.; Ludewig, B.; et al. Lymph Node Mesenchymal and Endothelial Stromal Cells Cooperate via the RANK-RANKL Cytokine Axis to Shape the Sinusoidal Macrophage Niche. Immunity 2019, 50, 1467–1481.e6. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.W.; Luo, W.; Lv, C.L. lncRNA-MIAT facilitates the differentiation of adipose-derived mesenchymal stem cells into lymphatic endothelial cells via the miR-495/Prox1 axis. Mol. Med. Rep. 2021, 23, 323. [Google Scholar] [CrossRef]
- Robering, J.W.; Weigand, A.; Pfuhlmann, R.; Horch, R.E.; Beier, J.P.; Boos, A.M. Mesenchymal stem cells promote lymphangiogenic properties of lymphatic endothelial cells. J. Cell. Mol. Med. 2018, 22, 3740–3750. [Google Scholar] [CrossRef]
- Nicenboim, J.; Malkinson, G.; Lupo, T.; Asaf, L.; Sela, Y.; Mayseless, O.; Gibbs-Bar, L.; Senderovich, N.; Hashimshony, T.; Shin, M.; et al. Lymphatic vessels arise from specialized angioblasts within a venous niche. Nature 2015, 522, 56–61. [Google Scholar] [CrossRef]
- Rodriguez-Niedenführ, M.; Papoutsi, M.; Christ, B.; Nicolaides, K.; Von Kaisenberg, C.; Tomarev, S.I.; Wilting, J. Prox1 is a marker of ectodermal placodes, endodermal compartments, lymphatic endothelium and lymphangioblasts. Anat. Embryol. 2001, 204, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.K.; Rauniyar, K.; Karpanen, T.; Leppänen, V.-M.; Brouillard, P.; Vikkula, M.; Alitalo, K.; Jeltsch, M. Efficient activation of the lymphangiogenic growth factor VEGF-C requires the C-terminal domain of VEGF-C and the N-terminal domain of CCBE1. Sci. Rep. 2017, 7, 4916. [Google Scholar] [CrossRef]
- Oh, S.-J.; Jeltsch, M.M.; Birkenhäger, R.; McCarthy, J.E.G.; Weich, H.A.; Christ, B.; Alitalo, K.; Wilting, J. VEGF and VEGF-C: Specific Induction of Angiogenesis and Lymphangiogenesis in the Differentiated Avian Chorioallantoic Membrane. Dev. Biol. 1997, 188, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Rauniyar, K.; Jha, S.K.; Jeltsch, M. Biology of Vascular Endothelial Growth Factor C in the Morphogenesis of Lymphatic Vessels. Front. Bioeng. Biotechnol. 2018, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, M.E.; Halford, M.M.; Roufail, S.; Williams, R.A.; Hibbs, M.L.; Grail, D.; Kubo, H.; Stacker, S.A.; Achen, M.G. Vascular endothelial growth factor D is dispensable for development of the lymphatic system. Mol. Cell. Biol. 2005, 25, 2441–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rissanen, T.; Markkanen, J.E.; Gruchala, M.; Heikura, T.; Puranen, A.; Kettunen, M.I.; Kholová, I.; Kauppinen, R.A.; Achen, M.; Stacker, S.; et al. VEGF-D Is the Strongest Angiogenic and Lymphangiogenic Effector Among VEGFs Delivered Into Skeletal Muscle via Adenoviruses. Circ. Res. 2003, 92, 1098–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef]
- Frara, N.; Abdelmagid, S.M.; Tytell, M.; Amin, M.; Popoff, S.N.; Safadi, F.F.; Barbe, M.F. Growth and repair factors, osteoactivin, matrix metalloproteinase and heat shock protein 72, increase with resolution of inflammation in musculotendinous tissues in a rat model of repetitive grasping. BMC Musculoskelet. Disord. 2016, 17, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, S.C., Jr.; Popoff, S.N. Bone cell biology: The regulation of development, structure, and function in the skeleton. Am. J. Anat. 1988, 183, 1–44. [Google Scholar] [CrossRef]
- Mellibovsky, L.; Diez, A.; Serrano, S.; Aubia, J.; Pérez-Vila, E.; Mariñoso, M.; Nogués, X.; Recker, R. Bone remodeling alterations in myelodysplastic syndrome. Bone 1996, 19, 401–405. [Google Scholar] [CrossRef]
- Landry, P.S.; Marino, A.A.; Sadasivan, K.K.; Albright, J.A. Bone injury response. An animal model for testing theories of regulation. Clin. Orthop. Relat. Res. 1996, 332, 260–273. [Google Scholar] [CrossRef]
- Leloup, G.; Lemoine, P.; Carmeliet, P.; Vaes, G. Bone resorption and response to calcium-regulating hormones in the absence of tissue or urokinase plasminogen activator or of their type 1 inhibitor. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 1996, 11, 1146–1157. [Google Scholar] [CrossRef]
- Tanoue, N.; Moedano, L.; Witte, M.; Montague, M.; Lukefahr, A.; Bernas, M. Primary versus trauma-induced Gorham-Stout disease. Lymphology 2018, 51, 18–27. [Google Scholar]
- Yerganyan, V.; Body, J.; Aubain, N.D.S.; Gebhart, M. Gorham–Stout disease of the proximal fibula treated with radiotherapy and zoledronic acid. J. Bone Oncol. 2015, 4, 42–46. [Google Scholar] [CrossRef]
- Prince, M.; Banerjee, C.; Javed, A.; Green, J.; Lian, J.B.; Stein, G.S.; Bodine, P.V.; Komm, B.S. Expression and regulation of Runx2/Cbfa1 and osteoblast phenotypic markers during the growth and differentiation of human osteoblasts. J. Cell. Biochem. 2001, 80, 424–440. [Google Scholar] [CrossRef]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Abdelmagid, S.M.; Belcher, J.Y.; Moussa, F.M.; Lababidi, S.L.; Sondag, G.R.; Novak, K.M.; Sanyurah, A.S.; Frara, N.A.; Razmpour, R.; Del Carpio-Cano, F.E.; et al. Mutation in Osteoactivin Decreases Bone Formation in Vivo and Osteoblast Differentiation in Vitro. Am. J. Pathol. 2014, 184, 697–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dworetzky, S.I.; Fey, E.G.; Penman, S.; Lian, J.B.; Stein, J.L.; Stein, G.S. Progressive changes in the protein composition of the nuclear matrix during rat osteoblast differentiation. Proc. Natl. Acad. Sci. USA 1990, 87, 4605–4609. [Google Scholar] [CrossRef] [Green Version]
- Yokota, K. [Inflammation and osteoclasts]. Nihon Rinsho Men’eki Gakkai Kaishi = Jpn. J. Clin. Immunol. 2017, 40, 367–376. [Google Scholar] [CrossRef]
- Safadi, F.F.; Xu, J.; Smock, S.L.; Kanaan, R.A.; Selim, A.-H.; Odgren, P.R.; Marks, S.C., Jr.; Owen, T.A.; Popoff, S.N. Expression of connective tissue growth factor in bone: Its role in osteoblast proliferation and differentiation in vitro and bone formation in vivo. J. Cell. Physiol. 2003, 196, 51–62. [Google Scholar] [CrossRef]
- Street, J.; Lenehan, B. Vascular endothelial growth factor regulates osteoblast survival—Evidence for an autocrine feedback mechanism. J. Orthop. Surg. Res. 2009, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deckers, M.M.L.; Karperien, M.; van der Bent, C.; Yamashita, T.; Papapoulos, S.E.; Löwik, C.W.G.M. Expression of Vascular Endothelial Growth Factors and Their Receptors during Osteoblast Differentiation. Endocrinology 2000, 141, 1667–1674. [Google Scholar] [CrossRef]
- Bodine, P.V.; Vernon, S.K.; Komm, B.S. Establishment and hormonal regulation of a conditionally transformed preosteocytic cell line from adult human bone. Endocrinology 1996, 137, 4592–4604. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, L.I.; Bivi, N. Chapter 3—Local Regulation of Bone Cell Function. In Basic and Applied Bone Biology; Burr, D.B., Allen, M.R., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 47–73. [Google Scholar]
- Marahleh, A.; Kitaura, H.; Ohori, F.; Kishikawa, A.; Ogawa, S.; Shen, W.-R.; Qi, J.; Noguchi, T.; Nara, Y.; Mizoguchi, I. TNF-α Directly Enhances Osteocyte RANKL Expression and Promotes Osteoclast Formation. Front. Immunol. 2019, 10, 2925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogawa, M.; Khalid, K.A.; Wijenayaka, A.R.; Ormsby, R.T.; Evdokiou, A.; Anderson, P.; Findlay, D.M.; Atkins, G.J. Recombinant sclerostin antagonizes effects of ex vivo mechanical loading in trabecular bone and increases osteocyte lacunar size. Am. J. Physiol. Cell Physiol. 2018, 314, C53–C61. [Google Scholar] [CrossRef]
- Sondag, G.R.; Mbimba, T.S.; Moussa, F.M.; Novak, K.; Yu, B.; Jaber, F.A.; Abdelmagid, S.M.; Geldenhuys, W.J.; Safadi, F.F. Osteoactivin inhibition of osteoclastogenesis is mediated through CD44-ERK signaling. Exp. Mol. Med. 2016, 48, e257. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.; Sondag, G.R.; Malcuit, C.; Kim, M.-H.; Safadi, F.F. Macrophage-Associated Osteoactivin/GPNMB Mediates Mesenchymal Stem Cell Survival, Proliferation, and Migration Via a CD44-Dependent Mechanism. J. Cell. Biochem. 2015, 117, 1511–1521. [Google Scholar] [CrossRef]
- Bateman, J.P.; Safadi, F.F.; Susin, C.; Wikesjö, U.M.E. Exploratory study on the effect of osteoactivin on bone formation in the rat critical-size calvarial defect model. J. Periodontal Res. 2011, 47, 243–247. [Google Scholar] [CrossRef]
- Hirayama, T.; Sabokbar, A.; Itonaga, I.; Watt-Smith, S.; Athanasou, N.A. Cellular and humoral mechanisms of osteoclast formation and bone resorption in Gorham-Stout disease. J. Pathol. 2001, 195, 624–630. [Google Scholar] [CrossRef]
- Zhang, Q.; Guo, R.; Lu, Y.; Zhao, L.; Zhou, Q.; Schwarz, E.M.; Huang, J.; Chen, D.; Jin, Z.-G.; Boyce, B.F.; et al. VEGF-C, a lymphatic growth factor, is a RANKL target gene in osteoclasts that enhances osteoclastic bone resorption through an autocrine mechanism. J. Biol. Chem. 2008, 283, 13491–13499. [Google Scholar] [CrossRef] [Green Version]
- Iacobas, I.; Adams, D.M.; Pimpalwar, S.; Phung, T.; Blei, F.; Burrows, P.; Lopez-Gutierrez, J.C.; Levine, M.A.; Trenor, C.C., III. Multidisciplinary guidelines for initial evaluation of complicated lymphatic anomalies—Expert opinion consensus. Pediatric Blood Cancer 2020, 67, e28036. [Google Scholar] [CrossRef]
- Dupond, J.-L.; Bermont, L.; Runge, M.; de Billy, M. Plasma VEGF determination in disseminated lymphangiomatosis–Gorham–Stout syndrome: A marker of activity? A case report with a 5-year follow-up. Bone 2010, 46, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Yuan, X.-G.; Hu, X.-Y.; Shen, F.-R.; Wang, J.-A. Gorham-Stout syndrome in mainland China: A case series of 67 patients and review of the literature. J. Zhejiang Univ. Sci. B 2013, 14, 729–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möller, G.; Priemel, M.; Amling, M.; Werner, M.; Kuhlmey, A.S.; Delling, G. The Gorham-Stout syndrome (Gorham’s massive osteolysis). A report of six cases with histopathological findings. J. Bone Jt. Surgery. Br. Vol. 1999, 81, 501–506. [Google Scholar] [CrossRef]
- Gordon, K.; Varney, R.; Keeley, V.; Riches, K.; Jeffery, S.; Van Zanten, M.; Mortimer, P.; Ostergaard, P.; Mansour, S. Update and audit of the St George’s classification algorithm of primary lymphatic anomalies: A clinical and molecular approach to diagnosis. J. Med Genet. 2020, 57, 653–659. [Google Scholar] [CrossRef]
- Wassef, M.; Blei, F.; Adams, D.; Alomari, A.; Baselga, E.; Berenstein, A.; Burrows, P.; Frieden, I.J.; Garzon, M.C.; Lopez-Gutierrez, J.-C.; et al. Vascular Anomalies Classification: Recommendations from the International Society for the Study of Vascular Anomalies. Pediatrics 2015, 136, e203–e214. [Google Scholar] [CrossRef] [Green Version]
- Sevick-Muraca, E.M.; King, P.D. Lymphatic vessel abnormalities arising from disorders of Ras signal transduction. Trends Cardiovasc. Med. 2013, 24, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Laguna, L.; Agra, N.; Ibáñez, K.; Oliva-Molina, G.; Gordo, G.; Khurana, N.; Hominick, D.; Beato, M.; Colmenero, I.; Herranz, G.; et al. Somatic activating mutations in PIK3CA cause generalized lymphatic anomaly. J. Exp. Med. 2018, 216, 407–418. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Cheng, C.; Chen, K.; Wu, Y.; Wu, Z. Recent Progress in Lymphangioma. Front. Pediatr. 2021, 9, 735832. [Google Scholar] [CrossRef]
- Mirzaa, G.; Timms, A.E.; Conti, V.; Boyle, E.A.; Girisha, K.M.; Martin, B.; Kircher, M.; Olds, C.; Juusola, J.; Collins, S.; et al. PIK3CA-associated developmental disorders exhibit distinct classes of mutations with variable expression and tissue distribution. JCI Insight 2016, 1, e87623. [Google Scholar] [CrossRef] [Green Version]
- Barclay, S.F.; Inman, K.W.; Luks, V.L.; McIntyre, J.B.; Al-Ibraheemi, A.; Church, A.J.; Perez-Atayde, A.R.; Mangray, S.; Jeng, M.; Kreimer, S.R.; et al. A somatic activating NRAS variant associated with kaposiform lymphangiomatosis. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Sepehr, N.H.; McCarter, A.L.; Helaers, R.; Galant, C.; Boon, L.M.; Brouillard, P.; Vikkula, M.; Dellinger, M.T. KRAS-driven model of Gorham-Stout disease effectively treated with trametinib. JCI Insight 2021, 6, e149831. [Google Scholar]
- Manevitz-Mendelson, E.; Leichner, G.S.; Barel, O.; Davidi-Avrahami, I.; Ziv-Strasser, L.; Eyal, E.; Pessach, I.; Rimon, U.; Barzilai, A.; Hirshberg, A.; et al. Somatic NRAS mutation in patient with generalized lymphatic anomaly. Angiogenesis 2018, 21, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.B.; Li, D.; March, M.E.; Sheppard, S.E.; Adams, D.M.; Hakonarson, H.; Dori, Y. Kaposiform lymphangiomatosis effectively treated with MEK inhibition. EMBO Mol. Med. 2020, 12, e12324. [Google Scholar] [CrossRef]
- Li, D.; March, M.; Gutierrez-Uzquiza, A.; Kao, C.; Seiler, C.; Pinto, E.; Matsuoka, L.S.; Battig, M.R.; Bhoj, E.J.; Wenger, T.L.; et al. ARAF recurrent mutation causes central conducting lymphatic anomaly treatable with a MEK inhibitor. Nat. Med. 2019, 25, 1116–1122. [Google Scholar] [CrossRef]
- Martinez-Corral, I.; Zhang, Y.; Petkova, M.; Ortsäter, H.; Sjöberg, S.; Castillo, S.D.; Brouillard, P.; Libbrecht, L.; Saur, D.; Graupera, M.; et al. Blockade of VEGF-C signaling inhibits lymphatic malformations driven by oncogenic PIK3CA mutation. Nat. Commun. 2020, 11, 2869. [Google Scholar] [CrossRef]
- Li, D.; Wenger, T.L.; Seiler, C.; March, M.E.; Gutierrez-Uzquiza, A.; Kao, C.; Bhoj, E.; Tian, L.; Rosenbach, M.; Liu, Y.; et al. Pathogenic variant in EPHB4 results in central conducting lymphatic anomaly. Hum. Mol. Genet. 2018, 27, 3233–3245. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, A.; Ozeki, M.; Niihori, T.; Suzui, N.; Miyazaki, T.; Aoki, Y. A somatic activating KRAS variant identified in an affected lesion of a patient with Gorham–Stout disease. J. Hum. Genet. 2020, 65, 995–1001. [Google Scholar] [CrossRef]
- Blesinger, H.; Kaulfuß, S.; Aung, T.; Schwoch, S.; Prantl, L.; Rössler, J.; Wilting, J.; Becker, J. PIK3CA mutations are specifically localized to lymphatic endothelial cells of lymphatic malformations. PLoS ONE 2018, 13, e0200343. [Google Scholar] [CrossRef] [Green Version]
- Ozeki, M.; Aoki, Y.; Nozawa, A.; Yasue, S.; Endo, S.; Hori, Y.; Matsuoka, K.; Niihori, T.; Funayama, R.; Shirota, M.; et al. Detection of NRAS mutation in cell-free DNA biological fluids from patients with kaposiform lymphangiomatosis. Orphanet J. Rare Dis. 2019, 14, 215. [Google Scholar] [CrossRef]
- Glaser, K.; Dickie, P.; Dickie, B.H. Proliferative Cells from Kaposiform Lymphangiomatosis Lesions Resemble Mesenchyme Stem Cell–like Pericytes Defective in Vessel Formation. J. Pediatr. Hematol./Oncol. 2018, 40, e495–e504. [Google Scholar] [CrossRef]
- Byrne, A.B.; Brouillard, P.; Sutton, D.L.; Kazenwadel, J.; Montazaribarforoushi, S.; Secker, G.A.; Oszmiana, A.; Babic, M.; Betterman, K.L.; Brautigan, P.J.; et al. Pathogenic variants in MDFIC cause recessive central conducting lymphatic anomaly with lymphedema. Sci. Transl. Med. 2022, 14, eabm4869. [Google Scholar] [CrossRef]
- Liu, Y.; Zhong, D.-R.; Zhou, P.-R.; Lv, F.; Ma, D.-D.; Xia, W.-B.; Jiang, Y.; Wang, O.; Xing, X.-P.; Li, M. Gorham-Stout disease: Radiological, histological, and clinical features of 12 cases and review of literature. Clin. Rheumatol. 2014, 35, 813–823. [Google Scholar] [CrossRef]
- Gorham, L.W.; Stout, A.P. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone): Its relation to hemangiomatosis. J. Bone Jt. Surg. Am. Vol. 1955, 37-a, 985–1004. [Google Scholar] [CrossRef]
- Kato, H.; Ozeki, M.; Fukao, T.; Matsuo, M. MR imaging findings of vertebral involvement in Gorham-Stout disease, generalized lymphatic anomaly, and kaposiform lymphangiomatosis. Jpn. J. Radiol. 2017, 35, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Buonuomo, P.S.; Battafarano, G.; Conforti, A.; Mariani, E.; Algeri, M.; Del Fattore, A.; D’Agostini, M.; Macchiaiolo, M.; De Vito, R.; et al. Dissecting the mechanisms of bone loss in Gorham-Stout disease. Bone 2020, 130, 115068. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, M.; Fujino, A.; Matsuoka, K.; Nosaka, S.; Kuroda, T.; Fukao, T. Clinical Features and Prognosis of Generalized Lymphatic Anomaly, Kaposiform Lymphangiomatosis, and Gorham-Stout Disease. Pediatr Blood Cancer 2016, 63, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, M.T.; Garg, N.; Olsen, B.R. Viewpoints on vessels and vanishing bones in Gorham-Stout disease. Bone 2014, 63, 47–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tena-Sanabria, M.E.; Jesús-Mejenes, L.Y.; Fuentes-Herrera, G.; Álvarez-Martínez, F.A.; Victorio-García, N.P.; Núñez-Enríquez, J.C. A report of two children with Gorham-Stout disease. BMC Pediatr. 2019, 19, 206. [Google Scholar] [CrossRef] [Green Version]
- Florchinger, A.; Bottger, E.; Claass-Bottger, F.; Georgi, M.; Harms, J. Gorham-Stout syndrome of the spine. Case report and review of the literature. Rofo 1998, 168, 68–76. [Google Scholar] [PubMed]
- Perschbacher, S.E.; Perschbacher, K.A.; Pharoah, M.J.; Bradley, G.; Lee, L.; Yu, E. Gorham’s disease of the maxilla: A case report. Dentomaxillofac. Radiol. 2010, 39, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 2003, 17, 1263–1293. [Google Scholar] [CrossRef]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, O.A.; Kjellin, I.; Zuppan, C.W. Thoracic Lymphangiomatosis in a Child. J. Pediatric Hematol./Oncol. 2004, 26, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Yabuta, M.; Nozaki, T.; Fukuda, T.; Suzuki, K.; Kurihara, Y.; Niimi, Y. Generalized Lymphatic Anomaly Associated with Multiple Paraspinal Arteriovenous Malformations and Renal Artery Aneurysms. J. Vasc. Interv. Radiol. 2018, 29, 1633–1635.e1. [Google Scholar] [CrossRef]
- Ricci, K.W.; Iacobas, I. How we approach the diagnosis and management of complex lymphatic anomalies. Pediatr. Blood Cancer 2021, 2021, e28985. [Google Scholar] [CrossRef] [PubMed]
- Croteau, S.E.; Kozakewich, H.P.; Perez-Atayde, A.R.; Fishman, S.J.; Alomari, A.I.; Chaudry, G.; Mulliken, J.B.; Trenor, C.C., 3rd. Kaposiform Lymphangiomatosis: A Distinct Aggressive Lymphatic Anomaly. J. Pediatr. 2013, 164, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Le Cras, T.D.; Goines, J.; Lakes, N.; Pastura, P.; Hammill, A.M.; Adams, D.M.; Boscolo, E. Constitutively active PIK3CA mutations are expressed by lymphatic and vascular endothelial cells in capillary lymphatic venous malformation. Angiogenesis 2020, 23, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Le Cras, T.D.; Mobberley-Schuman, P.S.; Broering, M.; Fei, L.; Trenor, C.C., 3rd; Adams, D.M. Angiopoietins as serum biomarkers for lymphatic anomalies. Angiogenesis 2017, 20, 163–173. [Google Scholar] [CrossRef]
- Ozeki, M.; Nozawa, A.; Kawamoto, N.; Fujino, A.; Hirakawa, S.; Fukao, T. Potential biomarkers of kaposiform lymphangiomatosis. Pediatr. Blood Cancer 2019, 66, e27878. [Google Scholar] [CrossRef]
- Crane, J.; Manfredo, J.; Boscolo, E.; Coyan, M.; Takemoto, C.; Itkin, M.; Adams, D.M.; Le Cras, T.D. Kaposiform lymphangiomatosis treated with multimodal therapy improves coagulopathy and reduces blood angiopoietin-2 levels. Pediatr. Blood Cancer 2020, 67, e28529. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Nurmi, H.; Appak, S.; Sabine, A.; Bovay, E.; Korhonen, E.A.; Orsenigo, F.; Lohela, M.; D’Amico, G.; Holopainen, T.; et al. Angiopoietin 2 regulates the transformation and integrity of lymphatic endothelial cell junctions. Genes Dev. 2014, 28, 1592–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, K.W.; Hammill, A.M.; Mobberley-Schuman, P.; Nelson, S.C.; Blatt, J.; Bender, J.L.G.; McCuaig, C.C.; Synakiewicz, A.; Frieden, I.J.; Adams, D.M. Efficacy of systemic sirolimus in the treatment of generalized lymphatic anomaly and Gorham–Stout disease. Pediatr. Blood Cancer 2019, 66, e27614. [Google Scholar] [CrossRef] [PubMed]
- Trenor, C.C.; Chaudry, G. Complex lymphatic anomalies. Semin. Pediatr. Surg. 2014, 23, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Boscolo, E.; Coma, S.; Luks, V.L.; Greene, A.K.; Klagsbrun, M.; Warman, M.L.; Bischoff, J. AKT hyper-phosphorylation associated with PI3K mutations in lymphatic endothelial cells from a patient with lymphatic malformation. Angiogenesis 2014, 18, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Luks, V.L.; Kamitaki, N.; Vivero, M.P.; Uller, W.; Rab, R.; Bovee, J.V.; Rialon, K.L.; Guevara, C.J.; Alomari, A.I.; Greene, A.K.; et al. Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J. Pediatr. 2015, 166, 1048–1054.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, O.; Walker, J.; Seet, C.; Ikram, M.; Kuchta, A.; Arnold, A.; Hernández-Vásquez, M.; Frye, M.; Vizcay-Barrena, G.; Fleck, R.A.; et al. Mutations in EPHB4 cause human venous valve aplasia. JCI Insight 2021, 6, e140952. [Google Scholar] [CrossRef]
- Monroy, M.; McCarter, A.L.; Hominick, D.; Cassidy, N.; Dellinger, M.T. Lymphatics in bone arise from pre-existing lymphatics. Development 2020, 147, dev184291. [Google Scholar] [CrossRef]
- Cao, Y.; Zhou, Z.; de Crombrugghe, B.; Nakashima, K.; Guan, H.; Duan, X.; Jia, S.-F.; Kleinerman, E.S. Osterix, a transcription factor for osteoblast differentiation, mediates antitumor activity in murine osteosarcoma. Cancer Res. 2005, 65, 1124–1128. [Google Scholar] [CrossRef] [Green Version]
- Hominick, D.; Silva, A.; Khurana, N.; Liu, Y.; Dechow, P.C.; Feng, J.Q.; Pytowski, B.; Rutkowski, J.; Alitalo, K.; Dellinger, M.T. VEGF-C promotes the development of lymphatics in bone and bone loss. eLife 2018, 7, e34323. [Google Scholar] [CrossRef]
- Lala, S.; Mulliken, J.B.; Alomari, A.I.; Fishman, S.J.; Kozakewich, H.P.; Chaudry, G. Gorham-Stout disease and generalized lymphatic anomaly—Clinical, radiologic, and histologic differentiation. Skelet. Radiol. 2013, 42, 917–924. [Google Scholar] [CrossRef]
- Ozeki, M.; Fukao, T. Generalized Lymphatic Anomaly and Gorham–Stout Disease: Overview and Recent Insights. Adv. Wound Care 2019, 8, 230–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.; Rana, I.; Buonuomo, P.S.; Battafarano, G.; Mariani, E.; D’Agostini, M.; Porzio, O.; De Martino, V.; Minisola, S.; Macchiaiolo, M.; et al. Dysregulated miRNAs in bone cells of patients with Gorham-Stout disease. FASEB J. 2021, 35, e21424. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, A.E.; Ralston, S.H.; Marken, J.; Bell, C.; MacPherson, H.; Wallace, R.G.; Van Hul, W.; Whyte, M.P.; Nakatsuka, K.; Hovy, L.; et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat. Genet. 2000, 24, 45–48. [Google Scholar] [CrossRef]
- Otero, K.; Shinohara, M.; Zhao, H.; Cella, M.; Gilfillan, S.; Colucci, A.; Faccio, R.; Ross, F.P.; Teitelbaum, S.L.; Takayanagi, H.; et al. TREM2 and beta-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J. Immunol. 2012, 188, 2612–2621. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, H.; Zhou, X.; Li, X.; Sun, W.; Dellinger, M.; Boyce, B.F.; Xing, L. Lymphatic Endothelial Cells Produce M-CSF, Causing Massive Bone Loss in Mice. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2017, 32, 939–950, Erratum in J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2019, 34, 2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sironi, M.; Conti, A.; Bernasconi, S.; Fra, A.M.; Pasqualini, F.; Nebuloni, M.; Lauri, E.; De Bortoli, M.; Mantovani, A.; Dejana, E.; et al. Generation and characterization of a mouse lymphatic endothelial cell line. Cell Tissue Res. 2006, 325, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Illeez, O.G.; Ozkan, K.; Ozkan, F.U.; Bostan, A.B.; Akpinar, F.; Bilgic, B.; Aktas, I. Zoledronic acid: Treatment option for Gorham-Stout disease. Der Orthop. 2018, 47, 1028–1031. [Google Scholar] [CrossRef]
- Stevens, J.; Flower, H.; Patton, J.T. What does vanishing bone disease look like? BMJ Case Rep. 2018, 2018, bcr2017224061. [Google Scholar] [CrossRef] [Green Version]
- Heffez, L.; Doku, H.C.; Carter, B.L.; Feeney, J.E. Perspectives on massive osteolysis. Report of a case and review of the literature. Oral Surg. Oral Med. Oral Pathol. 1983, 55, 331–343. [Google Scholar] [CrossRef]
- Edwards, J.R.; Williams, K.; Kindblom, L.G.; Meis-Kindblom, J.M.; Hogendoorn, P.C.; Hughes, D.; Forsyth, R.G.; Jackson, D.; Athanasou, N.A. Lymphatics and bone. Hum. Pathol. 2008, 39, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Weivoda, M.M.; Ruan, M.; Hachfeld, C.M.; Pederson, L.; Howe, A.; Davey, R.A.; Zajac, J.D.; Kobayashi, Y.; Williams, B.O.; Westendorf, J.J.; et al. Wnt Signaling Inhibits Osteoclast Differentiation by Activating Canonical and Noncanonical cAMP/PKA Pathways. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2016, 31, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, A.P.; McAlinden, A. The role of microRNAs in bone development. Bone 2020, 143, 115760. [Google Scholar] [CrossRef] [PubMed]
- Hatano, Y.; Nakahama, K.-I.; Isobe, M.; Morita, I. Tumor associated osteoclast-like giant cells promote tumor growth and lymphangiogenesis by secreting vascular endothelial growth factor-C. Biochem. Biophys. Res. Commun. 2014, 446, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wu, Y.; Liang, Y.; Tan, W.; Liu, Z.; Xiao, J. Regulation of expression level of fms-like tyrosine kinase-4 is related to osteoclast differentiation. Arch. Med. Sci. 2016, 12, 502–506. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.S.; Chen, C.C.; Chen, L.L.; Chua, K.V.; Hung, H.C.; Hsu, J.T.; Huang, T.S. Extracellular HSP90α Induces MyD88-IRAK Complex-Associated IKKα/β-NF-κB/IRF3 and JAK2/TYK2-STAT-3 Signaling in Macrophages for Tumor-Promoting M2-Polarization. Cells 2022, 11, 229. [Google Scholar] [CrossRef]
- Kim, D.; Mebius, R.E.; MacMicking, J.D.; Jung, S.; Cupedo, T.; Castellanos, Y.; Rho, J.; Wong, B.R.; Josien, R.; Kim, N.; et al. Regulation of peripheral lymph node genesis by the tumor necrosis factor family member TRANCE. J. Exp. Med. 2000, 192, 1467–1478. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.-Y.; Yoshida, H.; Sarosi, I.; Tan, H.-L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-Dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef]
- Berendam, S.J.; Koeppel, A.F.; Godfrey, N.R.; Rouhani, S.J.; Woods, A.N.; Rodriguez, A.B.; Peske, J.D.; Cummings, K.L.; Turner, S.D.; Engelhard, V.H. Comparative Transcriptomic Analysis Identifies a Range of Immunologically Related Functional Elaborations of Lymph Node Associated Lymphatic and Blood Endothelial Cells. Front. Immunol. 2019, 10, 816. [Google Scholar] [CrossRef]
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.-R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption. Int. J. Mol. Sci. 2020, 21, 5169. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Chosa, N.; Sawada, S.; Kondo, H.; Yaegashi, T.; Ishisaki, A. VEGF-C and TGF-beta reciprocally regulate mesenchymal stem cell commitment to differentiation into lymphatic endothelial or osteoblastic phenotypes. Int. J. Mol. Med. 2016, 37, 1005–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, C.; Niess, H.; Huss, R.; Huber, S.; von Luettichau, I.; Nelson, P.J.; Ott, H.C.; Jauch, K.W.; Bruns, C.J. Multipotent mesenchymal stem cells acquire a lymphendothelial phenotype and enhance lymphatic regeneration in vivo. Circulation 2009, 119, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redlich, K.; Smolen, J.S. Inflammatory bone loss: Pathogenesis and therapeutic intervention. Nat. Rev. Drug Discov. 2012, 11, 234–250. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Yang, H.-Y.; Huang, S.-W.; Ou, G.; Hsu, Y.-F.; Hsu, M.-J. Interleukin-6 Induces Vascular Endothelial Growth Factor-C Expression via Src-FAK-STAT3 Signaling in Lymphatic Endothelial Cells. PLoS ONE 2016, 11, e0158839. [Google Scholar] [CrossRef]
- Geleff, S.; Schoppmann, S.F.; Oberhuber, G. Increase in podoplanin-expressing intestinal lymphatic vessels in inflammatory bowel disease. Virchows Arch. 2003, 442, 231–237. [Google Scholar] [CrossRef]
- Xu, H.; Edwards, J.; Banerji, S.; Prevo, R.; Jackson, D.G.; Athanasou, N.A. Distribution of lymphatic vessels in normal and arthritic human synovial tissues. Ann. Rheum. Dis. 2003, 62, 1227–1229. [Google Scholar] [CrossRef] [Green Version]
- Kunstfeld, R.; Hirakawa, S.; Hong, Y.-K.; Schacht, V.; Lange-Asschenfeldt, B.; Velasco, P.; Lin, C.; Fiebiger, E.; Wei, X.; Wu, Y.; et al. Induction of cutaneous delayed-type hypersensitivity reactions in VEGF-A transgenic mice results in chronic skin inflammation associated with persistent lymphatic hyperplasia. Blood 2004, 104, 1048–1057. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Lu, Y.; Proulx, S.T.; Guo, R.; Yao, Z.; Schwarz, E.M.; Boyce, B.F.; Xing, L. Increased lymphangiogenesis in joints of mice with inflammatory arthritis. Arthritis Res. Ther. 2007, 9, R118. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhou, X.; Huang, D.; Ji, Y.; Kang, F. IL-6 Enhances Osteocyte-Mediated Osteoclastogenesis by Promoting JAK2 and RANKL Activity In Vitro. Cell. Physiol. Biochem. 2017, 41, 1360–1369. [Google Scholar] [CrossRef]
- Tamura, T.; Udagawa, N.; Takahashi, N.; Miyaura, C.; Tanaka, S.; Yamada, Y.; Koishihara, Y.; Ohsugi, Y.; Kumaki, K.; Taga, T.; et al. Soluble interleukin-6 receptor triggers osteoclast formation by interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 11924–11928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewiecki, E.M. Role of sclerostin in bone and cartilage and its potential as a therapeutic target in bone diseases. Ther. Adv. Musculoskelet. Dis. 2013, 6, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bélanger, L.F. Osteocytic osteolysis. Calcif. Tissue Res. 1969, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tsourdi, E.; Jähn, K.; Rauner, M.; Busse, B.; Bonewald, L.F. Physiological and pathological osteocytic osteolysis. J. Musculoskelet. Neuronal Interact. 2018, 18, 292–303. [Google Scholar] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solorzano, E.; Alejo, A.L.; Ball, H.C.; Magoline, J.; Khalil, Y.; Kelly, M.; Safadi, F.F. Osteopathy in Complex Lymphatic Anomalies. Int. J. Mol. Sci. 2022, 23, 8258. https://doi.org/10.3390/ijms23158258
Solorzano E, Alejo AL, Ball HC, Magoline J, Khalil Y, Kelly M, Safadi FF. Osteopathy in Complex Lymphatic Anomalies. International Journal of Molecular Sciences. 2022; 23(15):8258. https://doi.org/10.3390/ijms23158258
Chicago/Turabian StyleSolorzano, Ernesto, Andrew L. Alejo, Hope C. Ball, Joseph Magoline, Yusuf Khalil, Michael Kelly, and Fayez F. Safadi. 2022. "Osteopathy in Complex Lymphatic Anomalies" International Journal of Molecular Sciences 23, no. 15: 8258. https://doi.org/10.3390/ijms23158258
APA StyleSolorzano, E., Alejo, A. L., Ball, H. C., Magoline, J., Khalil, Y., Kelly, M., & Safadi, F. F. (2022). Osteopathy in Complex Lymphatic Anomalies. International Journal of Molecular Sciences, 23(15), 8258. https://doi.org/10.3390/ijms23158258